
Abstract
Purpose
Primary adrenal schwannoma (PAS) is a very rare benign tumor, and most of them have been described in case reports. This study aimed to analyze their distinct clinicopathologic features and follow-up data through the largest series yet.
Methods
Clinicopathologic features of 31 primary adrenal schwannomas were retrospectively studied. Imaging and histologic features were re-evaluated and summarized. Immunohistochemical markers were measured, including S100, SOX10, AE1/AE3, EMA, SMA, Desmin, HMB45, GLUT1, and Ki67. Follow-up of all cases was performed.
Results
All the tumors were clinically misdiagnosed as nonfunctioning adrenal adenoma (NAA; 23/31), aldosterone-producing adenoma/aldosteronoma (APA; 3/31), cortisol-producing adenoma (CPA; 3/31), or pheochromocytoma (PCC; 2/31). Some 87% (27/31) presented with adrenal incidentaloma, and 13% (4/31) had a clinical symptom or unregulated hormone levels. They comprised conventional (19/31), cellular (7/31), plexiform (2/31), ancient (1/31), epithelioid (1/31) and microcystic/reticular variants (1/31) and had various histologic features. Immunohistochemically, all tumors (31/31) were positive for S100 and Sox10, with a low Ki-67 proliferative index. In the long-term follow-up (mean, 53 mo.; median, 56 mo.), none had evidence of recurrence and metastasis. Univariate analysis showed that OS and DFS were not associated with age; sex; tumor side, size, or number; adrenal-related symptoms; gross feature (solid vs. cystic); or any histologic feature (P > 0.9999).
Conclusion
PAS is an extremely rare tumor and mostly appears as an incidentaloma. Clinically, it tends to be misdiagnosed as other common adrenal tumors. This tumor has a benign biologic behavior and prognosis, without correlations with clinical or histologic parameters.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Schwannoma, formerly known as neurilemmoma or neurinoma, is considered a benign tumor arising from a relatively uniform group of cells displaying schwannian differentiation [1, 2]. It affects patients of all ages, with a peak between 20 and 50 years [3]. The head and neck or flexor surface of the extremities are the most involved anatomic locations [4]. Schwannoma has various distinct histologic features, and hence, a collection of variants have been described, including cellular, ancient, plexiform, melanotic, epithelioid, gastric, microcystic/reticular, pseudoglandular, neuroblastoma-like variants [5,6,7,8,9,10,11,12,13]. Most schwannomas occur sporadically, but some may be associated with neurofibromatosis type 2 (NF2) or schwannomatosis [3].
Adrenal lesions are not uncommon in routine clinical practice. Among them, 92.5% are benign, and three of the most common, in descending order, are adenoma (63.1%), pheochromocytoma (13.8%) and nodular adrenal hyperplasia (11.6%) [14]. Most benign adrenal tumors manifest as “incidentaloma”, a radiologic term referring to an adrenal mass that is larger than 1 cm and incidentally discovered in the course of diagnostic evaluation or treatment of otherwise medical scenarios [15, 16].
Primary adrenal schwannoma (PAS) is extremely rare, accounting for 0.7% of adrenal lesions. Only ~80 cases around the world have been reported to date; however, most reports have been case reports or lacked a detailed description of clinicopathologic features in a large series and did not mention their correlations to the prognosis in the adrenal gland. Herein, we collected the largest group of PASs from two of the largest hospitals in China to investigate their clinicopathologic presentations, various pathologic features, and prognosis by long-term follow-up.
Materials and methods
Patients and case selection
This study was approved by the Institutional Review Board at the Department of Pathology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, and The First Affiliated Hospital of Zhengzhou University. Of 31 adrenal schwannomas, 11 cases were from Ruijin Hospital and 20 cases were from The First Affiliated Hospital of Zhengzhou University. One case from the latter (microcystic-reticular schwannoma) has been reported previously [11]. All cases were resected specimens with complete clinical data. According to the international clinical practice guidelines for adrenal incidentaloma, primary aldosteronism, and pheochromocytoma [17,18,19], as well as our hospitals’ practical experience, adrenal gland-related laboratory data were collected. The biochemical markers included blood potassium (K+) and sodium (Na+) concentrations, plasma adrenocorticotropic hormone (ACTH), plasma cortisol (CORT), 24-h urinary free cortisol (UFC), 24-h urinary norepinephrine (UNE), 24-h urinary epinephrine (UE), 24-h urinary dopamine (UDPA), plasma metanephrine (MN) and normetanephrine (NMN), plasma renin activity (PRA), plasma angiotensin II (PAT-II), plasma aldosterone concentration (PAC), urinary 17-hydroxycorticosteroid (17-OHCS), and urinary 17-ketosteroid (17-KS). All hematoxylin and eosin (HE)-stained slides were independently reviewed by 2 experienced pathologists (J. Z. and C.F.W.), and a diagnostic consensus of PAS was eventually reached on every case.
Pathologic evaluation
The gross features were collected through the routine archiving of pathologic samples. Histologically, each schwannoma was classified into concrete subtypes, including the conventional type and other variants mentioned above, according to the latest WHO classification and relevant classical paper [5,6,7,8,9,10, 12, 13, 20, 21]. The commonly distinct morphologic features were recorded, including the capsule, lymphoplasmacytic cuff, Antoni A and B areas, Verocay body, calcification, necrosis, hyalinization, hemorrhage, hyalinized vessels, glandular-like or microcystic structure, myxoid stroma, and foamy cells.
Immunohistochemistry (IHC)
Each surgical specimen was specifically resectioned. Four-micron-thick sections were taken from 10% formalin-fixed and paraffin-embedded tissue blocks, followed by immunohistochemical staining using commercially available antibodies as follows: S100 (polyclonal, prediluted; Dako, Glostrup, Denmark), SOX10 (EP268, 1:100; ZSGB-BIO, Beijing, China), AE1/AE3 (AE1/AE3, prediluted; Dako, Carpinteria, California, USA), EMA (E29, prediluted; Dako, Glostrup, Denmark), SMA (1A4, 1:200; ZSGB-BIO, Beijing, China), Desmin (D33, prediluted; Dako, Glostrup, Denmark), HMB45 (HMB45, prediluted; Dako, Glostrup, Denmark), Ki67 (MIB-1, prediluted; Dako, Glostrup, Denmark). Antibody binding was detected using the universal immunoperoxidase polymer method (Envision-kit; Dako, Carpinteria, CA, USA). A Dako automated immunohistochemistry system (Dako, Carpinteria, CA, USA) was applied according to the manufacturer’s protocol. The IHC results were independently interpreted by 2 experienced pathologists (J.Z. and C.F.W.).
Follow-up and statistical analysis
Follow-up was performed in the office setting or by telephone interview. Overall survival (OS) curve was plotted. Univariate analysis of OS and DFS (disease-free survival) was performed to evaluate the associations between prognosis and various factors, including clinical and pathologic features. These parameters included age (<50 vs. ≥50 years), sex (male vs. female), side (left vs. right), tumor number (single vs. multiple), size (<4 vs. ≥4 cm), symptom (adrenal related symptom vs incidentaloma), gross feature (solid vs. cyst), and histologic features (capsule, lymphoplasmacytic cuff, Antoni A/B, Verocay body, calcification, necrosis, hyalinization, hemorrhage, hyalinized vessels, glandular-like/microcystic or myxoid stroma, foamy cells). The grouping of patient age and tumor size accorded with the primarily published literature [22, 23]. Only the factors showing significance were taken into the multivariate analysis using Cox proportional hazard regression. P < 0.05 was considered statistically significant. The statistical analyses were performed with GraphPad Prism 7.0 (GraphPad Software, San Diego, CA).
Results
Clinical features
The main clinical features and adrenal-related laboratory test results of our series are summarized in Table 1 and Table 2, respectively. The 31 patients were 13 males and 18 females (male to female ratio: 1:1.4), ranging in age from 26 to 71 years (mean, 48.5 yr.; median, 47 yr.). The numbers of patients with left-side (n = 15) and right-side (n = 16) were not significantly different. The great majority of tumors were solitary (cases 1 and 19 were multiple), with the maximum diameter ranging from 1.0 to 12.1 cm (mean, 5.7 cm; median, 4.5 cm). No patients showed any evidence of a related familial history of schwannoma, and all had normal plasma K+ and Na+ concentrations.
Approximately 84% of cases (26/31) were accidentally found by imaging when patients underwent routine physical (24/31) or other disease examinations (2/31) (e.g., lithiasis). They were hence assigned to “incidentaloma”. Some 27% (7/26) of the incidentalomas had a drug-adjustable hypertension, all of which were chronic and seemed to have nothing to do with the adrenal lesions. Considering the presentation of abruptly increasing blood pressure upon an attempt to remove the tumor at a basic-level hospital in case 5 and intermittent facial flushing for 2 months in case 24, there were clinical suspicions of pheochromocytoma (PCC). However, neither of them showed any abnormal levels of plasma NMN, MN, 24-h UNE or 24-h UN, though there was slightly elevated plasma CORT at 4 p.m. (353 ng/ml; normal, 64–327 ng/ml) and decreased PRA when in the supine position (0.01 pg/mL/h; normal, 0.15–2.33 pg/mL/h) in case 5 (Table 2). Higher PAC (normal, supine, 30–160; stand, 70–300 pg/ml) was displayed in cases 11 (supine, 459 pg/ml stand, 468 pg/ml) and 12 (supine, 310 pg/ml; stand, 327 pg/ml) with incidentally discovered adrenal masses, resulting in the clinical suspicion of aldosterone-producing adenoma/aldosteronoma (APA). Other incidentalomas (85%, 22/26) were found without or with some nonspecific symptoms or irreverent other disease states, including lithiasis (case 2, 12), anemia (case 1), fatigue (case 1), headache (case 20), weight loss (case 20), hepatic/renal cyst (case 1, 2, 21), and pneumonia (case 21). As a result, their adrenal masses were clinically diagnosed as nonfunctioning adrenal adenoma (NAA).
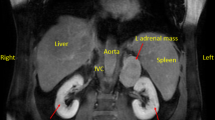
The remaining 5 patients (16%, 5/31) were admitted to the hospital with symptoms. Of these, 2 cases (cases 3 and 8) were clinically suspicious of the diagnosis of cortisol-producing adenoma (CPA). Case 3 and case 8 featured elevated adjustable blood pressure (up to 160/101 mmHg) or left waist pain in the short term (Table 1), with superimposed higher 24-h UFC (case 3, 438 nmol/d; case 8, 432 nmol/d [normal, 73–372 nmol/d]) and PAC in the supine position (case 3, 238 pg/ml; case 8, 163), suggesting the possibility of subclinical Cushing syndrome (CS). Patient 15 had an increased 24-h UFC (433 nmol/d) and obvious presentation, including thirst, polydipsia, polyuria, and weight loss in a short time, implying CS. Patient 10 had a history of chronic hypertension and nephrotic syndrome with elevated 24-h UNE (55 ng/d; normal, 0–50 ng/d), UE (24 ng/d; normal, 0–20 ng/d), and PAC (supine, 480 pg/ml; stand, 486 pg/ml), prompting the possible diagnosis of APA for adrenal lesions. Patient 26 was hospitalized for acute intermittent abdominal pain without any evidence of deregulated adrenal-relevant hormones and was diagnosed with NAA. All patients had normal concentrations of 17-OHCS and 17-KS, other than patients 8, 9, and 10, who had slightly higher levels (Table 2). X-ray plain-film and computed tomography (CT) scans showed all the tumors had a well-defined demarcation and soft-tissue intensity (Fig. 1a, b), and 7 cases displayed various degrees of cystic regions with (cases 2, 22, and 24) or without an air-fluid level. Contrast-enhanced CT showed mild to moderate heterogeneous enhancement in the venous phase (Fig. 1c).

Axial a and coronal CT b showed a well-defined mass (arrow) involved in the left adrenal gland; contract-enhanced CT (venous phage) from another case demonstrated a well-demarcated mass (arrow) with a mild enhancement c. Gross examination. The tumors were solitary d or multinodular e with a solid d, e or cystic appearance f and yellow-white or yellow in color (fixed by formalin, d, f; fresh specimen, e)
Pathologic features
Grossly, the tumors grew as discrete, round, well-circumscribed and encapsulated masses (Table 1). On the cross-section, they showed a solid, cystic-solid or cystic appearance with a yellow, gray-whitish, or tan color, in some of which hemorrhage was appreciable (Table 1, Fig. 1c, d). The histological features are listed in Table 3. Our series included 1 ancient, 1 microcystic/reticular, 1 epithelioid, 2 plexiform, 19 conventional, and 7 cellular schwannomas. In total, 94% (29/31) of the patients had uninodular tumors (all except the 2 with plexiform variants), and all of them were encased by intact fibrous capsules (Fig. 2a). The great majority of cases (87.1%, 27/31) had a lymphoplasmacytic cuff at the periphery of the tumor (Fig. 2a). The alternating Antoni A and Antoni B (Fig. 2b), a flagship growth pattern, could be appreciated in all the conventional, plexiform and ancient variants (71%, 22/31), but varied from case to case. However, the cellular-variant tumors were characterized by almost exclusively of hypercellular Antoni A areas composed of compact and slender spindle cells with twisted nuclei and eosinophilic cytoplasm (Fig. 2c). The microcystic-reticular variant lacking prominent Antoni A and B alternation was mainly composed of intersecting and anastomosing cords or strands of spindle cells with eosinophilic cytoplasm and oval nuclei (case 27). Verocay bodies principally presented in the Antoni A area of conventional schwannomas (58%, 11/19; Fig. 2d). Calcification and hyalinization were found in 22.6% (7/31) and 45.2% (11/31) of cases, respectively (Fig. 2e), and the latter was usually located in the Antoni B area. Most conventional (95%, 18/19) and cellular schwannomas (86%, 6/7) in our series had more or less hyalinized vessels, which could be found in both cellular and acellular regions (Fig. 2f). Only 2 cases presented with obvious necrosis (Fig. 2g); however, neither was coagulative tumor cell necrosis but resulted from degenerative changes. Hemorrhage (32%, 10/31) and myxoid stroma (16%, 5/31) were not infrequent (Fig. 2g). Glandular-like/microcystic structure was typically seen in the microcystic/reticular schwannoma (case 27), but it was also encountered in epithelioid (case 31) and in focal areas of some conventional (21%, 4/19) and cellular variants (15.8%, 3/19) (Fig. 2h). Foamy cells almost always appeared in the cellular variants (86%, 6/7) and in the cellular regions of some plexiform (50%, 1/2) and conventional (26%, 5/19) variants (Fig. 2i). Mitosis was absent in all of our cases, and prominent nuclear dysplasia was not seen except for some atypical cells scattered in the otherwise bland schwannoma in 1 ancient variant (Fig. 2j). One adrenal epithelioid variant was encountered in our series, which has never been reported. It was circumscribed and encapsulated; it typically showed a multinodular growth pattern and consisted of small round Schwann cells in clusters or cords with focal prominent myxoid stroma (Fig. 2k, l). The ganglion cells and neuroblasts were not appreciable. The one microcystic/reticular variant has been reported before, which showed strikingly anastomosing strands of spindle cells imparting a microcystic and reticular pattern with a focal transition to epithelioid nests [11]. Immunohistochemically, the tumor cells were consistently and diffusely positive for S-100 (Fig. 3a), SOX10 (Fig. 3b), but totally negative for AE1/AE3, EMA, SMA, Desmin, and HMB45. The proliferative index labeled by Ki67 was always low (range: 1–3%; mean, 1.5%; median, 1%; Fig. 3c).

The adrenal schwannomas were well circumscribed and encapsulated with a complete capsule and, frequently, a lymphoplasmacytic cuff (the center) (note the opposed normal adrenal tissues on the right side) (a); the alternating Antoni A (right side) and Antoni B areas (left side) were very common (b); cellular variants were characterized by almost exclusively hypercellular Antoni A areas, usually with foamy cells (c); Verocay bodies principally presented in the Antoni A of conventional schwannomas (d); calcification and hyalinization were found in some cases (e); hyalinized vessels were frequently found (f); this case presented with obvious necrosis and hemorrhage due to its degenerative changes (g, the upper region); a glandular-like/microcystic structure was appreciable in focal areas of some schwannomas (h); Foamy cells could be seen in cellular variants and the cellular regions of some plexiform and conventional variants (i); atypical cells scattered in the otherwise bland schwannoma in ancient schwannoma (j); Plexiform growth pattern was encountered in the plexiform or epithelioid variant (k); the epithelioid variant consisted of small, round Schwann cells in clusters or cords (l)

Immunohistochemical features of adrenal schwannoma. The tumor cells were consistently and diffusely positive for S100 and Sox10, with a low proliferative index labeled by Ki-67 (1%)
Treatment and prognosis
The mainstay treatment in our cases was laparoscopic removal of the adrenal tumor through partial adrenalectomy (55%, 17/31), entire gland removal (39%, 12/31) or merely tumor exenteration (6.5%, 2/31). No additional chemotherapy was administered. Abnormal hormone levels or obvious signs, such as short-run elevated hypertension and abdominal pain, gradually disappeared following monitoring. All follow-up data were available until this article was written. The follow-up time ranged from 7 to 115 months (mean, 53 mo.; median, 56 mo.; Fig. 4). Every patient has lived through a calm period without any evidence of recurrence or metastasis. Univariate analysis showed that none of the clinical parameters (age, sex, side, tumor number, size, symptoms), gross feature, or histologic features (capsule, lymphoplasmacytic cuff, Antoni A/B, Verocay body, calcification, necrosis, hyalinization, hemorrhage, hyalinized vessels, glandular-like/microcystic or myxoid stroma, foamy cells) affected the OS or DFS of the patients (P > 0.9999, Table 4).

The OS curve calculated by the Kaplan-Meier method showed that all patients were alive during the follow-up
Discussion
Retroperitoneal schwannomas are rare, accounting for only 0.7–5% of all schwannomas [24, 25]. Primary juxta-adrenal or adrenal schwannoma represents a very small proportion of retroperitoneal schwannomas [26], the latter being especially unusual. Distinguishing these two is arbitrary sometimes due to the lack of strict criteria to separate them. Some scholars have argued that adrenal schwannoma arises from the sheath of the peripheral sympathetic nerve dominating the adrenal medulla, and therefore, the adrenal schwannoma should be located in the medullary region [26,27,28]. However, in contrast to the small medullar mass obviously rimmed by cortical cells in either imaging or H&E-stained slides, a large enough one may markedly push away surrounding normal adrenal tissues, resulting in an unclear mutually histologic or anatomic relationship. In addition, other scholars hold that the tumors derived from retroperitoneal nerve tissue are located in the region with a close relation to the adrenal gland [26, 29]. From our perspective, if the tumor is microscopically close to the normal adrenal tissues on any one slide (under low power, × 40), it is reasonable to consider it a lesion arising in the adrenal gland. Therefore, in our case series, any encapsulated tumors were surrounded by or closely opposed to the normal adrenal tissues on at least one slide under low power and were considered derived from the adrenal gland.
The median age of adrenal schwannoma is 50 years, and it appears to be more common in females, with a male-to-female ratio of 1:1.8 [27]. The epidemiology is similar to the ratio in our series and further suggests its female predilection. The overwhelming majority of adrenal schwannomas are found incidentally when performing regular physical or other disease examinations (referred to as “incidentaloma”). A subset may present with clinical symptoms due to the mass effect, such as mild abdominal discomfort or pain [27, 30, 31]. Generally, clinicians would make the diagnosis of NAA for an adrenal mass without any evidence of anomalous hormone levels or significantly unusual symptoms. It is difficult to differentiate NAA from schwannoma in such a scenario through either clinical presentation or imaging features. Pathologic examination is likely to be the only way to make a finally correct diagnosis. CS is defined as the state of hypercortisolism that results from endogenous or exogeneous leukocortical excess [32]. The clinical manifestation of CS is variable, ranging from mild or nonspecific symptoms to obvious features due to profound cortisol excess [33]. Compared to the majority of ACTH-dependent hypercortisolism, the ACTH-independent subtype only accounts for 20%, most of which are adrenal CPA (55%), carcinoma (35%) and macronodular or micronodular hyperplasia (10%). Therefore, given the benign imaging presentation, it, most often leads to the diagnosis of CPA clinically. The 24-h UFC is still a classical screening laboratory marker for CS, though there are discrepancies in the reported sensitivity and specificity and variations in reported measurements [33]. Based on the observation of our cases, adrenal schwannomas with 24-h UFC abnormalities usually appear as subclinical CS without overt or with very limited signs of hypercortisolism. Moreover, adrenal schwannomas do not seem to have a very high concentration of 24-h UFC (only 1/6- to 1/7-fold elevation in our series) compared to the typical Cushing syndrome. Additionally, in contrast to some incidentalomas, the subclinical CS caused by schwannoma in our cases may not be associated with perturbation of the hypothalamic-pituitary-adrenal (HPA) axis. The cause of hypersecretion is largely unknown but may be associated with mechanical irritation, as in other rare, nonfunctional adrenal tumors, such as myelolipoma or lipoma [34]. A similar mechanism may also affect the adrenal medulla in some cases and result in some symptoms or evidence of increased catecholamines. In this circumstance, clinicians are likely to assign to these adrenal tumors the diagnosis of PCC. For the two patients suspected to have PCC in our series, although they had a limited subset of signs mimicking PCC (intermittent facial flushing or elevated hypertensive flushing during operation), they did not show typical deregulated hormone levels, including abnormal 24-h UNE, UE, MN, and NMN. Two clinical research groups have reported adrenal and retroperitoneal schwannomas with catecholamine hypersecretion, respectively [35, 36]. However, from the pathologist’s point of view, we hold that schwannomas do not have the capacity for catecholamine secretion per se based on their histologic components and pathologic features. Mechanical irritation affecting adjacent nerves or the adrenal medulla may be a plausible reason for the secretion, but the real truth should be further investigated. The most common causes of primary hyperaldosteronism are unilateral APA and bilateral adrenal hyperplasia [37]. Because most patients are asymptomatic, a biochemical test of the ratio of PAC to PRA (PAC [ng/dL] / PRA [ng/mL/hr]) is a classical diagnostic index with a threshold of 20 or 30, yielding a high sensitivity and specificity [18, 38]. The increased PAC in our series would also prompt the clinician to diagnose APA for the adrenal mass. Nonetheless, the normal values of plasma K+ and PRA and the PAC/PRA ratio far less than 20 are not consistent with the typical features of primary aldosteronism. In this scenario, other entities than APA should enter our consideration. Arriving at the correct diagnosis also depends on the pathologic examination.
Some tumors can grow larger with or without superimposed hemorrhage or cystic degeneration because the retroperitoneal cavity is flexible and space is limited. They may mimic other retroperitoneal tumors, such as pheochromocytoma, liposarcoma, paraganglioma, gastrointestinal stromal tumor or deep fibrous histiocytoma, on imaging [30, 39]. Radiologic study may play a limited role in telling them apart because of their overlapping imaging features, particularly in the setting of no endocrine-related clinical background [26]. Histologic examination is the gold standard for diagnosis. Resected masses are the most frequent samples, and therefore, the diagnosis of schwannoma is straightforward, even if it is likely to demonstrate various morphologic features. The diagnostic accuracy of fine-needle aspiration (FNA) biopsy is controversial and has been discussed by some publications [30]. As far as we are concerned, its utility depends on whether the pathologist has enough experience and his/her familiarity with the histologic characteristics of schwannoma. However, FNA biopsy should be avoided if pheochromocytoma or echinococcal parasitic cysts are suspected, since it would have potentially harmful effects [31]. Identical to schwannoma developing in other locations, adrenal schwannomas have various histologic features, including a complete capsule, prominent lymphoplasmacytic cuff or stroma, Antoni A and B areas, a Verocay body, hyalinized vessels, myxoid stroma, and foamy cells. A subset of tumors may grow larger and display ancient degeneration, hemorrhage, calcification or cystic changes [30]. Necrosis is rare, and most, if present, is associated with changes in degeneration. In addition, compared to schwannomas occurring in superficial soft tissues, cellular variants appear to be more frequent in the adrenal gland, according to our series (22.6%, 7/31). This is plausible because cellular schwannoma tends to arise in the retroperitoneal area or the mediastinum [5]. The atypical cells in ancient schwannoma or the cellularity and occasionally focal infiltration in cellular schwannoma may raise a clinical suspicion of malignancy. Awareness of their distinct features contributes to avoiding misdiagnosis. The epithelioid variant is usually small (0.3 to 22.7 cm; mean, 1.2 cm) and arises in the skin and the superficial soft tissues, particularly in the extremities; only 17% involves the visceral locations, but adrenal epithelioid schwannoma has not been reported yet. This epithelioid variant was described as developing in the adrenal gland for the first time in our series, demonstrating similar histologic features to those in other locations. Microcystic/reticular schwannoma is a recently reported variant with a predilection to arise in the visceral locations and has distinct morphologic features. Recognizing this variant facilitates the avoidance of unnecessary misdiagnosis. For further information, please refer to the study we reported in 2015 [11]. Immunohistochemically, S100 or Sox10 is used mainly for confirmation of the diagnosis. The low proliferative index of schwannomas suggests their indolent biologic nature.
For adrenal incidentaloma, the American Association of Clinical Endocrinologists/American Association of Endocrine Surgeons (AACE/AAES) and William F.Y. recommend that if the tumor is <4 cm with benign characteristics and no hormonally reactive evidence, then imaging and hormonal tests can be repeated; tumors >4 cm on CT scan should be removed [40, 41]. The NIH consensus statement concluded that tumors >6 cm should be excised, those <4 cm with radiologically benign features are generally not removed, and those between 4 and 6 cm can be either observed or resected [30, 42]. However, the conclusion above is generally applicable to adenoma, which may be easier to confirm. Almost all reported adrenal schwannomas are surgically removed because of the potential misdiagnosis on the imaging, regardless of their size [30]. In contrast to the typical CPAs, APAs and PCCs, patients of AS, if any, only have a limited hormonal disturbances or signs and be unnecessary to take any preoperative drug interventions [18, 19, 32]. To date, there have been no reported cases of adrenal schwannoma with malignant transformation. In our cases, the data showed no evidence of recurrence or metastasis in a relatively long-term follow-up, irrespective of their clinical and histologic backgrounds. This seemingly indicates their bona fide benign nature and that clinical and various morphologic parameters have no significant association with the patient’s prognosis.
In conclusion, primary adrenal schwannoma is an extremely rare tumor and mostly appears as an incidentaloma. Misdiagnosis as NAA, APA, or CA can be clinically made due to unfamiliarity with the clinicopathologic features of adrenal schwannoma. There are various histologic features and a relatively higher frequency of the cellular variant in the adrenal gland. It has a benign biologic behavior and a frankly good prognosis, which is not associated with its clinical and histologic parameters.
References
B.L.K. Wong, S. Bathala, D. Grant, Laryngeal schwannoma: a systematic review. Eur Arch Otorhinolaryngol 274(1), 25–34 (2017). https://doi.org/10.1007/s00405-016-4013-6
D.A. Hilton, C.O. Hanemann, Schwannomas and their pathogenesis. Brain Pathol 24(3), 205–220 (2014). https://doi.org/10.1111/bpa.12125
M. Tulli, S. Bondi, C.E. Smart, L. Giordano, M. Trimarchi, A. Galli, D. Di Santo, M. Biafora, M. Bussi, Diagnosis and Treatment of Laryngeal Schwannoma: A Systematic Review. Otolaryngol Head Neck Surg 158(2), 222–231 (2018). https://doi.org/10.1177/0194599817735508
A. Majbar, A. Hrora, A. Jahid, M. Ahallat, M. Raiss, Perineal schwannoma. BMC Res. Notes 9, 304 (2016). https://doi.org/10.1186/s13104-016-2108-1
J.M. Woodruff, T.A. Godwin, R.A. Erlandson, M. Susin, N. Martini, Cellular schwannoma: a variety of schwannoma sometimes mistaken for a malignant tumor. Am J Surg Pathol 5(8), 733–744 (1981)
D. Dayan, A. Buchner, A. Hirschberg, Ancient neurilemmoma (Schwannoma) of the oral cavity. J Craniomaxillofac Surg 17(6), 280–282 (1989)
W. White, M.H. Shiu, M.K. Rosenblum, R.A. Erlandson, J.M. Woodruff, Cellular schwannoma. A clinicopathologic study of 57 patients and 58 tumors. Cancer 66(6), 1266–1275 (1990)
R.P. Mennemeyer, K.O. Hallman, S.P. Hammar, J.E. Raisis, J.S. Tytus, D. Bockus, Melanotic schwannoma. Clinical and ultrastructural studies of three cases with evidence of intracellular melanin synthesis. Am. J. Surg. Pathol. 3(1), 3–10 (1979)
L.G. Kindblom, J.M. Meis-Kindblom, G. Havel, C. Busch, Benign epithelioid schwannoma. Am. J. Surg. Pathol. 22(6), 762–770 (1998)
Y.Y. Hou, Y.S. Tan, J.F. Xu, X.N. Wang, S.H. Lu, Y. Ji, J. Wang, X.Z. Zhu, Schwannoma of the gastrointestinal tract: a clinicopathological, immunohistochemical and ultrastructural study of 33 cases. Histopathology 48(5), 536–545 (2006). https://doi.org/10.1111/j.1365-2559.2006.02370.x
J. Zhou, D. Zhang, G. Wang, W. Li, J. Xu, Y. Ma, J. Zhang, Z. Li, Z. Zhao, Primary adrenal microcystic/reticular schwannoma: clinicopathological and immunohistochemical studies of an extremely rare case. Int. J. Clin. Exp. Pathol. 8(5), 5808–5811 (2015)
J.K. Chan, K.O. Fok, Pseudoglandular schwannoma. Histopathology 29(5), 481–483 (1996)
C. Fisher, M.E. Chappell, S.W. Weiss, Neuroblastoma-like epithelioid schwannoma. Histopathology 26(2), 193–194 (1995)
Gong, X., Yu, Y., Zhan, W, Ultrasonographic findings of 1385 adrenal masses: A Retrospective Study of 1319 Benign and 66 Malignant Masses. J. Ultrasound Med. (2017). https://doi.org/10.1002/jum.14471
A. Pinto, J.A. Barletta, Adrenal tumors in adults. Surg Pathol Clin 8(4), 725–749 (2015). https://doi.org/10.1016/j.path.2015.07.005
J.M. Lee, M.K. Kim, S.H. Ko, J.M. Koh, B.Y. Kim, S.W. Kim, S.K. Kim, H.J. Kim, O.H. Ryu, J. Park, J.S. Lim, S.Y. Kim, Y.K. Shong, S.J. Yoo, Korean Endocrine Society, C.f.C.P.G.: Clinical Guidelines for the Management of Adrenal Incidentaloma. Endocrinol. Metab. (Seoul) 32(2), 200–218 (2017). https://doi.org/10.3803/EnM.2017.32.2.200
M. Fassnacht, W. Arlt, I. Bancos, H. Dralle, J. Newell-Price, A. Sahdev, A. Tabarin, M. Terzolo, S. Tsagarakis, O.M. Dekkers, Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 175(2), G1–G34 (2016). https://doi.org/10.1530/EJE-16-0467
J.W. Funder, R.M. Carey, F. Mantero, M.H. Murad, M. Reincke, H. Shibata, M. Stowasser, W.F. Young Jr., The management of primary aldosteronism: case detection, diagnosis, and treatment: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 101(5), 1889–1916 (2016). https://doi.org/10.1210/jc.2015-4061
J.W. Lenders, Q.Y. Duh, G. Eisenhofer, A.P. Gimenez-Roqueplo, S.K. Grebe, M.H. Murad, M. Naruse, K. Pacak, W.F. Young Jr.; Endocrine, S., Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 99(6), 1915–1942 (2014). https://doi.org/10.1210/jc.2014-1498
B. Liegl, M.W. Bennett, C.D. Fletcher, Microcystic/reticular schwannoma: a distinct variant with predilection for visceral locations. Am. J. Surg. Pathol. 32(7), 1080–1087 (2008). https://doi.org/10.1097/PAS.0b013e318160cfda
C.R. Antonescu, A. Perry, J.M. Woodruff. schwannoma (including variants. WHO Classification of Tumours of Soft Tissue and Bone. C.D.M. Flethcer, J.A. Bridge, P.C.W. Hogendoorn, F. Mertens (eds) IARC, Lyon), 2013) 170–172.
J.C. Ip, T.C. Pang, A.R. Glover, P. Soon, S. Clarke, A. Richardson, P. Campbell, B.G. Robinson, S.B. Sidhu, Improving outcomes in adrenocortical cancer: an australian perspective. Ann. Surg. Oncol. 22(7), 2309–2316 (2015). https://doi.org/10.1245/s10434-014-4133-4
W.M. Rashed, A.M. Saad, M.J. Al-Husseini, A.M. Galal, A.M. Ismael, A.M. Al-Tayep, A. El Shafie, M.A. Ali, A.S. Alfaar, Incidence of adrenal gland tumor as a second primary malignancy: SEER-based study. Endocr Connect (2018). https://doi.org/10.1530/EC-18-0304
S. Caliskan, G. Gumrukcu, C. Kaya, Retroperitoneal ancient schwannoma: a case report. Rev. Urol. 17(3), 190–193 (2015)
J.Y. Song, S.Y. Kim, E.G. Park, C.J. Kim, D.G. Kim, H.K. Lee, I.Y. Park, Schwannoma in the retroperitoneum. J. Obstet. Gynaecol. Res. 33(3), 371–375 (2007). https://doi.org/10.1111/j.1447-0756.2007.00539.x
S.Q. Li, Y.S. Zhang, J. Shi, H.Z. Li, Clinical features and retroperitoneal laparoscopic resection of adrenal schwannoma in 19 patients. Endocr. Pract. 21(4), 323–329 (2015). https://doi.org/10.4158/EP14453.OR
A.K.Y. Lam, P.-A. Just, E. Lack, F. Tissier, L.M. Weiss. Mesenchymal and stromal tumors: schwannoma. WHO Classification of Tumors of Endocrine Organs. R.V. Lloyd, O.R. Y., K. G., J. Rosai (eds.) IARC, Lyon), 2013) 176
J.D. Jakowski, P.E. Wakely Jr., R.E. Jimenez, An uncommon type of adrenal incidentaloma: a case report of a schwannoma of the adrenal medulla with cytological, histological, and ultrastructural correlation. Ann. Diagn. Pathol. 12(5), 356–361 (2008). https://doi.org/10.1016/j.anndiagpath.2008.06.003
Y.M. Zhang, P.F. Lei, M.N. Chen, X.F. Lv, Y.H. Ling, P.Q. Cai, J.M. Gao, CT findings of adrenal schwannoma. Clin. Radiol. 71(5), 464–470 (2016). https://doi.org/10.1016/j.crad.2016.01.010
Y. Mohiuddin, M.G. Gilliland, Adrenal schwannoma: a rare type of adrenal incidentaloma. Arch. Pathol. Lab. Med. 137(7), 1009–1014 (2013). https://doi.org/10.5858/arpa.2012-0291-RS
C. Xiao, B. Xu, H. Ye, Q. Yang, L. Wang, Y.H. Sun, Experience with adrenal schwannoma in a Chinese population of six patients. J. Endocrinol. Invest. 34(6), 417–421 (2011). https://doi.org/10.1007/BF03346705
A. Lacroix, R.A. Feelders, C.A. Stratakis, L.K. Nieman, Cushing’s syndrome. Lancet 386(9996), 913–927 (2015). https://doi.org/10.1016/S0140-6736(14)61375-1
J.M. Pappachan, C. Hariman, M. Edavalath, J. Waldron, F.W. Hanna, Cushing’s syndrome: a practical approach to diagnosis and differential diagnoses. J. Clin. Pathol. 70(4), 350–359 (2017). https://doi.org/10.1136/jclinpath-2016-203933
A.K. Lam, Lipomatous tumours in adrenal gland: WHO updates and clinical implications. Endocr. Relat. Cancer. 24(3), R65–R79 (2017). https://doi.org/10.1530/ERC-16-0564
J. Hou, L. Zhang, Y. Guo, H. Chen, W. Wang, Primary adrenal schwannoma with catecholamine hypersecretion. Arch. Med. Sci. 12(3), 681–683 (2016). https://doi.org/10.5114/aoms.2016.59942
T. Hori, K. Yamagiwa, S. Yagi, T. Iida, K. Taniguchi, C. Yamamoto, Y. Eshita, Y. Kozuka, H. Takaki, T. Kato, K. Saito, M. Torii, S. Isaji, S. Uemoto, Noradrenalin-secreting retroperitoneal schwannoma resected by hand-assisted laparoscopic surgery: report of a case. Surg. Today. 36(12), 1108–1113 (2006). https://doi.org/10.1007/s00595-006-3304-8
W.F. Young, Primary aldosteronism: renaissance of a syndrome. Clin. Endocrinol. (Oxf) 66(5), 607–618 (2007). https://doi.org/10.1111/j.1365-2265.2007.02775.x
F.H. Perschel, R. Schemer, L. Seiler, M. Reincke, J. Deinum, C. Maser-Gluth, D. Mechelhoff, R. Tauber, S. Diederich, Rapid screening test for primary hyperaldosteronism: ratio of plasma aldosterone to renin concentration determined by fully automated chemiluminescence immunoassays. Clin. Chem. 50(9), 1650–1655 (2004). https://doi.org/10.1373/clinchem.2004.033159
Y. Shen, Y. Zhong, H. Wang, L. Ma, Y. Wang, K. Zhang, Z. Sun, H. Ye, MR imaging features of benign retroperitoneal paragangliomas and schwannomas. BMC Neurol. 18(1), 1 (2018). https://doi.org/10.1186/s12883-017-0998-8
M.A. Zeiger, G.B. Thompson, Q.Y. Duh, A.H. Hamrahian, P. Angelos, D. Elaraj, E. Fishman, J. Kharlip, American Association of Clinical, E., American Association of Endocrine, S, The American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons medical guidelines for the management of adrenal incidentalomas. Endocr. Pract. 15(Suppl 1), 1–20 (2009). https://doi.org/10.4158/EP.15.S1.1
W.F. Young Jr., Clinical practice. The incidentally discovered adrenal mass. N. Engl. J. Med. 356(6), 601–610 (2007). https://doi.org/10.1056/NEJMcp065470
NIH state-of-the-science statement on management of the clinically inapparent adrenal mass (“incidentaloma”). NIH Consens State Sci. Statements 19(2), 1–25 (2002).
Funding
The study was supported by the Shanghai science and technology commission (17ZR1417500).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhou, J., Zhang, D., Li, W. et al. Primary adrenal schwannoma: a series of 31 cases emphasizing their clinicopathologic features and favorable prognosis. Endocrine 65, 662–674 (2019). https://doi.org/10.1007/s12020-019-01992-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-019-01992-z