
Abstract
Purpose
Patients with anaplastic thyroid cancer (ATC) have a very poor prognosis. Immunotherapy is a potential treatment, while the current outcome is limited which may be due to the complicated tumor microenvironment (TME). Tumor associated macrophages (TAMs) is the most abundant cell in the TME of ATC. We aimed to clarify the novel indicators based on TAM in ATC.
Methods
Transcriptome files were downloaded from the Gene Expression Omnibus (GEO) dataset. Weighted gene co-expression network analysis, cox regression, support vector machine, and random forest were utilized to identify TAM-related prognostic genes. Consensus clustering and principal component analysis were performed for integrated analysis. Moreover, external validation (Fudan University Shanghai Cancer Center cohort) was conducted in 23 ATC samples via immunohistochemistry.
Results
ATC patients with an abundance of TAMs had a poorer prognosis. Four TAM related genes (FZD6, RBBP8, PREX1, HSD3B7) were identified and a TAM-related prognostic index (TAMRPI) was constructed with high area under the curve (AUC). Next, high TAMRPI was related to the higher level of TAM infiltration and upregulation of several pathways, such as E2F targets, IL6-JAK-STAT3, and G2M checkpoint. Immune checkpoint TIM-3 and CSF1R were positively associated with TAMRPI, and dysfunction of T cells was increased in high TAMRPI subset. Moreover, in the external validation of protein level, strong expression of TAM related genes was related to poorer prognosis, which was further supported by time-dependent AUC analysis.
Conclusion
TAM is negatively correlated to the prognosis of ATC. FZD6, RBBP8, PREX1, and HSD3B7 are potential biomarkers of ATC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although it accounts for just 1-2 percent of all thyroid cancers, anaplastic thyroid carcinoma (ATC) is one of the most aggressive cancers, with a median overall survival of 3–6 months [1]. For inoperable and metastatic ATCs, tumor debulking surgery before or after local radiation and systemic therapy, is the most common therapy [2]. Recently, immune checkpoint inhibitor (ICI) treatment has been shown to be effective and has even resulted in long-term remission in cancer patients [3, 4]. However, a major limitation is the low response rate of patients to ICI therapy, which may be explained by the complicated interaction of multiple cells in the tumor microenvironment (TME) [5]. Notably, tumor associated macrophages (TAM) is found at an increased level in more aggressive ATC compared with poorly differentiated thyroid cancer (PDTC) and papillary thyroid cancer (PTC) and commonly constitute more than 50% of the tumor mass in ATC [6]. TAM plays an extremely important role in the biology of malignant neoplasms [7], and generally can be divided into two main subtypes: M1 macrophages, displaying “classic” activation, and M2 macrophages, which are narrowly defined TAMs committed to immunosuppression [8]. Unfortunately, effective prognostic and therapeutic predictors of TME still remains unclear. We believe indicators based on TAM infiltration would be helpful. In the present study, we analyzed public mRNA expression profiles and identified TAM-related prognostic gene signatures to distinguish activation and suppression of the immune status in ATC patients.
Materials & methods
Data procession
GSE76039, GSE29265, GSE65144 and GSE33630 are microarray datasets of bulk tumors and were downloaded from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). To obtain a pooled GEO cohort, batch effect was removed among different microarray datasets. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013).
Estimation of the abundance of TAMs
The expression data of ATC patients were uploaded to CIBERSORT (https://cibersort.stanford.edu/) to estimate the abundance of 22 types of immune cells [9]. Furthermore, the quantified enrichment scores of the involved cell types in each ATC sample were obtained by XCell (https://xcell.ucsf.edu/) [10].
Weighted Gene Co-Expression Network Analysis (WGCNA)
WGCNA is a useful algorithm to find modules of highly correlated genes and to explore the significant correlation between modules and external sample traits. In the present study, a scale-free co-expression network was constructed for ATC. A power of β = 5 and a scale-free R2 = 0.90 were selected as soft-threshold parameters. Eleven nongray modules were identified by setting the merging threshold function at 0.25. The module eigengenes (MEs) were defined as the principal component of the gene expression matrix of each corresponding module. The significant module was identified from the correlation between MEs and tumor microenvironment (TME) cell abundance.
Identification of TAM-related prognostic genes
Univariate Cox regression was performed with all genes in the 3 modules depicting the significant correlation TME to find TAM-related prognostic genes. Furthermore, 150 candidate TAM-related prognostic gene features were screened via the support vector machine recursive feature elimination (SVM-RFE) (linear kernel) and random forest recursive feature elimination (RF-RFE) algorithm based on leave-one-out cross-validation. SVM-RFE and RF-RFE were two popular supervised embedded approach and can be performed to rank variables associated with the TAM density in ATC.
Molecular subtype identification
The K-means algorithm is an unsupervised, nonparametric and nonlinear method of clustering. K-means-based consensus clustering was conducted utilizing the combined GEO transcriptome data of 13 TAM-related gene signatures. The consensus matrix plots were illustrated based on the k-value. Cumulative distribution function (CDF) plots revealed the consensus distributions for each k. In the CDF curve of a consensus matrix, the lower left portion represents sample pairs rarely clustered together, the upper right portion represents those almost always clustered together, and the middle segment represents those with ambiguous assignments in different clustering runs. The k-value with a flatter middle segment has a lower rate of discordant assignments across permuted clustering runs, which would be prior and appropriate to be the selected one.
Differential gene expression analysis and gene set enrichment analysis (GSEA)
The differentially expressed genes (DEGs) between two ATC clusters were identified when the absolute log2-fold change was greater than 1 and the false discovery rate (FDR) was less than 0.05. Moreover, to observe the effect of gene expression on tumors, the enrichment of HALLMARK pathways in the one over two cluster was analyzed using GSEA.
TAM-related prognostic index (TAMRPI) construction
Principal component analysis (PCA) is an often-applied strategy of reducing dimensionality for high-dimensional data. In this analysis, PCA was used to calculate a TAM-related prognostic index (TAMRPI) indicating TAM cell infiltration. TAMRPI was derived from the first principal component of top four genes from the overlapped features by SVM-RFE and RF-RFE.
Clinical samples and immunohistochemistry (IHC)
Samples from 23 patients with anaplastic thyroid carcinoma, who underwent thyroidectomy from August 2006 to February 2019 at Fudan University Shanghai Cancer Center (FUSCC), were collected and immunohistochemistry was performed. This study was approved by the Ethics Committee of the FUSCC (050432-4-1911D), and informed consent was provided for each patient. Briefly, deparaffinization of histological sample slides were performed by xylene and different concentrations of ethanol and washed with phosphate buffer saline three times. Then the slides were incubated with 3% hydrogen peroxidase at room temperature for 20 min and 5% goat serum was applied to block the nonspecific binding of antibody. Immunohistochemical staining was performed at 4 °C overnight using a monoclonal antibody directed against FZD6 (37296, 1:200, Signalway Antibody, Maryland, USA), HSD3B7 (36537, 1:100, Signalway Antibody, Maryland, USA), PREX1 (75706, 1:100, Cell Signaling Technology, MA, USA), RBBP8 (25120, 1:200, Signalway Antibody, Maryland, USA). Following day, the slides washing was done with PBS for three times and were probed with secondary antibody for 2 h at 37 °C. Subsequently, slides were stained with DAB and hematoxylin, and were dehydrated and mounted. Staining intensity was classified as follows: negative (0), weak (1), moderate (2), or strong (3). The percentage of positive tumor cells was also recorded at the same time: <5% (0), 5–30% (1), 30–50% (2), ≥50% (3). The final immunoreactivity score of each sample was calculated by multiplying intensity and proportion scores, ranged from 0 to 9. The average scores were calculated using 5–7 cores from each sample, with outliers excluded.
Cell culture and treatment
Three human ATC cell lines including BHT-101, C643, and CAL-62, obtained from the cell bank at the Chinese Academy of Sciences (Shanghai, People’s Republic of China) were used, and one normal human thyroid epithelial cell line Nthy-ori 3-1, were purchased from Sigma and used as the normal control of gene expression compared to other ATC cell lines. C643 and Nthy-ori 3-1 were cultured in RPMI-1640 medium (Gibco, USA) while BHT-101 and CAL-62 were cultured in DMEM (Gibico, USA). All the medium was supplemented with 10% fetal bovine serum (FBS; Gibco, USA), 1% penicillin/streptomycin (Gibco, USA), and 2 mM Glutamine (Gibco, USA) (Nthy-ori 3-1 only). All the cell lines were routinely cultured at 37 °C with 5% CO2.
In order to silence RBBP8 gene, BHT-101 and C643 cells were transfected with RBBP8-specific siRNA (si-RBBP8; Ribobio, China) using Lipofectamine RNAiMAX (Thermo Fisher Scientific, USA). For overexpression, the RBBP8 sequence was synthesized and subcloned into a GV492 vector (GENECHEM Co., Ltd., Shanghai, China). Transfection was performed according to the manufacturer’s protocol and the efficiency was confirmed by western blot.
Cell proliferation and migration assay
Cell proliferation was detected using a cell-counting kit-8 (CCK-8) assay according to the manufacturer’s protocol. Briefly, at the indicated time point, 10 µL of MTT solution (5 mg/mL) was added into per well of 96-well dishes and the plate was incubated for 1 h at 37 °C. Then, the absorbance at 450 nm was assessed using a spectrophotometer.
For migration assay, ATC cells were re-suspended in serum-free media and seeded on the top of the membranes (CAL62: 3 × 105/mL; BHT-101:2 × 105/mL; C643 knockdown: 3 × 105/mL, C643 overexpression: 1 × 105/mL) in 24-transwell inserts (8 µm pore size, BD Biosciences). These inserts were placed in wells containing 10% FBS media. After incubation, cells which did not pass the pores were removed. Then, the filters were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet, where cells on the lower surface of the membrane were scored. Each experiment was performed in triplicate.
Western blot
Protein lysates were obtained from cultured cells with RIPA buffer. Approximately 15 μg protein lysates were extracted from each sample, separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After 1 h blocking in 5% bovine serum albumin (BSA, Sigma, USA), the interested protein was probed with primary antibody against human RBBP8(D76F7, 1:1000 dilution, Cell signaling technology, USA) and β-Tubulin (66240, 1:5000, Proteintech, USA) at 4 °C overnight and then incubated with goat anti-rabbit IgG or goat anti-mouse IgG (1:5000 dilution for both; Proteintech, USA) at room temperature for 1 h and visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore Sigma, United States) and detected by the Alpha Imager (Alpha Innotech, San Leandro, CA, United States). The protein expression level was quantified and normalized to β-Tubulin protein expression by densitometry using Image J.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cells using TRIzol Reagent (Invitrogen, USA) according to manufacturer’s instructions. Next, a total of 1 μg of RNA was reverse transcribed using a PrimeScript RT reagent Kit (Takara, Dalian, China). For qRT-PCR, cDNA was amplified using SYBR Green Premix Ex Taq (Takara, Dalian, China) following the manufacturer’s instructions. For normalization, β-Actin was introduced as an internal reference. The relative expression of interested genes was calculated by the 2−ΔΔCT formula.
Statistical analysis
For quantitative data, the statistical significance of comparisons between two groups was estimated by the Wilcoxon rank-sum test. Univariate survival analysis was performed by univariate Cox regression. The overall survival (OS) curves were drawn by the Kaplan-Meier method and compared using log-rank tests. All comparisons were two-sided with an alpha level of 0.05. All statistical analyses were performed with R software (version 4.0.3). In the figures, statistical significance is represented as asterisks (ns: not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Results
Proportion of tumor-infiltrating immune cells in thyroid cancer
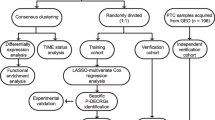
A schematic diagram of bioinformatic analysis in this study is presented (Fig. 1). The transcriptome data of 52 ATCs and 81 normal thyroid tissues (NT) from GEO genomic data sets were analyzed to explore the abundance of immune cell subpopulations in each sample (Fig. 2A). The immune cell characteristics of the TME were evaluated between ATC and NT (Fig. 2B). We found that macrophages were more abundant in tumor tissues, whereas CD8 T cells, CD4 T cells, and NK cells were less enriched. Macrophages were the most abundant cells in ATCs, accounting for approximately 50% of cells in the TME. Furthermore, ATC patients with a higher enrichment of M2 macrophages had a worse survival (Fig. 2C).

Schematic diagram of the informatic study. TAM density and its association with survival were analyzed based on public data we collected. Then weighted gene co-expression network (WGCNA), along with univariate Cos regression and two kinds of recursive feature elimination (RFE), were used to further find TAM related genes. Then, molecular subtypes and prognostic index were analyzed subsequently

Immune landscape in different thyroid cancers. A Proportions of tumor microenvironment (TME) cells for patients with anaplastic thyroid carcinoma (ATC) and normal tissue (NT). B The proportions of TME cells in ATC and NT. The scattered dots represent the immune scores of the two subgroups (CIBERSORTx). The thick lines represent the median value. The bottom and top of the boxes are the 25th and 75th percentiles (interquartile range), respectively. Significant differences between the four groups were assessed (ns: not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). C Kaplan-Meier plot of the M2 score groups
TAM-related gene identification
To further understand the potential roles of TAMs in ATC, WGCNA was performed to construct a scale-free co-expression network, where a power of β = 5 was selected as the soft-threshold parameter (Fig. 3A). A total of 12 modules were identified with average linkage hierarchical clustering (Fig. 3B). Three modules were chosen out of 11 nongray modules according to their significant correlation to the M2 macrophage scores (p < 0.05), among which the yellow module had the highest association (r = 0.53, p = 6e-5) (Fig. 3C). Module membership was used to calculate the correlation between gene expression profiles and module eigenvalues, and the gene significance represented the association of individual genes with M2 macrophage scores. The scatter diagram showed a positive correlation between gene significance and module membership in the module (Fig. 3D), indicating that these modules were correlated with M2 macrophages. Genes in the M2 macrophage-related modules were filtered out by univariate Cox regression analysis with a p value less than 0.05. A total of 150 candidate genes were identified and subjected to SVM-RFE and RF-RFE to investigate the most important genes associated with M2 macrophage scores through leave-one-out cross-validation. Finally, top four were extracted as TAM-related prognostic parameters (Fig. 3E, Supplementary Table 1).

Identification of modules significantly correlated to TAM infiltration. A Analysis of the scale-free network coefficient R2 for the soft threshold (β) and the mean connectivity for the soft threshold. A power of β = 5 and a scale-free R2 = 0.90 were selected as soft-threshold parameters. B A cluster dendrogram was built based on the dissimilarity of the topological overlap, which presents 15 network modules. C Heatmap demonstrating the correlation between module eigengenes and different subtypes of macrophages. Three modules (yellow, brown, turquoise) depict significant correlations with M2 macrophage abundance. D The gene significance and module membership of the genes in the modules exhibited positive correlations. E Importance measures for 150 TAM-related genes using recursive feature elimination based on support vector machine and random forest
Integrated analysis of molecular subtype
Unsupervised and nonparametric consensus clustering was applied to classify ATCs based on the four TAM-related prognostic gene signatures. The CDF plot shows the consensus matrix for each k, from 2 to 6, which is a quantification of how the consensus matrices are distributed within a range from 0 to 1 (Fig. 4A). We chose k = 2 as the best-suited parameter due to its optimal classification ability and relatively low cluster number (Fig. 4B). Subsequently, M2 macrophage infiltration of two clusters were shown and the cluster one had a higher TAM infiltration than the cluster two (Fig. 4C). Differentially expressed gene analysis was conducted, and the combined GEO cohort was divided into two different subgroups (adjusted p value < 0.05, log2 (FC) > 1). A total of 254 DEGs were identified, of which 31 genes were upregulated and 223 genes were downregulated. The heatmap indicated the DEGs between the two clusters (Fig. 4D). The volcano plot revealed several top upregulated and downregulated genes in the cluster one over two (Fig. 4E). Enrichment analysis was carried out via hallmark gene sets. Upregulated pathways included E2F targets, IL6-JAK-STAT3 signaling, G2M checkpoint, MYC targets V2, and TNFA signaling via NFKB, which indicated the positive activation of tumor cell proliferation, invasion, and metastasis. In the meanwhile, the downregulated pathways were related to KRAS signaling, p53 pathway, protein secretion, and TGF-β signaling (Fig. 4F).

Molecular subtype analysis. A Cumulative distribution function plot showing the cumulative distribution functions of the consensus matrix for each k (from 2 to 6). B Consensus matrix (k = 2). C M2 abundance in different clusters compared through the Wilcoxon test (***p < 0.001). D The differentially expressed genes between two molecular subtypes (cluster 1 versus cluster 2). E Volcano plot for differentially expressed genes. Red dots represent upregulated genes, and blue dots represent downregulated genes. F Upregulated Hallmark pathways in gene set enrichment analysis (GSEA) (top). Downregulated Hallmark pathways in GSEA (bottom)
TAMRPI construction based on PCA
Due to the limited sample number, multivariate Cox regression was not appropriate for the fitting scores of the TAM-related gene signatures. We utilized the PCA algorithm to reduce the variable dimension. TAMRPI was calculated based on the 4 gene expression levels for each ATC patient using the PCA algorithm (Fig. 5A). Patients with high TAMRPI had a worse prognosis than those within the low TAMRPI group in the GSE76039 set (p < 0.05) (Fig. 5B), where a significantly different infiltration of M2 macrophages was found (Fig. 5C). In the GSE76039 set, the AUCs of the TAMRPI for 3-, 6-, 9-, and 12-months survival prediction were 0.806, 0.700, 0.783, and 0.938, respectively (Fig. 5D). In the combined set (GSE76039, GSE29265, GSE65144 and GSE33630), there was an abundant proportion of myeloid cells in the high TAMRPI group, such as mast cells, macrophages, monocytes, neutrophiles (Fig. 5E).

Construction and association analysis of prognostic index. A PCA of the four TAM related genes. B Kaplan-Meier survival analysis of the tumor associated macrophage related prognostic index (TAMRPI) subgroups (GSE76039). C M2-TAM abundance in different TAMRPI subgroups (GSE76039). D The AUCs of the TAMRPI for 3-, 6-, 9-, and 12-months survival prediction were 0.806, 0.700, 0.783, and 0.938, respectively. E Heatmap of enrichment scores (XCell) of tumor microenvironment (TME) cells in two TAMRPI subgroups. F Pearson correlation matrix of TAMRPI and the expression of immune checkpoints. Red, blue, and ‘X’ represented positive, negative, and insignificant correlations, respectively. G T cell dysfunction and exclusion score were compared between TAMRPI subgroups. Wilcoxon test was used for the analysis (ns: not significant, *p < 0.05, **p < 0.01)
Correlation between TAMRPI and immune profile
The relationships between immune checkpoint genes (ICGs) and each TAM-related prognostic gene as well as TAMRPI are visualized in the correlation matrix (Pearson test). PD-1 had a positive association with TAMRPI, suggesting that higher TAM abundance in the TME of ATC was associated with higher transcriptome levels of PD-1. However, PD-L1 and PD-L2 had no significant correlation with TAMRPI. Furthermore, B7-H3, TIM-3, CD47, and CSF1R were statistically associated with TAMRPI (Fig. 5E). Additionally, dysfunction and exclusion score of T cell were calculated online (http://tide.dfci.harvard.edu/). The TAMRPI-high subgroup had a higher T cell dysfunction score, while there was no difference in T cell exclusion between the two subgroups (Fig. 5F, G).
External validation of the prognostic prediction of four TAM related genes
In order to further validate the protein level expression of FZD6, HSD3B7, and PREX1, as well as their predictive significance of survival in ATC, IHC was conducted utilizing TMAs from 23 patients with ATC. Although there were no significant differences, it was found that FZD6, HSD3B7, PREX1, and RBBP8 indicated a shorter OS in ATC patients (Fig. 6A–D). However, additional co-expression analysis revealed that poorer prognosis was associated with increased co-expression of these four genes, with a p value of 0.01 among the entire three groups (Fig. 6E) and p values of 0.088 and 0.0059 in the comparison of single positive subset and co-positive subset against all-negative subset, respectively (data was not shown in K-M plot). Furthermore, RBBP8, FZD6, and PREX1 demonstrated good predictive abilities of ATC prognosis in both the short and long term, whereas HSD3B7 demonstrated a low AUC value (1-year AUC: RBBP8 = 0.706, FZD6 = 0.626, PREX1 = 0.728, HSD3B7 = 0.415; 3-years AUC: RBBP8 = 0.671, FZD6 = 0.803, PREX1 = 0.708, HSD3B7 = 0.550). AUC values were low in age, tumor size (cm), and TNM stages. Basic information of ATC patients was shown in Supplementary Table 2.

External validation of collected genes in protein level and their clinical significance. A Typical immunohistochemical staining of positive expression of FZD6, B HSD3B7, C RBBP8, D PREX1 in ATC (400X), followed by Kaplan-Meier plot of overall survival (OS) in the below panel. E Kaplan-Meier plot of OS stratified by the co-expression status of FZD6, HSD3B7, RBBP8, and PREX1 of patients with anaplastic thyroid carcinoma (ATC). The AUCs of age, TNM stage (IVA, IVB, and IVC), and four TAM related biomarkers for one year survival prediction. F The time-dependent AUCs of age, tumor size (cm), TNM stage (IVA, IVB, and IVC), and four TAM related biomarkers for overall survival prediction
RBBP8 promotes the proliferation and migration of ATC cells
According to GEO data, of the four TAM-related prognostic genes, FZD6 and RBBP8 were expressed at higher levels than HSD3B7 and PREX1, however FZD6 had no significant difference in transcriptome level between ATC malignant and para-cancerous tissues (Supplementary Fig. 1). It was shown that RBBP8 had a good predictive value in the FUSCC ATC cohort (Fig. 6F). Furthermore, when the three ATC cell lines were compared to normal follicular epithelial cell lines, it was discovered that RBBP8 expression was greatly enhanced in ATC cells (Supplementary Fig. 2), thus we concentrated on continuing functional experiments on the RBBP8 gene.
As shown in Fig. 7, after knocking down RBBP8 in BHT-101 and C643 in vitro (confirmed by western blot), both proliferation and migration of these two ATC cell lines were significantly inhibited compared to control cells. On the contrary, overexpression of RBBP8 promoted the proliferation and migration in CAL62 and C643, respectively. This outcome suggested the ability of RBBP8 to promote tumorigenesis in ATC, which was in line with the previous result that RBBP8 was acting as a risk factor in the FUSCC validated cohort (Fig. 6C).

Bio-functional investigation of RBBP8 in vitro. A Knockdown (left) and overexpression (right) of RBBP8 were confirmed by western blot. B Knockdown of RBBP8 significantly inhibited proliferation (left panel) and migration (right panel) in BHT-101 and C643 cells. C Overexpression of RBBP8 significantly promoted proliferation (left panel) and migration (right panel) in C643 and CAL62 cells. The left panel shows representative images and quantitative analysis of migration were shown representatively. NC Negative control. Statistically significant differences are indicated: *p < 0.05; **p < 0.01; ***p < 0.001
Discussion
Anaplastic thyroid cancer is a rare but lethal malignancy with a median overall survival of 3–6 months [11,12,13]. In decades, there has been no efficient and safe way to overcome this problem. Traditional surgery combined with chemoradiotherapy has only slight benefits and cannot prolong survival. Cancer cells have the ability to adapt and circumvent the immune system, which has been found to be a hallmark of cancer [14]. To date, immunotherapy provides a potential and alternative treatment choice for patients with ATC. However, the overall response is still uncertain. Therefore, identification of potential prognostic biomarkers correlated to treatment benefit can allow individualization of immunotherapy for ATC patients.
TAMs, a critical component in the TME, have an important role in cancer development, angiogenesis, metastasis, drug resistance, and antitumor immunosuppression [7, 15]. There are two main types of activation and polarization states in mammals: M1 and M2 [16]. M2 macrophages are an anti-inflammatory subtype of macrophages and have been noticed to be abundant in many cancer tissues [17,18,19]. This kind of cell has been extensively studied and has been shown to play a key role in the subversion of adaptive immunity and in inflammatory circuits that promote tumor growth and progression [20,21,22,23]. Consistent with previous findings, our results also showed an enrichment of M2 macrophages in ATCs, which indicated a poorer survival [24].
In this study, we identified M2-like TAM-related prognostic genes by WGCNA, Cox regression, SVM-RFE, and RF-RFE. Next, differentially expressed genes were observed in high M2 infiltrating subgroup over low M2 enrichment subgroup classified by clustering algorithm. For example, we found that COLEC12 was upregulated in M2 enrichment group (Fig. 4E). One study reported that COLEC12 was related to the progression from well differentiated thyroid carcinoma to ATC [25]. GSEA also showed a possible activation of cell proliferation and invasion in high M2 group. Subsequently, TAMRPI was further calculated based on the PCA algorithm, and poor overall survival outcomes were found in the high TAMRPI group. Correlation analysis was performed between TAMRPI and biotargets of immunotherapy. ATC patients with a high TAMRPI, indicating an abundance of M2 macrophages and poor prognosis, would have overexpression of a series of immune checkpoints, including PD-1, B7-H3, TIM-3, CSF1R. Moreover, in the protein level, immunostaining suggested one or more positive-expressed features correlated to poor prognosis. Thus, ATC patients within TAMRPI-high group, especially expressing PD-1, B7-H3, TIM-3, or CSF1R, may benefit more from the targeted ICI.
ICI therapy has generated considerable interest within solid tumors. Blockades of PD-1 and PD-L1 improve T cell exhaustion. Histopathologic studies have shown that PD-L1 and PD-1 proteins are upregulated in ATC tumors and tumor infiltrating lymphocytes, demonstrating a potential chance for immune checkpoint therapy in ATC [26]. However, the clinical outcome of PD-1 was limited. A clinical study using spartalizumab reported a 17% overall response rate in ATC patients. Integrated analysis of the immune profile is required to better understand the immune treatment response in ATC. Recently, a more complex classification of the cancer immune contexture was proposed, including four major immune coordination profiles: hot, altered–immunosuppressed, altered–excluded, and cold [27]. The study conducted by Giannini et al suggested that ATC has two main subtypes, hot (34%) and altered–immunosuppressed tumors (50%) [28]. This result indicates that ATC patients with different subtypes of TMEs would have totally diverse outcomes of ICI treatments. In addition to T cell infiltration, M2-like TAM accounts for more than 50% of nucleated cells in the TME of ATC, and this group of cells might play an even more important role than T cells do in ATC [29]. Currently, there are some novel immune checkpoints of TAMs under exploration and validation, both experimentally and clinically. For example, CD47 is an immunoglobulin family member protein overexpressed on the surface of many cancer types and works as a “don’t eat me” signal by combining its ligand (SIRPα) in macrophages, resulting in tumor immune escape [30, 31]. A study validated that the application of an anti-CD47 antibody enhances tumor phagocytosis in ATCs both in vitro and in vivo [19].
Conclusions
Conclusively, TAM-related prognostic genes were found to predict ATC patient outcomes. TAMRPI was established and might be helpful for classifying immune activation and immunosuppression in ATCs, demonstrating that it might be a potential prognostic indicator of ICI treatment. Nevertheless, further studies are required to validate this point and explore the underlying molecular mechanisms.
References
G. Nagaiah, A. Hossain, C.J. Mooney, J. Parmentier, S.C. Remick, Anaplastic thyroid cancer: A review of epidemiology, pathogenesis, and treatment. J. Oncol. 2011, 542358 (2011)
R.I. Haddad, W.M. Lydiatt, D.W. Ball, N.L. Busaidy, D. Byrd, G. Callender, P. Dickson, Q.Y. Duh, H. Ehya, M. Haymart, C. Hoh, J.P. Hunt, A. Iagaru, F. Kandeel, P. Kopp, D.M. Lamonica, J.C. McCaffrey, J.F. Moley, L. Parks, C.D. Raeburn, J.A. Ridge, M.D. Ringel, R.P. Scheri, J.P. Shah, R.C. Smallridge, C. Sturgeon, T.N. Wang, L.J. Wirth, K.G. Hoffmann, M. Hughes, Anaplastic Thyroid Carcinoma, Version 2.2015. J. Natl Compr. Canc Netw. 13, 1140–1150 (2015)
P. D.M., The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012)
C.G. Drake, E.J. Lipson, J.R. Brahmer, Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 11, 24–37 (2014)
M. Nishino, N.H. Ramaiya, H. Hatabu, F.S. Hodi, Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 14, 655–668 (2017)
M. Ryder, R.A. Ghossein, J.C.M. Ricarte-Filho, J.A. Knauf, J.A. Fagin, Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr.-Relat. Cancer 15, 1069–1074 (2008)
A. Mantovani, A. Sica, Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr. Opin. Immunol. 22, 231–237 (2010)
S. Gordon, Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35 (2003)
A.M. Newman, C.L. Liu, M.R. Green, A.J. Gentles, W. Feng, Y. Xu, C.D. Hoang, M. Diehn, A.A. Alizadeh, Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015)
D. Aran, Z. Hu, A.J. Butte, xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220 (2017)
L. Zhang, Z. Ren, Z. Su, Y. Liu, T. Yang, M. Cao, Y. Jiang, Y. Tang, H. Chen, W. Zhang, R. Gong, T. Wei, Y. Peng, B. Liu, W. Zhang, L. Yang, Y. Hu, Z. Li, J. Zhu, H. Xu, Y. Shu, H. Luo, Novel recurrent altered genes in Chinese patients with anaplastic thyroid cancer. J. Clin. Endocrinol. Metabol. 106, 988–998 (2021)
I. Abe, A.K. Lam, Anaplastic thyroid carcinoma: Updates on WHO classification, clinicopathological features and staging. Histol Histopathol. 36, 239–248 (2020)
A.M. Mirabile, L. Biafora, G. Giordano, G. Arrigoni, M.G. Cangi, I. Dell'Oca, D. Lira Luce, D. Di Santo, A. Galli, M. Tulli, R. Mellone, D. Valsecchi, V. Gregorc, M. Bussi, Uncommon Site of Metastasis and Prolonged Survival in Patients with Anaplastic Thyroid Carcinoma: A Systematic Review of the Literature. Cancers (Basel) 12, 2585 (2020)
D. Hanahan, A. Robert, Weinberg, hallmarks of cancer: The next generation. Cell 144, 646–674 (2011)
D. Chen, X. Zhang, Z. Li, B. Zhu, Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics 11, 1016–1030 (2021)
P.J. Murray, Macrophage polarization. Annu Rev. Physiol. 79, 541–566 (2017)
X.-T. Fu, Z. Dai, K. Song, Z.-J. Zhang, Z.-J. Zhou, S.-L. Zhou, Y.-M. Zhao, Y.-S. Xiao, Q.-M. Sun, Z.-B. Ding, J. Fan, Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J. Oncol. 46, 587–596 (2015)
M.L. Pinto, E. Rios, C. Durães, R. Ribeiro, J.C. Machado, A. Mantovani, M.A. Barbosa, F. Carneiro, M.J. Oliveira, The Two Faces of Tumor-Associated Macrophages and Their Clinical Significance in Colorectal Cancer. Front Immunol. 10, 1875 (2019)
C.M. Schürch, M.A. Roelli, S. Forster, M.-H. Wasmer, F. Brühl, R.S. Maire, S. Di Pancrazio, M.-D. Ruepp, R. Giger, A. Perren, A.M. Schmitt, P. Krebs, R.-P. Charles, M.S. Dettmer, Targeting CD47 in anaplastic thyroid carcinoma enhances tumor phagocytosis by macrophages and is a promising therapeutic strategy. Thyroid 29, 979–992 (2019)
A. Mantovani, S. Sozzani, M. Locati, P. Allavena, A. Sica, Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555 (2002)
H. Ye, Q. Zhou, S. Zheng, G. Li, Q. Lin, L. Wei, Z. Fu, B. Zhang, Y. Liu, Z. Li, R. Chen, Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 9, 453 (2018)
M. Shen, Y. Chen, L. Xu, R. Zhu, X. Xue, Y. Tsai, P.C. Keng, S.O. Lee, Y. Chen, Increased infiltration of macrophages to radioresistant lung cancer cells contributes to the development of the additional resistance of tumor cells to the cytotoxic effects of NK cells. Int J. Oncol. 53, 317–328 (2018)
Y. Chen, Y. Song, W. Du, L. Gong, H. Chang, Z. Zou, Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 26, 78 (2019)
D.I. Kim, E. Kim, Y.A. Kim, S.W. Cho, J.A. Lim, Y.J. Park, Macrophage densities correlated with CXC chemokine receptor 4 expression and related with poor survival in anaplastic thyroid cancer. Endocrinol. Metab. (Seoul.) 31, 469–475 (2016)
J. Espinal-Enríquez, S. Muñoz-Montero, I. Imaz-Rosshandler, A. Huerta-Verde, C. Mejía, E. Hernández-Lemus, Genome-wide expression analysis suggests a crucial role of dysregulation of matrix metalloproteinases pathway in undifferentiated thyroid carcinoma. BMC Genomics 16, 207 (2015)
S. Ahn, T.H. Kim, S.W. Kim, C.S. Ki, H.W. Jang, J.S. Kim, J.H. Kim, J.H. Choe, J.H. Shin, S.Y. Hahn, Y.L. Oh, J.H. C., Comprehensive screening for PD-L1 expression in thyroid cancer. Endocr. Relat. Cancer 24, 97–106 (2017)
M. Camus, M. Tosolini, B. Mlecnik, F. Pages, A. Kirilovsky, A. Berger, A. Costes, G. Bindea, P. Charoentong, P. Bruneval, Z. Trajanoski, W.H. Fridman, J. Galon, Coordination of intratumoral immune reaction and human colorectal cancer recurrence. Cancer Res 69, 2685–2693 (2009)
R. Giannini, S. Moretti, C. Ugolini, E. Macerola, E. Menicali, N. Nucci, S. Morelli, R. Colella, M. Mandarano, A. Sidoni, M. Panfili, F. Basolo, E. Puxeddu, Immune profiling of thyroid carcinomas suggests the existence of two major phenotypes: An ATC-Like and a PDTC-Like. J. Clin. Endocrinol. Metab. 104, 3557–3575 (2019)
B. Caillou, M. Talbot, U. Weyemi, C. Pioche-Durieu, A. Al Ghuzlan, J.M. Bidart, S. Chouaib, M. Schlumberger, C. Dupuy, Tumor-associated macrophages (TAMs) form an interconnected cellular supportive network in anaplastic thyroid carcinoma. PLoS One 6, e22567 (2011)
P.A. Oldenborg, A. Zheleznyak, Y.F. Fang, C.F. Lagenaur, H.D. Gresham, F.P. Lindberg, Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054 (2000)
C.M. Schürch, S. Forster, F. Brühl, S.H. Yang, E. Felley-Bosco, E. Hewer, The “don’t eat me” signal CD47 is a novel diagnostic biomarker and potential therapeutic target for diffuse malignant mesothelioma. Oncoimmunology 7, e1373235 (2017)
Acknowledgements
We thank the colleagues for their useful comments for the improvement and modification of the study.
Author contributions
Conceptualization: Y.L.; methodology: Y.L. and Y.Y.; analysis of data: Y.L., W.X., and B.M.; writing: Y.L.; supervision, project administration: T.L. and Y.W.
Funding
This work was supported by the National Natural Science Foundation of China (82072951).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing interests.
Ethical approval
The research ethics committee of FUSCC has confirmed that no ethical approval is required (050432-4-1911D).
Informed consent
Informed consent was obtained from all individual participants.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Luo, Y., Yang, YC., Ma, B. et al. Integrated analysis of novel macrophage related signature in anaplastic thyroid cancer. Endocrine 78, 517–530 (2022). https://doi.org/10.1007/s12020-022-03179-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-022-03179-5