
Abstract
Purpose
Obestatin and ghrelin are peptides encoded by the preproghrelin gene. Obestatin inhibits food intake, in addition to regulation of glucose and lipid metabolism. Here, we test the ability of obestatin at improving metabolic control and liver function in type 2 diabetic animals (type 2 diabetes mellitus).
Methods
The effects of chronic obestatin treatment of mice with experimentally induced type 2 diabetes mellitus on serum levels of glucose and lipids, and insulin sensitivity are characterized. In addition, alterations of hepatic lipid and glycogen contents are evaluated.
Results
Obestatin reduced body weight and decreased serum glucose, fructosamine, and β-hydroxybutyrate levels, as well as total and low-density lipoprotein fractions of cholesterol. In addition, obestatin increased high-density lipoproteins cholesterol levels and enhanced insulin sensitivity in mice with type 2 diabetes mellitus. Moreover, obestatin diminished liver mass, hepatic triglycerides and cholesterol contents, while glycogen content was higher in livers of healthy and mice with type 2 diabetes mellitus treated with obestatin. These changes were accompanied by reduction of increased alanine aminotransferase, aspartate aminotransferase, and gamma glutamyl transpeptidase in T2DM mice with type 2 diabetes mellitus. Obestatin increased adiponectin levels and reduced leptin concentration. Obestatin influenced the expression of genes involved in lipid and carbohydrate metabolism by increasing Fabp5 and decreasing G6pc, Pepck, Fgf21 mRNA in the liver. Obestatin increased both, AKT and AMPK phosphorylation, and sirtuin 1 (SIRT1) protein levels as well as mRNA expression in the liver.
Conclusion
Obestatin improves metabolic abnormalities in type 2 diabetes mellitus, restores hepatic lipid contents and decreases hepatic enzymes. Therefore, obestatin could potentially have a therapeutic relevance in treating of insulin resistance and metabolic dysfunctions in type 2 diabetes mellitus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obestatin is a 23 amino-acids peptide isolated from rat stomach in 2005 by Zhang [1]. It arises as a result of posttranslational processing of amino acids 76–98 of the prepropeptide preproghrelin—obestatin name comes from the Latin word “obedere” meaning to devour, and “statin” meaning to suppress. Initially, it was described that obestatin is able to activate G-protein-coupled receptor 39 (GPR39) [1], however, others have questioned this result [2]. In 2008 Granata et al. suggested that glucagon-like peptide 1 receptor (GLP-1R) may be involved in obestatin action [3]. Initially it was shown that obestatin behaves as a physiological opponent to ghrelin, by reducing body weight, food intake and delaying gastric empting [1, 4]. Recently it was also shown that obestatin promotes cell survival and proliferation, increases insulin secretion from pancreatic beta cells, and reduces apoptosis of pancreatic islets [3, 5]. Moreover, in adipose tissue obestatin stimulates GLUT4 translocation to plasma membranes and increases the uptake of free fatty acids and glucose. In addition, obestatin decreases insulin resistance in mice and enhances recovery of myocardial dysfunction in type 1 diabetes mellitus (T1DM) [6, 7]. Based on these and other studies, obestatin was suggested as a potential therapeutic agent in type 2 diabetes mellitus (T2DM) [8].
Taking into account the above, we investigated the long-term effects of obestatin administration on metabolic and hormonal profiles and NASH-like liver dysfunction in mice with experimentally induced T2DM.
Material and methods
Animals
Male Balb/c mice (15 ± 3 g; n = 12 per group) were purchased from Academic Experimental Animal House (Research and Service Center, University of Gdansk, Poland) and housed under standard conditions (12/12 h light/dark cycle, 21 ± 1 °C). The experiments were approved by the Local Ethics Commission for Investigation on Animals, Poznań University of Life Sciences. Prior to conducting experiments, animals were fed standard laboratory diet (Labofed–B, Kcynia, Poland) for 3 weeks and had free access to water.
Induction of diabetes
Diabetes was induced as previously described [9]. After 3 weeks of recovery, standard diet was replaced by a high-fat diet (HFD), containing 60% energy from fat (cat. no. C 1090-60, Altromin, Germany). Mice were fed HFD diet ad libitum for 10 weeks, followed by administration of a single dose of streptozotocin (STZ) (50 mg/kg body weight). After 7 days, animals with blood glucose levels above11 mmol/l were considered diabetic.
Obestatin injections
Diabetic and healthy animals were injected daily (about 5 p.m.) with obestatin (Tocris Bioscience, Bristol, UK) at a dose of 75 nmol/kg b.w. or vehicle (0.9% NaCl) ip for consecutive 30 days. Experimental design is shown in supplementary data.
Real Time PCR
Tripure isolation reagent was used for isolation of total RNA. For the synthesis of cDNA, 1 μg of total RNA and High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, USA) were used. Real Time PCR reaction was performed using gene-specific intron-spanning primers: (supplementary: Table 1), 5× HOT FIREPol® EvaGreen® qPCR Mix Plus (ROX) on QuantStudio 12 K Flex™ Real-time PCR system (Life Technologies, Grand Island, NY, USA). Real Time program included 15 min of initial activation and a three-step amplification program: denaturation at 95 °C for 15 s, annealing at 61.5 °C for 35 s, and elongation at 72 °C for 20 s. The specificity of reaction products was tested by determining the melting points (0.1 C/s transition rate). Relative gene expression was evaluated by Delta Delta CT (ΔΔCT) with Gapd as a reference.
Western blot
Western blot analyses were performed as previously described [10]. In brief, liver fragments were homogenized in radioimmunoprecipitation assay buffer containing: 50 mmol Tris–HCl, pH 8.0 with 150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 10 mM NaF and 1 mM Na3(VO4), supplemented with protease and phosphatase inhibitors (Roche Diagnostics, Mannheim, Germany). Next, liver homogenates were centrifuged at 12,000 × g for 12 min and protein concentration was determined using BCA Protein Assay Kit (Thermo Scientific, USA). 30 μg of protein was loaded, resolved on 12% Tris-HCl SDS-PAGE gel and blotted onto a nitrocellulose membrane (BioRad, USA). Membranes were incubated for 2 h using 3% bovine serum albumin in TBST buffer. Then, membranes were incubated with primary antibodies, diluted to 1:1000 for 24 h at 4 °C (SirT1 cat. no 9475, AMPK cat. no 5831, phospho-AMPK cat. no 2535, phospho-Akt cat. no 4060, Akt cat. no 9272, β-Actin cat. no 3700, Cell Singaling USA). After 24 h, membrane was washed 3 times using TBST buffer and incubated with a secondary antibody for 1 h, and washed again. Signals were visualized using Amersham ECL prime Western Blotting Detection Reagent (GE Healthcare Life Sciences, UK) on VersaDoc system (BioRad, USA). Between quantification of the phosphorylation and determination of the levels of total proteins, membranes were stripped using mild stripping buffer containing: 200 mM glycine, 0.1% SDS, 1% Tween20, pH to 2.2.
Liver triglycerides, cholesterol and glycogen determination
Triglycerides, glycogen and cholesterol contents in liver were determined using the same liver fragments. The values were normalized per mg of liver tissue.
Determination of triglycerides was performed using liver saponification in an ethanol—KOH mixture, as described [11, 12]. In brief, liver fragments were transferred to eppendorf tubes, pre-filled with ethanol—KOH mixture, and incubated overnight at 55 °C. Next, the digested tissue was added to 1 ml EtOH:H2O (1:1) and the mixture was centrifuged for 5 min at 10,000 g. The supernatant was diluted to 1.2 ml with 50% ethanol and mixed. Then, 200 µl of the upper phase was transferred to new tubes, and mixed with 215 µl of 1 M MgCl2. The mixture was vortexed, incubated on ice for 10 min, and then centrifuged for 5 min at 10,000 g. Glycerol concentration in the supernatant was analyzed using Free Glycerol Reagent (F6428, Sigma-Aldrich, Germany).
Liver glycogen content was determined using Glycogen Assay Kit (MAK016, Sigma-Aldrich, Germany), according to manufacturer’s protocol.
Liver cholesterol was analyzed after lipid extraction using cholesterol kit (Pointe Scientific, USA), as described [13, 14].
Intraperitoneal glucose tolerance test (ipGTT)
Glucose tolerance test was performed 5 days before decapitation. After 8 h fasting, glucose (2 g/kg body weight) was injected ip to animals. Glucose concentration was measured in blood drawn from tails 5 min before glucose injection, and at the indicated time points (5, 15, 30, 45, 60, 90, and 120 min.) after glucose injections. Glucose concentration was measured using glucometers AccuCheck Active (Roche Diagnostics GmbH, Mannheim, Germany). The trapezoidal rule was used to determine the area under the curve (AUC).
Intraperitoneal insulin tolerance test (ipITT)
After 2.5 h of fasting in the morning and 5 min before insulin injection, blood glucose concentration was determined. Insulin (1 U/kg body weight) (Novolin,Novo Nordisk, Bagsværd, Denmark) was injected ip into mice at the t = 0 min. time point. Glucose was measured 5, 15, 30, 45, and 60 min after insulin injection. ipITT (n = 5 per group) and ipGTT (n = 5 per group) tests were performed using different cohorts of animals from the same group. The trapezoidal rule was used to determine the AUC.
Metabolic profile
Triglycerides, NEFA, cholesterol (total, high-density lipoproteins (HDL) and low-density lipoproteins (LDL) fraction), β-hydroxybutyrate, fructosamine and glucose levels in serum were determined using colorimetric assays (Pointe Scientific, USA). Serum levels of aminotransferases and transpeptidase were determined using commercial kits (Pointe Scientific, USA). Optical density of samples was measured using a microplate reader Synergy 2 (Biotek, USA). All samples were assayed in duplicates.
Hormone profile and liver cytokine level
Serum concentration of hormones was determined using mouse specific ELISA or RIA kits: mouse insulin ELISA (DRG International Inc, USA), glucagon RIA (Merck-Millipore, USA), mouse adiponectin ELISA (Raybiotech, USA), mouse/rat leptin Quantikine ELISA Kit (R&D Systems, USA) and ghrelin (rat, mouse) EIA kit (Phoenix Pharmaceuticals, Inc. USA). For quantification of the hepatic IL-6 and TNF-α levels, liver fragments were homogenized by sonication in CelLytic™ MT Cell Lysis Reagent (Sigma-Aldrich, Germany). Protein protein concentration was determined using BCA protein Assay Kit (Thermo Scientific, USA). TNF-α and interleukin-6 levels in liver homogenates were measured using Mouse TNF-alpha Quantikine ELISA Kit (R&D Systems, USA) and Mouse IL-6 ELISA Kit (Merck-Millipore, USA). The levels of IL-6 and TNF-α were expressed as pg/mg protein.
HOMA, QUICKI
QUICKI (quantitative insulin sensitivity check index) and HOMA-IR (homeostasis model assessment: insulin resistance) were calculated based on fasting glucose (G0) and insulin (I0) levels using the following formulas: HOMA-IR = (G0 × I0)/22.5; QUICKI = 1/[log(G0) + log(I0)] [15, 16].
Statistical analysis
Results are presented as the arithmetic mean ± SEM (Graph Pad Prism, GraphPad Software, Inc., USA). Statistical analysis was performed using One Way ANOVA followed by the Bonferroni post hoc test. Statistical significance was accepted if *p < 0.05, **p < 0.01 and ***p < 0.001. Changes comparing T2DM groups with healthy controls without obestatin treatment are marked as #p < 0.05, ##-p < 0.01 and † p < 0.05 and †† p < 0.01 shows changes comparing T2DM groups with healthy, obestatin-treated mice.
Results
Obestatin accelerates glucose utilization and enhances insulin sensitivity in healthy and T2DM mice
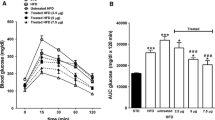
After 30 days of obestatin administration, ipGTT and ipITT were performed (Fig. 1a–d). Insulin sensitivity increased in both, healthy (Fig. 1c; p < 0.01) as well as T2DM mice (Fig. 1c; p < 0.001). Additionally, the rate of glucose utilization was faster in animals treated with obestatin, in both animal groups (Fig. 1a; p < 0.05 non-diabetic; Fig. 1a; p < 0.01 T2DM). The AUC is shown in the Fig. 1b, d.

Effect of obestatin administration on body weight, glucose metabolism and insulin sensitivity in healthy and T2DM mice. (a) ipGTT performed in healthy (black lines) and diabetic (red lines) mice (top figure) and blood glucose. (b) AUC calculated for individual animals during ipGTT. (c) Blood glucose during ipITT in healthy (black lines) and diabetic animals (red lines). (d) AUC calculated for individual animal during ipITT. (e) effects of obestatin on body weight in animals. (f) concentrations of insulin and (g) glucagon at the end of the study. (h) Changes of blood glucose concentrations after experiment in healthy and T2DM mice. Influence of obestatin on insulin resistance/sensitivity calculated by (i) HOMA–IR and (j) QUICKI indexes. Results are showed as means ± SEM, (figure a–d, n = 5 per group and in figure e–j n = 10 per group). Statistically significant changes are marked *p < 0.05, **p < 0.01 and ***p < 0.001
Moreover, body weight decreased after 30 days of obestatin administration in healthy and T2DM mice (Fig. 1e; p < 0.05 and p < 0.01).
In healthy animals, serum insulin levels were higher in obestatin group as compared to vehicle-injected controls (Fig. 1f; p < 0.05), whereas in T2DM mice insulin levels were lower in obestatin group, as compared to vehicle-treated controls (Fig. 1f; p < 0.05). A significant decrease of serum glucagon and blood glucose concentrations were observed in the obestatin-treated diabetic group in comparison to vehicle-treated T2DM group (Fig. 1g; p < 0.05). T2DM mice injected with obestatin had lower levels of serum glucose compared to the control group (Fig. 1h; p < 0.01). In healthy mice such differences were not detectable.
Furthermore, also a long-term parameter of diabetes—fructosamine—was measured in serum. After 30 days, obestatin decreased the concentration of fructosamine in T2DM mice (Table 1; p < 0.05). Furthermore, HOMA-IR was lower in T2DM group after chronic obestatin injections, (Fig. 1i; p < 0.05) whereas QUICKI increased in this animal group (Fig. 1j; p < 0.01). Additionally, we measured cumulative food intake in every cage. We noted a small decrease in food intake after obestatin treatment in both investigated animal groups, however these changes were statistically insignificant (Supp. Fig. 3a).
Obestatin decreases liver mass and modulates hepatic triglycerides, cholesterol and glycogen contents in mice with T2DM
In T2DM animals groups we observed increase in liver mass as well as liver/body weight ratio compared to healthy animals. Liver mass and liver/body weight ratios were lower in obestatin-treated T2DM mice (Fig. 2a; p < 0.01 and Fig. 2b; p < 0.05). Quantification of hepatictriglycerides, cholesterol and glycogen contents showed that obestatin decreased triglycerides (Fig. 2c, d; p < 0.05) and cholesterol content (Fig. 2d; p < 0.01) in T2DM animals. In contrast, hepatic glycogen content in obestatin-treated healthy and T2DM animals increased (Fig. 2e; p < 0.05).

Obestatin’s effect on liver mass (a), liver mass/body weight ratio (b). Changes in hepatic triglycerides (c), cholesterol (d) and glycogen (e) after 30 days of obestatin treatment in healthy and T2DM mice. Results are showed as means ± SEM, n = 10 per group. Statistically significant changes are marked *p < 0.05, **p < 0.01 and ***p < 0.001
Effect of obestatin on alanine aminostransferase (ALT), aspartate aminostransferase (AST) and gamma glutamyl transferase (γGT) levels in serum and hepatic cytokines contents
Next, we investigated the influence of obestatin on serum ALT, AST, γGT, and hepatic TNF-α, and IL-6 levels (as surrogate parameters of steatohepatitis, NASH) in the liver. In T2DM animals, serum levels of ALT, AST, and γGT increased. Moreover, hepatic TNF-α and IL-6 increased in T2DM animals indicating the presence of inflammation in livers.
ALT, AST (Fig. 3a, b; p < 0.05) and γGT (Fig. 3c; p < 0.01) serum levels were lower in obestatin treated T2DM group compared to vehicle treatment Moreover, obestatin lowered hepatic TNF-α (Fig. 3d; p < 0.01) and IL-6 (Fig. 3e; p < 0.05) concentrations in T2DM group. In healthy mice, obestatin administration did not affect both cytokines.

Changes in ALT (a), AST (b), and γGT (c) serum levels after 30 days of obestatin administration in healthy and T2DM mice. Effect of obestatin treatment on hepatic TNF-α (d) and IL-6 (e) protein level. Results are shown as means ± SEM. Statistically significant changes are marked *p < 0.05, **p < 0.01 and ***p < 0.001. n = 10 animals per group
Obestatin increases adiponectin and decreases circulating leptin and ghrelin
Obestatin increased serum adiponectin levels in healthy (Fig. 4a; p < 0.05) and T2DM mice (Fig. 4a; p < 0.01). Obestatin administration reduced leptin levels in T2DM mice (Fig. 4b; p < 0.05). Serum concentrations of ghrelin were lower in obestatin-treated T2DM mice (Fig. 4c; p < 0.05). We noted a small but statistically insignificant decrease in obestatin levels in T2DM mice after obestatin treatment (supp. Fig. 3b).

Adiponectin (a), leptin (b) and ghrelin (c) serum concentrations at the end of the study Results are showed as means ± SEM, n = 10 per group. Statistically significant changes are marked *p < 0.05, **p < 0.01 and ***p < 0.001
Effect of obestatin on lipids and ketones in blood serum
Serum triglycerides and NEFA concentrations were comparable in all experimental groups (Table 1; p < 0.43). However, obestatin modified the levels of total, LDL and HDL cholesterol. Total cholesterol (Table 1; p < 0.01) and LDL cholesterol (Table 1; p < 0.05) concentrations decreased, while HDL cholesterol level increased (Table 1; p < 0.05) in obestatin-treated T2DM group as compared to control mice.
Next, the effects on ketone bodies were studied. obestatin reduced β-hydroxybutyrate levels in T2DM group (Table 1; p < 0.01).
To rule out any distress associated with injections of obestatin serum cortisol was measured. All animals had comparable serum levels of cortisol (Table 1).
Obestatin modulates genes expression in liver
We investigated the effects of obestatin administration on mRNA expression of putative obestatin receptors: Gpr39 and Glp-1r as well as the expression of genes involved in carbohydrate and lipid metabolism: fatty acid binding protein 1 (Fabp1), fatty acid binding protein 4 (Fabp4), fatty acid binding protein 5 (Fabp5), phosphoenolpyruvate carboxykinase (Pepck), glucose-6-phosphatase, catalytic subunit (G6pc).
We found an increase of GPR39 mRNA expression after obestatin administration in healthy and T2DM mice (Fig. 5a; p < 0.05). obestatin increased Glp-1r mRNA expression in livers in T2DM group, only (Fig. 5b; p < 0.05). Moreover, obestatin reduced mRNA expression of Fgf21 (Fig. 5c; p < 0.05), G6pc (Fig. 5f; p < 0.05), Pepck (Fig. 5g; p < 0.01) in T2DM group, whereas mRNA expression of Fabp5 in T2DM was higher as compared to controls (Fig. 5e; p < 0.05). All other tested genes were not altered in obestatin -treated animals.

Hepatic mRNA expression of Gpr39 (a), Glp-1r (b), Fgf21 (c), fatty acid binding proteins: Fabp1 (d), Fabp4 (e), Fabp5 (f) and G6pc (g), Pepck (h), Got1 (i). Results are shown as means ± SEM, n = 6–7 per group. Statistically significant changes are marked *p < 0.05, **p < 0.01 and ***p < 0.001
Obestatin increases both, sirtuin 1 (Sirt1) protein and mRNA expression as well as AKT, and AMPK phosphorylation
Obestatin increased Sirt1 mRNA expression in T2DM group (Fig. 6a; p < 0.05) as well as SIRT1 protein production in both, healthy (Fig. 6b; p < 0.05) and T2DM mice (p < 0.01) (Fig. 6b). Moreover, obestatin increased AMPK and AKT phosphorylation in both animal groups (non-diabetic; p < 0.05 and T2DM; p < 0.01).

Effect of obestatin on hepatic Sirt1 mRNA expression (a) and sirtuin protein levels (b) as well as AMPK (c) and AKT (d) phosphorylation. Results are shown as means ± SEM, n = 8 per group. Statistically significant changes are marked *p < 0.05, **p < 0.01 and ***p < 0.001
Discussion
Obesity, T1DM or T2DM are associated with changes of the receptor GPR39 expression profile [1, 10, 17, 18]. Recently, it was shown that obestatin modulates body weight, food intake, lipid and carbohydrate metabolism [19]. Therefore, in the present study we investigated the role of obestatin in regulating body weights, insulin resistance/sensitivity, lipid and carbohydrate metabolism, and its impact on liver functions in a mouse model of T2DM.
Our results indicate that a chronic obestatin administration over a period of 30 days improves insulin sensitivity, decreases body weight and influences lipid and glucose metabolism in healthy and T2DM mice. We noted a faster glucose clearance as well as an improvement of the insulin sensitivity. These results are in agreement with the results reported by others [6].
In healthy mice, obestatin increased serum insulin concentration in our study, whereas in T2DM animals treated with obestatin insulin levels were lower compared to vehicle-treated controls. It is known that obestatin increases serum insulin concentration in healthy and obese rats, and mice [6, 20]. Our results confirm these reports; however, after 30 days treatment with obestatin, 4-h fasting insulin levels were lower in T2DM mice. Additionally, we found that blood glucose concentration was also lower in obestatin-treated T2DM animals. These data suggest that obestatin increases insulin sensitivity in T2DM mice. Calculation of HOMA-IR and QUICKI confirmed our assumption. Moreover, we observed a decrease of body weight in both animal groups, which may be due to possible reduction of fat mass, as showed earlier by Granata’s lab [6]. Moreover, we measured cumulative food intake in every cage containing n = 10 animals. and not for individual animals. We found a small reduction in the amount of food consumed in mice receiving obestatin. However, the lack of statistical differences could be due to the lack of food intake measurement per animal housed individually. Some research groups showed that obestatin injection is able to reduce food intake in humans and rodents [1, 21, 22]. On the other hand, some studies did not show any inhibitory effects of obestatin on the food consumption [23, 24]. The lack of the monitoring of food intake per animal provides a limitation of this study. However, we suggest that there is a tendency towards reduced food intake after obestatin administration which is reflected by the reduced body weights. These changes could be relevant in the context of metabolic improvements inT2DM mice treated with obestatin
As components of the metabolic syndrome, obesity and T2DM are directly linked to metabolic dysfunctions of the liver, such as non-alcoholic fatty liver disease (NAFLD) or steatohepatitis (NASH) [25, 26]. In the pathogenesis of NAFLD/NASH an excessive lipid accumulation, as well as lipotoxicity due to free fatty acids and high serum level of cholesterol LDL are highly relevant [27]. Additionally, several studies showed that cytokines such as TNF-α and IL-6 are key factors in the development of NASH in humans and animals. In T2DM animals, liver weights were increased. Obestatin reduced liver weights in T2DM mice (p < 0.01) as well as the liver weight/body mass ratios (p < 0.05). Our results are in line with the findings of Gutierrez-Grobe et al. who showed that high obestatin and ghrelin levels correlate with low risk of developing NAFLD [27]. The vast part of data indicate that an increase in ALT, AST and GGT serum levels in T2DM is related to NAFLD in the most cases [28]. In that context we evaluated the impact of obestatin on serum levels of ALT, AST and γGT.
Obestatin decreased ALT, AST and γGT levels in serum in T2DM mice, which may indicate a protective effect of obestatin on NAFLD/NASH. Moreover, it was shown that hepatic IL-6 as well as TNF-α play important roles in pathophysiology of NASH, and that a clear correlation exists between the levels of these cytokines and NAFLD [29,30,31,32,33,34]. Other experiments demonstrated that mice lacking TNF-α and TNF receptors or obese, and a treatment with anti-IL-6 antibodies improves insulin sensitivity [35, 36]. As previously shown, obestatin protects pancreatic islets as well as insulin-producing INS-1E and HIT-T15 beta cell lines from cytokine-induced apoptosis [3]. Our results confirm that in T2DM mice, hepatic IL-6 and TNF-α protein are higher in comparison to control groups. Moreover, we found that the long-term obestatin treatment decreases the hepatic protein levels of both cytokines, which is in line with the concept of obestatin protection against NAFLD in T2DM.
Adipokines, like leptin and adiponectin play important roles at inducing insulin resistance and diabetes [37,38,39]. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase [40]. Decreased serum adiponectin levels correlate with insulin resistance and the severity of diabetes [41]. It is known that in contrast to adiponectin, leptin serum levels are higher in diabetes [42]. Moreover, high leptin levels in T2DM are related to the high fat mass in obese subjects. In addition, leptin may lead to lipid dysregulation and deterioration of glucose control [43, 44]. Several studies demonstrated that Obestatin affects adiponectin and leptin levels, thereby protecting from diet-induced insulin resistance [6]. Therefore, we measured the effect of obestatin on leptin and adiponectin concentrations in serum. Obestatin increased adiponectin serum level in both healthy and diabetic animals, whereas the decrease of the leptin concentration was observed in T2DM animals, only. These data suggest that obestatin regulates carbohydrate and lipid metabolism.
Ghrelin is important in the context of the regulation of other important metabolic events. Ghrelin is derived from the same precursor gene as obestatin, however, in contrast to obestatin, ghrelin stimulates appetite [45]. Ghrelin regulates liver functions by increasing triglycerides content and by increasing G6pc, Acc, Fas mRNA levels [46]. In our study, obestatin lowered serum ghrelin concentration, hepatic triglycerides content as well as G6pc mRNA of expression. Vestergaard et al. showed also that ghrelin infusion induces insulin resistance in humans [47]. Concordantly, acute administration of ghrelin in gastrectomized patients decreased insulin-stimulated glucose utilization [48]. A decrease of ghrelin levels by obestatin could also explain the reduction of insulin resistance in T2DM mice [47].
As mentioned above, persistent high levels of LDL cholesterol in serum may lead to liver dysfunction. In the present study, we found that obestatin administration had no effect on triglycerides or NEFA concentrations in serum. In 2011, Agnew and coworkers showed that N-terminally PEGylated obestatin decreased rat serum triglycerides (TG) level. However, there was no difference in the concentration of TG, when the regular obestatin was used [49]. Moreover, results obtained by Ren et al. confirmed that a long-term obestatin as well as TAT-obestatin injection had no effect on serum TG in mice [50]. However, obestatin decreased total as well as LDL cholesterol concentrations, and simultaneously increased HDL cholesterol level which is in line with results of obestatin-treated STZ-diabetic rats [51]. Moreover, we investigated the effect of obestatin on expression of genes involved in lipid, carbohydrate and amino-acids metabolism. It is known that G6pc and Fgf21 are involved in regulation of gluconeogenesis and glycogenolysis [52]. We found that obestatin administration modulates G6pc, Pepck, Fabp5 and Fgf21 mRNA expression levels.
As demonstrated in the present study, obestatin reduces the glycogen content in the liver. This effect can be explained by an influence of obestatin of the expression of above named genes which are relevant for glucose metabolism. Thus, it cannot be excluded that obestatin normalizes alterations of lipid and carbohydrate metabolism in diabetic NASH.
Sirtuin 1 (Sirt1) mRNA and protein, AKT (protein kinase B) and AMPK (5′AMP-activated protein kinase) phosphorylation decreased in T2DM, which is relevant in the context of development of insulin resistance and impaired regulation of hepatic glucose metabolism [53,54,55]. Recent studies suggest that obestatin can regulate Sirt1 expression and activate AKT and AMPK signaling pathways [6, 18, 19, 56]. Moreover, impairments of these signaling pathways are related to T2DM, and their reactivation improves glucose metabolism [53, 54, 57, 58]. Additionally, stimulation of AMP-activated protein kinase in the liver is considered as one of the potential strategy for the treatment of hepatic disorders and T2DM [54, 59]. In our study, we found that obestatin phosphorylates both, AKT and AMPK. Furthermore, obestatin increases Sirt1 mRNA and protein production in liver.
Our study is focused on the characterization of obestatin impact on various liver functions in T2DM, however, we are aware that the regulation of metabolic abnormalities by obestatin in T2DM is more complex. Additional limitation of our study is the use of a single animal model. However, STZ-diabetes in animals is a well-established model of T1DM and T2DM (in conjunction with HFD feeding) [9, 60, 61]. This is a rationale that justifies the use of a HFD/STZ diabetic animal model. This model has some advantages as compared to other genetically determined animal models, e. g., db/db, ob/ob mice or ZDF rats. In HFD/STZ mice diabetes is associated with obesity and a beta cell failure. OBST is a peptide involved in regulation of food intake and lipid metabolism. Moreover, this model of diabetes is accompanied by fatty liver, allowing us to study the effects of OBST on these diabetic complications resembling the human NASH-like pathology [62,63,64].
In summary, we found that the chronic obestatin treatment for 30 days of animals with T2DM leads to decreased body and liver weights, improves liver functions, carbohydrate and lipid metabolism, as well as insulin-resistance. Additionally, we found that obestatin increases Sirt1 mRNA and protein expression, and activates AKT, AMPK related signaling pathways, which are relevant in the context of improved insulin sensitivity. These data indicate that obestatin may alleviate diabetes associated NASH, however further in-depth studies focusing entirely on this aspect are needed. Our results suggest that obestatin can be further evaluated as a potential promising target for therapy of T2DM.
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- AKT:
-
Protein kinase B
- ALT:
-
Alanine aminostransferase
- AMPK:
-
5′AMP-activated protein kinase
- AST:
-
Aspartate aminostransferase
- FABP1:
-
Fatty acid binding protein 1
- FABP4:
-
Fatty acid binding protein 4
- FABP5:
-
Fatty acid binding protein 5
- FAS:
-
Fatty acids synthase
- FGF-21:
-
Fibroblastic growth factor 21
- G6Pc:
-
Glucose-6-phosphatase catalytic subunit G6Pc
- GAPD:
-
Glyceraldehyde-3-phosphate dehydrogenase
- γGT:
-
Gamma glutamyl transferase
- GLP-1R:
-
Glucagon like peptide-1 receptor
- GLUT4:
-
Glucose transporter 4
- GOT:
-
Glutamic-oxaloacetic transaminase
- GPR39:
-
G-protein-coupled receptor 39
- HFD:
-
High fat diet
- IL-6:
-
interleukin 6
- IP:
-
Intraperitoneally
- OBST:
-
Obestatin
- PEPCK:
-
Phosphoenolpyruvate carboxykinase
- STZ:
-
streptozotocin
- T1DM:
-
Type 1 diabetes mellitus
- T2DM:
-
Type 2 diabetes mellitus
- TG:
-
Triglycerides
- TNF-α:
-
Tumor necrosis factor alpha
References
J. Zhang, P. Ren, O. Avsian-kretchmer, C. Luo, R. Rauch, Obestatin, a peptide encoded by the Ghrelin gene, opposes Ghrelin’s effects on food intake. Science (80−.) 310, 996–999 (2005)
N. Chartrel, R. Alvear-Perez, J. Leprince, X. Iturrioz, A. Reaux-Le Goazigo, V. Audinot, P. Chomarat, F. Coge, O. Nosjean, M. Rodriguez, J. Galizzi, J. Boutin, H. Vaudry, C. Llorens-Cortes, Comment on“ obestatin, a peptide encoded by the Ghrelin gene, opposes Ghrelin’s effects on food intake.” Science (80–.) 315, 766 (2007)
R. Granata, F. Settanni, D. Gallo, L. Trovato, L. Biancone, V. Cantaluppi, R. Nano, M. Annunziata, P. Campiglia, E. Arnoletti, Obestatin promotes survival of pancreatic bCells and human islets and induces expression of genes involved in the regulation of b-Cell mass and function. Diabetes 57, 967–979 (2008)
B. De Smet, T. Thijs, T.L. Peeters, I. Depoortere, Effect of peripheral obestatin on gastric emptying and intestinal contractility in rodents. Neurogastroenterol. Motil. 19, 211–217 (2007)
R. Granata, M. Volante, F. Settanni, C. Gauna, C. Ghe, M. Annunziata, B. Deidda, I. Gesmundo, T. Abribat, A.J. Van Der Lely, G. Muccioli, E. Ghigo, M. Papotti, Unacylated ghrelin and obestatin increase islet cell mass and prevent diabetes in streptozotocin-treated newborn rats. J. Mol. Endocrinol. 45, 9–17 (2010)
R. Granata, D. Gallo, R.M. Luque, a Baragli, F. Scarlatti, C. Grande, I. Gesmundo, J. Cordoba-Chacon, L. Bergandi, F. Settanni, G. Togliatto, M. Volante, S. Garetto, M. Annunziata, B. Chanclon, E. Gargantini, S. Rocchietto, L. Matera, G. Datta, M. Morino, M.F. Brizzi, H. Ong, G. Camussi, J.P. Castano, M. Papotti, E. Ghigo, Obestatin regulates adipocyte function and protects against diet-induced insulin resistance and inflammation. FASEB J. 26, 3393–3411 (2012)
M. Aragno, R. Mastrocola, C. Ghé, E. Arnoletti, E. Bassino, G. Alloatti, G. Muccioli, Obestatin induced recovery of myocardial dysfunction in type 1 diabetic rats: underlying mechanisms. Cardiovasc. Diabetol. 11, 129 (2012)
E. Cowan, K.J. Burch, B.D. Green, D.J. Grieve, Obestatin as a key regulator of metabolism and cardiovascular function with emerging therapeutic potential for diabetes. Br. J. Pharmacol. 44, 2165–2181 (2016)
A. Vater, S. Sell, P. Kaczmarek, C. Maasch, K. Buchner, E. Pruszynska-Oszmalek, P. Kolodziejski, W.G. Purschke, K.W. Nowak, M.Z. Strowski, S. Klussmann, A. Mixed Mirror-image, DNA/RNA aptamer inhibits glucagon and acutely improves glucose tolerance in models of type 1 and type 2 diabetes. J. Biol. Chem. 288, 21136–21147 (2013)
P.A. Kolodziejski, E. Pruszynska-Oszmalek, M. Sassek, P. Kaczmarek, D. Szczepankiewicz, M. Billert, P. Maćkowiak, M. Z. Strowski, W. Nowak, K. Changes, in obestatin gene and receptor-GPR39 expression in peripheral tissues of rat models of obesity, type 1 and type 2 diabetes. J. Diabetes 9, 353–361 (2017)
A.Z. Caron, X. He, W. Mottawea, E.L. Seifert, K. Jardine, D. Dewar-Darch, G.O. Cron, M.E. Harper, A. Stintzi, M.W. McBurney, The SIRT1 deacetylase protects mice against the symptoms of metabolic syndrome. FASEB J. 28, 1306–1316 (2014)
B. Emanuelli, S.G. Vienberg, G. Smyth, C. Cheng, K.I. Stanford, M. Arumugam, M.D. Michael, A.C. Adams, A. Kharitonenkov, C.R. Kahn, Interplay between FGF21 and insulin action in the liver regulates metabolism. J. Clin. Invest. 124, 515–527 (2014)
C. Allain, L. Poon, Enzymatic determination of total serum cholesterol. Clin. Chem. 20, 470–475 (1974)
J. Folsh, M. Less, G. Sloane Stanley, A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 (1957)
D.R. Matthews, J.P. Hosker, A.S. Rudenski, B.A Naylor, D.F. Treacher, R.C. Turner, Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 28, 412–419 (1985)
A. Katz, S.S. Nambi, K. Mather, A. Baron, D. Follmann, G. Sullivan, M.J. Quon, Quantitative insuln sensitivity check index: a simple, accurate methof for assessing insulin sensitivity in humans. J. Clin. Endocrinol. Metab. 85, 2402–2410 (2000)
E. Pruszynska-Oszmalek, D. Szczepankiewicz, I. Hertig, M. Skrzypski, M. Sassek, P. Kaczmarek, P.A. Kolodziejski, P. Mackowiak, K.W. Nowak, M.Z. Strowski, T. Wojciechowicz, Obestatin inhibits lipogenesis and glucose uptake in isolated primary rat adipocytes. J. Biol. Regul. Homeost. Agents. 27, 23–33 (2013)
U. Gurriarán-Rodríguez, O. Al-Massadi, A. Roca-Rivada, A.B. Crujeiras, R. Gallego, M. Pardo, L.M. Seoane, Y. Pazos, F.F. Casanueva, J.P. Camiña, Obestatin as a regulator of adipocyte metabolism and adipogenesis. J. Cell. Mol. Med. 15, 1927–1940 (2011)
I. Gesmundo, D. Gallo, E. Favaro, E. Ghigo, R. Granata, Obestatin: A new metabolic player in the pancreas and white adipose tissue. IUBMB Life. 65, 976–982 (2013)
E.M. Egido, R. Hernandez, J. Marco, R.A. Silvestre, Effect of obestatin on insulin, glucagon and somatostatin secretion in the perfused rat pancreas. Regul. Pept. 152, 61–66 (2009)
B.D. Green, N. Irwin, P.R. Flatt, Direct and indirect effects of obestatin peptides on food intake and the regulation of glucose homeostasis and insulin secretion in mice. Peptides. 28, 981–987 (2007)
E. Bresciani, D. Rapetti, F. Donà, I. Bulgarelli, L. Tamiazzo, V. Locatelli, A. Torsello, Obestatin inhibits feeding but does not modulate GH and corticosterone secretion in the rat. J. Endocrinol. Invest. 29, 2–4 (2006)
G. Gourcerol, M. Million, D.W. Adelson, Y. Wang, L. Wang, J. Rivier, D.H. St-Pierre, Y. Taché, Lack of interaction between peripheral injection of CCK and obestatin in the regulation of gastric satiety signaling in rodents. Peptides. 27, 2811–2819 (2006)
P. Zizzari, R. Longchamps, J. Epelbaum, M.T. Bluet-Pajot, Obestatin partially affects ghrelin stimulation of food intake and growth hormone secretion in rodents. Endocrinology 148, 1648–1653 (2007)
K. Cusi, Nonalcoholic fatty liver disease in type 2 diabetes mellitus. Curr. Opin. Endocrinol. Diabetes. Obes. 16, 141–149 (2009)
L. Bozzetto, A. Prinster, S. Cocozza, A.A. Rivellese, G. Annuzzi, Role of type 2 diabetes mellitus in nonalcoholic fatty liver disease. Eur. J. Clin. Invest. 41, 1368–1368 (2011)
Y. Gutierrez-Grobe, I. Villalobos-Blasquez, K. Sánchez-Lara, A.R. Villa, G. Ponciano-Rodríguez, M.H. Ramos, N.C. Chavez-Tapia, M. Uribe, N. Méndez-Sánchez, High ghrelin and obestatin levels and low risk of developing fatty liver. Ann. Hepatol. 9, 52–57 (2010)
J.M. Clark, F.L. Brancati, A.M. Diehl, The prevalence and etiology of elevated aminotransferase levels in the United States. Am. J. Gastroenterol. 98, 960–967 (2003)
S.S. Abiru, K. Migita, Y. Maeda, M. Daikoku, M. Ito, K. Ohata, S. Nagaoka, T. Matsumoto, Y. Takii, K. Kusumoto, M. Nakamura, A. Komori, K. Yano, H. Yatsuhashi, K. Eguchi, H. Ishibashi, Serum cytokine and soluble cytokine receptor levels in patients with non-alcoholic steatohepatitis. Liver Int. 26, 39–45 (2006)
J.W. Haukeland, J.K. Damas, Z. Konopski, E.M. Løberg, T. Haaland, I. Goverud, P.A. Torjesen, K. Birkeland, K. Bjøro, P. Aukrust, Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2 *. J. Hepatol. 44, 1167–1174 (2006)
J. Crespo, A. Cayón, P. Fernández-Gil, M. Hernández-Guerra, M. Mayorga, A. Domínguez-Díez, J. Fernández-Escalante, F. Pons-Romero, Gene Expression of Tumor Necrosis Factor α and TNF-Receptors, p55 and p75, in Nonalcoholic Steatohepatitis Patients. Hepatology 34, 1158–1163 (2000)
A. Wieckowska, B. Papouchado, Z. Li, R. Lopez, N. Zein, A. Feldstein, Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am. J. Gastroenterol. 103, 1372–1379 (2008)
A.J. Wigg, I.C. Roberts-Thomson, R.B. Dymock, P.J. McCarthy, R.H. Grose, A.G. Cummins, The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor α in the pathogenesis of non-alcoholic steatohepatitis. Gut. 48, 206–211 (2001)
Y. Wang, L.M. Ausman, R.M. Russell, A.S. Greenberg, X.-D. Wang, Increased apoptosis in high-fat diet-induced nonalcoholic steatohepatitis in rats is associated with c-Jun NH2-terminal kinase activation and elevated proapoptotic bax. J. Nutr. 138, 1866–1871 (2008)
K. Uysal, S. Wiesbrock, M. Marino, G. Hotamisligil, Protection from obesity- induced insulin resistance in mice lacking TNF- alpha function. Nature 389, 610–614 (1997)
P.J. Klover, A.H. Clementi, R.A. Mooney, Interleukin-6 depletion selectively Improves hepatic insulin action in obesity. Endocrinology. 146, 3417–3427 (2016)
M. Maffei, J. Halaas, E. Ravussin, R.E. Pratley, G.H. Lee, Y. Zhang, H. Fei, S. Kim, R. Lallone, S. Ranganathan, Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1, 1155–1161 (1995)
K. Hotta, T. Funahashi, Y. Arita, M. Takahashi, M. Matsuda, Y. Okamoto, H. Iwahashi, H. Kuriyama, N. Ouchi, K. Maeda, M. Nishida, S. Kihara, N. Sakai, T. Nakajima, K. Hasegawa, M. Muraguchi, Y. Ohmoto, T. Nakamura, S. Yamashita, Adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 20, 1595–1599 (2000)
M. Matsubara, S. Maruoka, S. Katayose, Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur. J. Endocrinol. 147, 173–180 (2002)
T. Yamauchi, J. Kamon, Y. Minokoshi, Y. Ito, H. Waki, S. Uchida, S. Yamashita, M. Noda, S. Kita, K. Ueki, K. Eto, Y. Akanuma, P. Froguel, F. Foufelle, P. Ferre, D. Carling, S. Kimura, R. Nagai, B.B. Kahn, T. Kadowaki, Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 8, 1288–1295 (2002)
K. Hotta, T. Funahashi, Y. Arita, M. Takahashi, M. Matsuda, Y. Okamoto, H. Iwahashi, H. Kuriyama, N. Ouchi, K. Maeda, M. Nishida, S. Kihara, N. Sakai, T. Nakajima, K. Hasegawa, M. Muraguchi, Y. Ohmoto, T. Nakamura, S. Yamashita, Adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. 20, 1595–1599 (2000)
S. Fischer, M. Hanefeld, S.M. Haffner, C. Fusch, U. Schwanebeck, C. Kohler, K. Fucker, U. Julius, Insulin-resistant patients with type 2 diabetes mellitus have higher serum leptin levels independently of body fat mass. Acta Diabetol. 39, 105–110 (2002)
S. Zhang, Q. Zhang, L. Zhang, C. Li, H. Jiang, Expression of ghrelin and leptin during the development of type 2 diabetes mellitus in a rat model. Mol. Med. Rep. 7, 223–228 (2013)
B.P. Cummings, Leptin therapy in type 2 diabetes. Diabetes. Obes. Metab. 15, 607–612 (2013)
A. Wren, L. Seal, M. Cohen, A. Brynes, G. Frost, K. Murphy, W. Dhillo, M. Ghatei, S. Bloom, Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 86, 5992–5995 (2001)
R. Barazzoni, A. Bosutti, M. Stebel, M.R. Cattin, E. Roder, L. Visintin, L. Cattin, G. Biolo, M. Zanetti, G. Guarnieri, Ghrelin regulates mitochondrial-lipid metabolism gene expression and tissue fat distribution in liver and skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 288, 228–235 (2005)
E.T. Vestergaard, L.C. Gormsen, N. Jessen, S. Lund, T.K. Hansen, N. Moller, J. Otto, L. Jorgensen, Ghrelin infusion in humans induces acute insulin resistance and lipolysis independent of growth hormone signaling. Diabetes 57, 3205–3210 (2008)
S.S. Damjanovic, N.M. Lalic, P.M. Pesko, M.S. Petakov, A. Jotic, D. Miljic, K.S. Lalic, L. Lukic, M. Djurovic, V.B. Djukic, Acute effects of Ghrelin on insulin secretion and glucose disposal rate in Gastrectomized patients. J. Clin. Endocrinol. Metab. 91, 2574–2581 (2016)
A. Agnew, D. Calderwood, O.P. Chevallier, B. Greer, D.J. Grieve, B.D. Green, Chronic treatment with a stable obestatin analog significantly alters plasma triglyceride levels but fails to influence food intake; fluid intake; body weight; or body composition in rats. Peptides. 32, 755–762 (2011)
G. Ren, Z. He, P. Cong, H. Chen, Y. Guo, J. Yu, Z. Liu, Q. Ji, Z. Song, Y. Chen, Peripheral administration of TAT-obestatin can influence the expression of liporegulatory genes but fails to affect food intake in mice. Peptides. 42, 8–14 (2013)
S. El Sawy, R. El-Sherbiny, M. El-Saka, R. El-Shaer, Effect of obestatin on normal, diabetic, and obese male albino rats. Tanta Med. J. 44, 16 (2016)
C. Wang, J. Dai, M. Yang, G. Deng, S. Xu, Y. Jia, Silencing of FGF-21 expression promotes hepatic gluconeogenesis and glycogenolysis by regulation of the. FEBS J. 281, 2136–2147 (2014)
M. Kitada, D. Koya, SIRT1 in type 2 diabetes: mechanisms and therapeutic potential. Diabetes Metab. J 37, 315–325 (2013)
K. Coughlan, R. Valentine, N. Ruderman, A. Saha, AMPK activation: A therapeutic target for type 2 diabetes? Diabetes, Metab. Syndr. Obes. Targets Ther. 7, 241–253 (2014)
B.K. Smith, K. Marcinko, E.M. Desjardins, J.S. Lally, R.J. Ford, G.R. Steinberg, Treatment of nonalcoholic fatty liver disease : role of AMPK. Am. J. Physiol. 311(4), 730–740 (2016)
C.J.P. Alvarez, M. Lodeiro, M. Theodoropoulou, J.P. Camiña, F.F. Casanueva, Y. Pazos, Obestatin stimulates Akt signalling in gastric cancer cells through beta-arrestin-mediated epidermal growth factor receptor transactivation. Endocr. Relat. Cancer. 16, 599–611 (2009)
A.S. Banks, N. Kon, C. Knight, M. Matsumoto, R. Gutierrez-Juarez, L. Rossetti, W. Gu, D. Accili, SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 8, 333–341 (2008)
C. Sun, F. Zhang, X. Ge, T. Yan, X. Chen, X. Shi, Q. Zhai, SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 6, 307–319 (2007)
B. Viollet, M. Foretz, B. Guigas, S. Horman, R. Dentin, L. Bertrand, L. Hue, F. Andreelli, Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J. Physiol. 1, 41–53 (2006)
W.J. Schnedl, S. Ferber, J.H. Johnson, C.B. Newgard, STZ transport and cytotoxicity: specific enhancement in GLUT2-expressing cells. Diabetes. 43, 1326–1333 (1994)
T. Szkudelski, The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol. Res. 50, 537–546 (2001)
L. Pozzo, A. Vornoli, I. Coppola, C.M. Croce, Della, L. Giorgetti, P.G. Gervasi, V. Longo, Effect of HFD/STZ on expression of genes involved in lipid, cholesterol and glucose metabolism in rats. Life Sci. 166, 149–156 (2016)
A. Teufel, T. Itzel, W. Erhart, M. Brosch, X.Y. Wang, Y.O. Kim, W. von Schönfels, A. Herrmann, S. Brückner, F. Stickel, J.-F. Dufour, T. Chavakis, C. Hellerbrand, R. Spang, T. Maass, T. Becker, S. Schreiber, C. Schafmayer, D. Schuppan, J. Hampe, Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology 151, 513–525 (2016)
H. Nakagawa, Recent advances in mouse models of obesity- and nonalcoholic steatohepatitis-associated hepatocarcinogenesis. World J. Hepatol. 7, 2110–2118 (2015)
Acknowledgements
P.A.K. is the recipient of a 2013 fellowship program UE Human Capital. This study forms part of the PhD thesis of P.A.K. This study was partially supported by the National Science Centre, Poland 2015/19/N/NZ4/00572 PRELUDIUM grant. M.Z.S. was supported by the Deutsche Forschungsgemeinschaft and Deutsche Diabetes Stiftung.
Author contributions
P.A.K. designed the study, obtained the data and wrote the manuscript. E.P.O., contributed to the study design, experiment performing, edited, supported and critically revised the manuscript and contributed to the discussion. M.Z.S., K.W.N., contributed to the study design, edited, supported and critically revised the manuscript and contributed to the discussion. All authors have given final approval to the current version to be published.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Kołodziejski, P.A., Pruszyńska-Oszmałek, E., Strowski, M.Z. et al. Long-term obestatin treatment of mice type 2 diabetes increases insulin sensitivity and improves liver function. Endocrine 56, 538–550 (2017). https://doi.org/10.1007/s12020-017-1309-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-017-1309-2