
Abstract
Glucocorticoid-induced osteoporosis is the most frequent form of secondary osteoporosis caused by chronic exposure to glucocorticoid excess. Pathogenesis of glucocorticoid-induced osteoporosis is multifactorial including direct effects of glucocorticoids on bone cells and indirect effects of glucocorticoids on several neuroendocrine and metabolic pathways. Fragility fractures occur early in glucocorticoid-induced osteoporosis and anti-osteoporotic drugs along with calcium and vitamin D should be started soon after exposure to glucocorticoid excess. This paper summarizes some of the main topics discussed during the 9th Glucocorticoid-Induced Osteoporosis Meeting (Rome, April 2016) with a specific focus on the role of growth hormone/insulin-like growth factor-1 and parathyroid hormone/vitamin D axes in the pathogenesis of glucocorticoid-induced osteoporosis and the controversial aspects concerning therapeutic approach to skeletal fragility in this clinical setting.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucocorticoid-induced osteoporosis (GIO) is the most frequent form of secondary osteoporosis in men as well as in women [1]. GIO is almost always caused by exogenous glucocorticoids which are widely used in the treatment of autoimmune, pulmonary and gastrointestinal disorders, as well as in patients after organ transplantation and with neoplastic diseases. Fracture risk increases rapidly after starting oral corticosteroid treatment and is also related to the dose and duration of glucocorticoid exposure [2].
Endogenous hypercortisolism is less frequently a cause of GIO [3, 4] and fragility fractures can be the presenting manifestation of endogenous hypercortisolism, either clinical or subclinical [5, 6].
This paper summarizes some of the main topics discussed during the 9th GIO Meeting (Rome, April 2016) on pathophysiological and treatment aspects of GIO.
Pathophysiology of GIO: focus on growth hormone/insulin-like growth factor-1 (GH/IGF-1) and parathyroid hormone (PTH)/vitamin D axes
GH/IGF-I axis
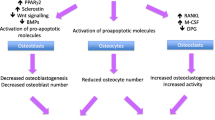
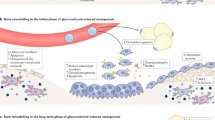
The central pathophysiological mechanism of GIO is reduced bone formation, due to actions of glucocorticoid excess on osteoblast differentiation and function [7]. Moreover, glucocorticoids may affect the survival, metabolism and function of osteocytes, increasing apoptosis and modifying the elastic modulus surrounding osteocyte lacunae [8]. The negative effects of glucocorticoids on osteoblasts and osteocytes account for chronic impairment of bone quality and disproportionate loss of bone strength in relation to bone mass in GIO [9]. However, during the first phases of glucocorticoid excess exposure, a significant increase in bone resorption may occur, ultimately leading to the observed early increase in the risk of fractures [1, 2].
Besides the direct effects on bone cells, glucocorticoids may also have indirect effects mediated by derangements in neuroendocrine signals. Glucocorticoids modulate GH secretion by various and competing effects on the hypothalamus and pituitary gland, with final effects depending on hormone concentrations and time of exposure [10]. Glucocorticoids are among the most prominent peripheral hormones involved in the regulation of GH secretion [10]. Specifically, glucocorticoids were shown to be essential for the differentiation and maturation of GH-secreting pituitary cells [11]. Moreover, glucocorticoids stimulate GH secretion either by direct effects on pituitary cells or by increasing their GH secretory response to GH-releasing hormone [12, 13]. Low circulating levels of glucocorticoids cause a functional impairment of GH secretion which is rapidly reverted by restoring normal cortisol values [14–16]. However, when glucocorticoid levels exceed the physiological range, an increase in hypothalamic somatostatin tone may occur with consequent impairment of GH secretion [17–20]. This inhibitory effect on pituitary GH secretion was observed even when the glucocorticoid excess was mild, as in patients treated with inhaled corticosteroids [21] and in those with “subclinical” endogenous hypercortisolism [22]. Moreover, glucocorticoid excess may suppress the peripheral expression of GH receptors impairing the GH-mediated synthesis of IGF-1 and thus amplifying the effects of functional GH deficiency (GHD) on target tissues [23]. On the other hand, there is cross-talking between glucocorticoids and the GH-IGF-I axis, since the peripheral metabolism of glucocorticoids by 11-β-hydroxysteroid dehydrogenase (11-βHSD) is modulated by GH and activation of cortisone to cortisol in target tissues is amplified by GHD [24]. The real impact of GHD on fracture risk in GIO is still not clearly known. However, since GH is anabolic for bone, one could argue that GHD may contribute to the impairment in bone quality in patients exposed to glucocorticoid excess [10, 25]. In fact, both exposure to glucocorticoid excess and GHD are associated with a “low-turnover osteoporosis” and high vertebral fracture risk [26, 27]. Theoretically, due to the kinetics of bone markers in subjects treated with glucocorticoids and GH, a window of opportunity may exist for GH treatment in this clinical context, although data are still few and not conclusive [10]. In adults with GIO, short-term (7 days) recombinant human GH treatment was able to significantly increase bone turnover markers potentially leading to favorable chronic effects on bone remodeling [28]. Moreover, GH treatment was shown to exert positive effects on the protein wasting syndrome caused by glucocorticoid excess with potential favorable effects on the glucocorticoid-induced sarcopenia [29].
In children with Cushing’s syndrome (CS), it is well recognized that growth at diagnosis, linear growth and development after cure are usually adversely affected [30–35]. Magiakou et al. [36] reported growth retardation at diagnosis in 83 % of paediatric CS patients—mean height before surgery −1.3 SDS and mean height at one year after treatment −1.2 SDS. According to Magiakou et al. [36], the major mechanism for growth retardation has been considered the glucocorticoid-induced resistance of target tissues to IGF-I and other growth factors. However, they reported that some patients that had not completed their growth at the time of diagnosis increased in height the year after surgical cure. They showed that patients with Cushing’s disease (CD) have marked GH suppression when the disease was active, which, in their cohort, did not appear to be a major contributor to the growth retardation reported in this condition. In their population GH hyposecretion continued for at least a year during convalescence, in spite of significant increases in the growth rate in patients that had not completed their growth. The high prevalence of growth failure in CD at presentation were also confirmed in the study of Savage et al. [37], reporting this as a symptom at diagnosis in 74 % of patients, with a mean height of −1.81 SDS. The same group [38] reported that 9/10 patients treated for CD failed to show clear evidence of catch-up growth after successful cortisol-lowering therapy. However, in 8/10 there was evidence of GHD, and hGH therapy was started early after transsphenoidal surgery (TSS) or radiotherapy. Assessment after one year of hGH therapy showed an increase of height SD score compared with the target height. They confirmed the high frequency of GH deficiency after treatment in pediatric CD, both by surgery alone or associated with pituitary radiotherapy and emphasized the importance for investigation of GHD. According to their results, early treatment with hGH appears to be indicated in the majority of patients, and appropriate treatment will lead to a considerable improvement in final height. Chan et al. also confirmed the high incidence of GHD in children receiving external beam radiotherapy after failed surgery; according to their population, at one year 5/6 patients showed GHD but they also demonstrated that it may recover with normalization of cortisol (3/4 patients at 9 years follow-up) [39].
Hypercortisolism in both adults and children is associated with loss of skeletal mass, osteopenia and osteoporosis, and can lean to an increased vertebral fracture risk [40–48]. Limited data are available regarding bone mineral density (BMD) in children with CD, but they suggest a high prevalence of reduced with associated osteopenia and osteoporosis [49].
Scommegna et al. [50] evaluated BMD in 2 groups of patients with CD: group 1 comprised 8 patients at diagnosis while group 2 comprised 11 patients 4.5 years after cure, of whom 8/9 with GHD were treated with hGH. Patients in group 1 showed a mean lumbar spine (LS) BMD Z score of −1.04 SD, while in group 2 it was −0.38 SD. They showed variability in BMD at diagnosis and near normal BMD after cure of pediatric CD, demonstrating that the mild reduction in BMD in pediatric CD is reversible after replacement of pituitary hormone deficiencies that are frequently present.
Interestingly, Leong et al. reported the first case of an identical twin who at age 15 years was successfully treated by TSS for CD: the bone density at the LS was compared to the healthy twin at baseline (−3.2 vs. −0.1 SD) and after 27 months after surgery (−1.9 vs. 0), the Cushingoid twin achieving a final height of 21 cm less compared to the normal twin [51]. Subsequently, they prospectively analysed 14 children and adolescents with CD, successfully treated, during regular follow-up for at least 3 years of remission. They showed that BMD scores progressively increased from a baseline of −1.79 SD to −0.46 SD during the first 2 years of follow-up in LS; from −1.4 SD to 0.4 SD during the first 2 years of follow-up in total hip and from −1.2 SD to −0.49 SD during the first 2 years of follow-up in the femoral neck (FN) and without a significant increase during the third year of follow-up [52].
Lodish et al. [53] retrospectively evaluated BMD in 35 children with CD before and after TSS, 16 of them with a follow-up scan 13–18 months after TSS. They showed that preoperatively 38 and 23 % of patients had osteopenia at the LS and FN, respectively. In their population, BMD Z scores improved from a baseline of −1.6 SD to −0.84 SD at follow-up at LS and from a baseline of −1.04 SD to 0.15 SD at follow-up at FN. They demonstrated that vertebral BMD was more severely affected than femoral BMD, and was independent of the degree or duration of hypercortisolism. Cortisol normalization after TSS led to an improvement in BMD in the LS for children with CD, and osteopenia in this group could be reversible. However, removal of the cause of endogenous hypercortisolism does not always induce complete recovery of bone health. Moreover, guidelines for the management of osteoporosis induced by endogenous hypercortisolism are not available and data of the literature do not allow an evidence-based approach, a single-case evaluation being often needed [4, 6].
PTH/vitamin D axis
Interactions between both parathyroid hormone (PTH) and vitamin D with steroid therapy have been a matter of intensive research.
The most recent studies have shown that vitamin D enhances glucocorticoid responses in human peripheral blood mononuclear cells [54]. Indeed, vitamin D stimulates glucocorticoid induction of mitogen-activated protein kinase phosphatase; furthermore, vitamin D enhances glucocorticoid inhibition of lipopolysaccharide-induced interleukin-6. Granulocyte-macrophage colony stimulating factor, found in culture supernatants from CD14 cells, was shown to mediate vitamin D enhancement of increased production of the mediator complex subunit 14. These data therefore show synergistic actions of glucocorticoid and vitamin D on inflammation even in human monocytes.
These studies lend support to the clinical observations that, according to some authors, vitamin D deficiency has a negative impact on autoimmune rheumatic disease, including initial disease development and worsening of the disease once present. In this context, a recent study by Tse and coworkers [55] demonstrated a dose-dependent effect of intermittent oral corticosteroid use on bone mineral accretion in boys with asthma, which was significantly modified by basal values of vitamin D levels. The negative effects of steroids are indeed exacerbated by low vitamin D values, suggesting that children with asthma and frequent exacerbations, thus requiring steroid treatment, might benefit from vitamin D status assessment; in these circumstances, vitamin D supplementation might help to preserve bone density.
A recent meta-analysis [56] addressed the hypothetical issue of alterations of vitamin D status in patients receiving steroid treatment. The authors found that most adults receiving steroid therapy have suboptimal vitamin D levels, independently of the threshold utilized [56]. Multiple factors may account for this finding including, for example, the disease for which steroids were prescribed, an increase of 25(OH)-vitamin D catabolism and the significant weight gain determining vitamin D sequestration in body fat which decreases bioavailability [57, 58].
It is now well established that steroid therapy reduces intestinal calcium absorption, mainly thorough diminished duodenal transient receptor potential vanilloid type 6 expression and calbidin D9k [59]. This process takes places independent of blood 1,25(OH)2D3 levels.
The reduced circulating levels of calcidiol might have stimulatory effects on PTH secretion owing to direct [60] or indirect actions secondary to decreased calcium absorption or increased urinary calcium excretion. However, the majority of papers indicate that PTH levels are not elevated in glucocorticoid-treated patients [61]. With this background, the results obtained by Bonadonna and coworkers [62] are particularly intriguing. Indeed, they demonstrated that steroid therapy does not affect basal PTH hormone levels but induces a redistribution of spontaneous PTH secretory dynamics by reducing the amount released in tonic fashion and increasing the amount released as pulses. Therefore, glucocorticoids may have effects that govern more the secretory behavior of PTH than the actual amount secreted over a period of time. These results have also practical implications, concerning both the pathophysiology of glucocorticoid-induced bone loss (1) and its treatment [63]. We also believe that some of the inconsistencies of the results found in the literature concerning PTH values might be also ascribed to a number of factors such as, for example, the dose of steroids administered, the heterogeneity of patients studied and assay specificity.
Finally, it is also conceivable that circulating levels of PTH might depend on the balance between two divergent driving forces. Indeed, while decreased calcium absorption and increased urinary calcium excretion tend to reduce serum ionized calcium levels (thus rising PTH level), the increased bone resorption has an opposing effect on the secretion of PTH by parathyroid glands (reducing PTH levels). From a theoretical point of view, the balance between these two opposite effects might be also complicated by a possible effect of steroids on the set point of parathyroid gland.
Treatment of GIO
Use of glucocorticoids constitutes the most important secondary cause of osteoporosis. This fact underscores the need for all practitioners to recognize the potential of these drugs to wreak havoc on the skeleton while appreciating as well their great potential to be therapeutically beneficial in a wide variety of disorders. Glucocorticoids should be used at the lowest dose for the shortest period of time, in order to reduce the risk of this complication. Another important point to bear in mind is that the decision to use glucocorticoids is often due to an underlying disease that has its own bone loss phenotype, such as rheumatoid arthritis and chronic obstructive pulmonary disease.
Nutritional principles
All patients receiving glucocorticoids should receive sufficient calcium. The guidelines offered by several organizations for all individuals are 1200–1500 mg, daily intake, from all sources [64, 65]. If patients have this amount of calcium in their diet, either through dairy products (the primary source of dietary calcium) or through calcium-fortified foods, there is no need for supplementation. A recommended upper limit of daily calcium intake has been set by the Institute of Medicine at 2.5 grams [66]. Another nutritional principle pertains to vitamin D sufficiency. This is a particularly important point because glucocorticoids have anti-vitamin D actions in the gastrointestinal tract as noted above. Thus, the controversy over what level of 25-hydroxyvitamin D should be considered adequate—20 ng/mL (50 nmol/l) or 30 ng/mL (75 nmol/l) [66, 67]—would not seem to be pertinent in this situation. Aiming for a minimal level of 30 ng/ml (75 nmol/l) is a reasonable clinical goal. The amount of vitamin D that one has to ingest to reach this level will vary from patient to patient. It is highly unusual for an individual to reach and maintain a level of 25-hydroxyvitamin D > 30 ng/mL (75 nmol/l) without supplemental vitamin D. There are many ways to achieve and to maintain vitamin D sufficiency. Daily, weekly, or monthly dosing of vitamin D can be effective, but empirical adjustments are often necessary to arrive at the correct oral dosing regimen. In general, for every additional 1000 IU of vitamin D a patient ingests on a daily basis, the 25-hydroxyvitamin D level will increase by approximately 10 ng/mL (25 nmol/l) when the new steady state is reached, in approximately 10–12 weeks.
Pharmacological principles
Similar to the adage above referencing the amount and duration of glucocorticoid use, the use of drugs to prevent and/or treat GIO should be for the shortest period of time possible. Consideration of duration of therapy takes into account a set of concerns that differ from those related to duration of therapy for postmenopausal osteoporosis. When these agents are used to prevent glucocorticoid- induced bone loss, they are no longer needed after the patient ends the period of glucocorticoid use. On the other hand, for patients who are being treated for osteoporosis due to glucocorticoids, therapy may need to be continued after glucocorticoids are stopped, in order to restore bone mass and strength. The following drugs can be used for the prevention and/or treatment of GIO:
Bisphosphonates
The data with bisphosphonates clearly indicate that they increase BMD at the lumbar spine and the hip. Reduction in fracture incidence has also been reported [68–70] although many studies have focused on changes in BMD of the lumbar spine as the primary endpoint [71]. Depending upon the design of the study, the bisphosphonate has been given a specific indication, prevention and/or treatment. The choice of bisphosphonate depends more on dosing schedules and administration routes than documented differences in efficacy.
RANK ligand inhibition
There are few, small and relatively short-term studies showing an improvement of BMD during denosumab treatment in patients under chronic glucocorticoid therapy [72–74]. A phase III randomized-double blind active-control two-year study to evaluate the effects on BMD of denosumab compared with risedronate in glucocorticoid-treated individuals is still ongoing (NCT01575873) and the results of which will not be known until the second part of 2016.
Teriparatide
Earlier concepts related to the pathogenesis of GIO would have made this an odd therapeutic choice because it was believed that in this setting, PTH levels were elevated. Abundant evidence now makes it clear that there is not a secondary hyperparathyroidism associated with glucocorticoid use [75]. Moreover, glucocorticoids are associated with a reduction in bone formation, an effect that would be countered by the administration of teriparatide, the foreshortened amino-terminal fragment of PTH. Thus, there is a clear rationale for considering teriparatide as a therapy of GIO. The work of Saag et al. has clearly shown that teriparatide is efficacious [76–78]. The study enrolled men and women with osteoporosis whose average T-score was −2.5, almost half of whom had evidence for non-vertebral fractures. The study directly compared daily teriparatide at 20 µg/day with daily alendronate at 10 mg/day for 18 months followed by an 18-month extension period. With respect to bone density, gains were statistically significantly greater in the teriparatide group than in the alendronate group at the lumbar spine, the total hip, and the femoral neck sites. While not a prespecified endpoint, there was a significantly greater reduction in vertebral fractures as seen by X-ray, in the teriparatide group compared with the alendronate group. This statistically significant difference was present at both 18 and 36 months. Clinically evident vertebral fractures were also statistically lower in incidence at 36 months in the teriparatide group vs. the alendronate group. The results were independent of the amount of glucocorticoid exposure [78]. Subgroup analyses showed densitometric changes favoring teriparatide among men and premenopausal women in the same manner as postmenopausal women [79]. Similar results have been obtained in a study comparing risedronate with teriparatide [80]. Additionally, by Finite Element Analysis, Gluer et al. showed greater effects with teriparatide on anterior bending, axial compression, and axial torsion [80]. In addition to the favorable effects on bone structure and strength, an improvement of glucose homeostasis was recently demonstrated in patients with diabetes and GIO undergoing treatment with teriparatide [81].
Guidelines for the use of pharmacological agents to prevent and to treat osteoporosis
Approaches to the management and treatment of GIO have been included into several available clinical guidelines such as those recently published by Grossman et al. and Lackawasem et al. [64, 65]. The American College of Rheumatology (ACR) guidelines consider a number of variables such as duration and amount of glucocorticoid use, menopausal status as part of risk stratification [64]. They are complicated guidelines and for many practitioners difficult to follow. A revised set of guidelines from the ACR is expected sometime during the 2016 calendar year. A more simplified set of guidelines is offered by other Societies [65, 82]. Examples of specific guidelines are provided in Table 1. While there are differences among the guidelines, they generally agree to assess fracture risk, identify those at highest risk, treat according to risk with conservative treatment (calcium, vitamin D, fall risk reduction) and when indicated with pharmacologic treatment (anti-resorptive and anabolic agents) based upon the following suggestions:
-
Bone protective treatment should be started at the onset of glucocorticoid therapy in patients at increased risk of fracture.
-
Alendronate, etidronate, risedronate, zoledronic acid, and teriparatide are the front-line therapeutic options for the majority of patients
-
If glucocorticoid therapy is subsequently stopped, withdrawal of bone protective agent may be considered.
-
In those who continue to take glucocorticoids long-term, treatment should be continued.
-
In patients treated with teriparatide, anti-resorptive therapy should be considered following the permitted treatment duration of 24 months
They also agree that all GIO patients need to be followed over time with updated fracture risk assessments. However, several uncertainties remain in the care of GIO patients, some of which have been summarized by Hansen et al. [63].
Despite several guidelines and consensus recommendations stating that patients are at remarkably increased fracture risk, little attention is still paid to the risk: clinicians do not follow the guidelines [83]. There have been many attempts to improve care of GIO patients. Videos for patients [84], specific clinician education [85], and clinician education with a specific audit of practices [86] have had little impact on management of GIO. Automatic order sets for calcium and vitamin D using the electronic medical record led to a modest improvement of practice [87]. A potential partial answer, however, is possible. The Geisinger Clinic, located in Pennsylvania in the United States, started a comprehensive osteoporosis program that included the equivalent of a Fracture Liaison Service (FLS) but also identified patients prescribed oral glucocorticoids [88]. Thus, the FLS nurse became the GIO nurse. In a closed system such as the Geisinger system, a list of patients on oral glucocorticoids could be extracted from the electronic medical record [88]. The osteoporosis nurse could then, as was done with fracture patients, contact the patients, order a BMD, and start conservative management with calcium, vitamin D, and fall risk reduction. Working with the patient’s primary care clinician, the nurse could have appropriate patients started on pharmacologic treatment. Follow-up could also be arranged.
Just as with an FLS program, a GIO Service requires that clinicians providing ongoing care for patients accept the help of the GIO nurse. Having an FLS and GIO nurse requires upfront investment but it leads to lower costs in the future. Preventing fractures saves money, not to mention suffering, morbidities, and death. It is up to each country to find ways to justify the investment in preventing fractures in these two groups of patients at high risk: those who have already fractured (via the FLS) and those on oral glucocorticoids (the GIO Service). These two jobs can be done by the same person: in the long run overall costs will decline. Studies are underway to determine the long term savings from FLS. We should do the same with prevention of fractures in GIO.
Conclusions
Based on the topics discussed at the 9th GIO, which was held in Rome in 2016 and summarized in this paper, we may conclude that glucocorticoid-mediated derangement in GH/IGF-1 axis is relevant to bone damage not only in childhood, but also in adults and it may have perspective treatment implications in both age groups. There may also be clinical relevance as to the negative impact of glucocorticoids on the PTH-vitamin D axis. Treatment algorithms for GIO are available but they tend to be complicated. Newer guidelines, which are expected imminently, will hopefully simplify our approach to the prevention and treatment of GIO.
References
E. Canalis, G. Mazziotti, A. Giustina, J.P. Bilezikian, Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos. Int. 18, 1319–1328 (2007)
A. Canalis, A. Giustina, Glucocorticoid-induced osteoporosis: summary of a workshop. J. Clin. Endocrinol. Metab. 86, 5681–5685 (2001)
T. Mancini, M. Doga, G. Mazziotti, A. Giustina, Cushing’s syndrome and bone. Pituitary 7, 1–4 (2005)
G. Mazziotti G, A. Delgado, F. Maffezzoni, A. Formenti, A. Giustina, Skeletal fragility in endogenous hypercortisolism. Front. Horm. Res. 46, 66–73 (2016)
P. Vestergaard, J. Lindholm, J.O. Jørgensen, C. Hagen, H.C. Hoeck, P. Laurberg, L. Rejnmark, K. Brixen, L.O. Kristensen, U. Feldt-Rasmussen, L. Mosekilde, Increased risk of osteoporotic fractures in patients with Cushing’s syndrome. Eur. J. Endocrinol. 146, 51–56 (2002)
A. Scillitani, G. Mazziotti, C. Di Somma, S. Moretti, A. Stigliano, R. Pivonello, A. Giustina, A. Colao; ABC group, treatment of skeletal impairment in patients with endogenous hypercortisolism: when and how? Osteoporos. Int. 25, 441–446 (2015)
G. Mazziotti, A. Angeli, J.P. Bilezikian, E. Canalis, A. Giustina, Glucocorticoid-induced osteoporosis: an update. Trends Endocrinol. Metab. 17, 144–149 (2006)
N.E. Lane, W. Yao, M. Balooch, R.K. Nalla, G. Balooch, S. Habelitz, J.H. Kinney, L.F. Bonewald, Glucocorticoid treated mice have localized changes in trabecular bone material properties and osteocyte lacunar size that are not observed in placebo-treated or estrogen-deficient mice. J. Bone Miner. Res. 21, 466–476 (2006)
T.P. Van Staa, R.F. Laan, I.P. Barton, S. Cohen, D.M. Reid DM, C. Cooper, Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum. 48, 3224–3229 (2003)
G. Mazziotti, A. Giustina, Glucocorticoids and the regulation of growth hormone secretion. Nat. Rev. Endocrinol 9, 265–276 (2013)
C.E. Dean, B. Morpurgo, T.E. Porter, Induction of somatotroph differentiation in vivo by corticosterone administration during chicken embryonic development. Endocrine 11, 151–156 (1999)
A. Giustina, J.D. Veldhuis, Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr. Rev. 19, 717–797 (1998)
A. Tamaki, M. Sato, S. Matsubara, Y. Wada, J. Takahara, Dexamethasone increases growth hormone (GH)-releasing hormone (GHR) receptor mRNA levels in culture rat anterior pituitary cells. Neuroendocrinology 8, 475–480 (1996)
A. Giustina, G. Romanelli, R. Candrina, G. Giustina, Growth hormone deficiency in patients with idiopathic adrenocorticotropin deficiency resolves during glucocorticoid replacement. J. Clin. Endocrinol. Metab. 68, 120–124 (1989)
Y. Hattori, T. Takeda, M. Fujii, J. Taura, Y. Ishii, H. Yamada, Dioxin-induced fetal growth retardation: the role of a preceding attenuation in the circulating level of glucocorticoid. Endocrine 47, 572–580 (2014)
A. Giustina, G. Mazziotti, Impaired growth hormone secretion associated with low glucocorticoid levels: an experimental model for the Giustina effect. Endocrine 47, 354–356 (2014)
W.B. Wehrenberg, P.J. Bergman, L. Stagg, J. Ndon, A. Giustina, Glucocorticoid inhibition of growth in rats: partial reversal with somatostatin antibodies. Endocrinology 127, 2705–2708 (1990)
G. Tulipano, D. Soldi, M. Bagnasco, M.D. Culler, J.E. Taylor, D. Cocchi, A. Giustina, Characterization of new selective somatostatin receptor subtype-2 (sst2) antagonists, BIM-23627 and BIM-23454. Effects of BIM-23627 on GH release in anesthetized male rats after short-term high-dose dexamethasone treatment. Endocrinology 143, 1218–1224 (2002)
A. Giustina, S. Bossoni, C. Bodini, A. Girelli, G.P. Balestrieri, G. Pizzocolo, W.B. Wehrenberg, Arginine normalizes the growth hormone (GH) response to GH-releasing hormone in adult patients receiving chronic daily immunosuppressive glucocorticoid therapy. J. Clin. Endocrinol. Metab. 74, 1301–1305 (1992)
A. Giustina, M. Doga, C. Bodini, A. Girelli, F. Legati, S. Bossoni, G. Romanelli, Acute effects of cortisone acetate on growth hormone response to growth hormone-releasing hormone in normal adult subjects. Acta Endocrinol. 122, 206–210 (1990)
M. Malerba, S. Bossoni, A. Radaeli, E. Mori, S. Bonadonna, A. Giustina, C. Tantucci, Growth hormone response to growth hormone-releasing hormone is reduced in adult asthmatic patients receiving long-term inhaled corticosteroid treatment. Chest 127, 515–521 (2005)
M. Terzolo, S. Bossoni, A. Alı, M. Doga, G. Reimondo, G. Milani, P. Peretti, F. Manelli, A. Angeli, A. Giustina, Growth hormone (GH) responses to GH-releasing hormone alone or combined with arginine in patients with adrenal incidentaloma: evidence for enhanced somatostatinergic tone. J. Clin. Endocrinol. Metab. 85, 1310–1315 (2000)
Z. Hochberg, Mechanisms of steroid impairment of growth. Horm. Res. 58, S33–S38 (2002)
H. Filipsson, G. Johannsson, GH replacement in adults: interactions with other pituitary hormone deficiencies and replacement therapies. Eur. J. Endocrinol. 161, S85–S95 (2009)
A. Giustina, G. Mazziotti, E. Canalis, Growth hormone, insulin-like growth factors, and the skeleton. Endocr. Rev. 29, 535–559 (2008)
A. Angeli, G. Guglielmi, A. Dovio, G. Capelli, D. de Feo, S. Giannini, R. Giorgino, L. Moro, A. Giustina, High prevalence of asymptomatic vertebral fractures in post-menopausal women receiving chronic glucocorticoid therapy: a cross-sectional outpatient study. Bone 39, 253–259 (2006)
G. Mazziotti, M. Doga, S. Frara, F. Maffezzoni, T. Porcelli, L. Cerri, R. Maroldi, A. Giustina, Incidence of morphometric vertebral fractures in adult patients with growth hormone deficiency. Endocrine 52, 103–110 (2016)
A. Giustina, A.R. Bussi, C. Jacobello, W.B. Wehrenberg, Effects of recombinant human growth hormone (GH) on bone and intermediary metabolism in patients receiving chronic glucocorticoid treatment with suppressed endogenous GH response to GH-releasing hormone. J. Clin. Endocrinol. Metab. 80, 122–129 (1995)
F.F. Horber, M.W. Haymond, Human growth hormone prevents the protein catabolic side effects of prednisone in humans. J. Clin. Invest. 86, 265–272 (1990)
M.A. Magiakou, G.P. Chrousos GP, Cushing’s syndrome in children and adolescents: current diagnostic and therapeutic strategies. J. Endocrinol. Invest. 25, 181–194 (2002)
R.G. McArthur, M.D. Cloutier, A.B. Hayles, R.G. Sprague, Cushing’s disease in children. Findings in 13 cases. Mayo. Clin. Proc. 47, 318–326 (1972)
D.H. Streeten, F.H. Faas, M.J. Elders, T.G. Dalakos, M. Voorhess, Hypercortisolism in childhood: shortcomings of conventional diagnostic criteria. Pediatrics 56, 797–803 (1975)
C.G. Thomas Jr., A.T. Smith, J.M. Griffith, F.B. Askin, Hyperadrenalism in childhood and adolescence. Ann. Surg. 199, 538–548 (1984)
M.A. Magiakou, G. Mastorakos, G.P. Chrousos, Final stature in patients with endogenous Cushing’s syndrome. J. Clin. Endocrinol. Metab. 79, 1082–1085 (1994)
N.R. Hughes, C.A. Lissett, S.M. Shalet, Growth hormone status following treatment for Cushing’s syndrome. Clin. Endocrinol. (Oxf). 51, 61–66 (1999)
M.A. Magiakou, G. Mastorakos, M.T. Gomez, S.R. Rose, G.P. Chrousos, Suppressed spontaneous and stimulated growth hormone secretion in patients with Cushing’s disease before and after surgical cure. J. Clin. Endocrinol. Metab. 78, 131–137 (1994)
M.O. Savage, A. Lienhardt, M.C. Lebrethon, L.B. Johnston, A. Huebner, A.B. Grossman, F. Afshar, P.N. Plowman, G.M. Besser, Cushing’s disease in childhood: presentation, investigation, treatment and long-term outcome. Horm. Res. 55(Suppl 1), 24–30 (2001)
M.C. Lebrethon, A.B. Grossman, F. Afshar, P.N. Plowman, G.M. Besser, M.O. Savage, Linear growth and final height after treatment for Cushing’s disease in childhood. J. Clin. Endocrinol. Metab. 85, 3262–3265 (2000)
L.F. Chan, H.L. Storr, P.N. Plowman, L.A. Perry, G.M. Besser, A.B. Grossman, M.O. Savage, Long-term anterior pituitary function in patients with paediatric Cushing’s disease treated with pituitary radiotherapy. Eur. J. Endocrinol. 156, 477–482 (2007)
V. Khanine, J.J. Fournier, E. Requeda, J.P. Luton, F. Simon, J. Crouzet, Osteoporotic fractures at presentation of Cushing’s disease: two case reports and a literature review. Joint Bone Spine 67, 341–345 (2000)
L. Futo, J. Toke, A. Patocs, A. Szappanos, I. Varga, E. Glaz, Z. Tulassay, K. Racz, M. Toth, Skeletal differences in bone mineral area and content before and after cure of endogenous Cushing’s syndrome. Osteoporos. Int. 19, 941–949 (2008)
K. Godang, T. Ueland, J. Bollerslev, Decreased bone area, bone mineral content, formative markers, and increased bone resorptive markers in endogenous Cushing’s syndrome. Eur. J. Endocrinol. 141, 126–131 (1999)
A.R. Hermus, A.G. Smals, L.M. Swinkels, D.A. Huysmans, G.F. Pieters, C.F. Sweep, F.H. Corstens, P.W. Kloppenborg PW, Bone mineral density and bone turnover before and after surgical cure of Cushing’s syndrome. J. Clin. Endocrinol. Metab. 80, 2859–2865 (1995)
A. Kawamata, M. Iihara, T. Okamoto, T. Obara, Bone mineral density before and after surgical cure of Cushing’s syndrome due to adrenocortical adenoma: prospective study. World J. Surg. 32, 890–896 (2008)
A.W. van der Eerden, M. den Heijer, W.J. Oyen, A.R. Hermus, Cushing’s syndrome and bone mineral density: lowest Z scores in young patients. Neth. J. Med. 65, 137–141 (2007)
C. Di Somma, R. Pivonello, S. Loche, A. Faggiano, P. Marzullo, A. Di Sarno, M. Klain, M. Salvatore, G. Lombardi, A. Colao, Severe impairment of bone mass and turnover in Cushing’s disease: comparison between childhood-onset and adulthood-onset disease. Clin. Endocrinol. 56, 153–158 (2002)
D.J. Devoe, W.L. Miller, F.A. Conte, S.L. Kaplan, M.M. Grumbach, S.M. Rosenthal, C.B. Wilson, S.E. Gitelman, Long-term outcome in children and adolescents after transsphenoidal surgery for Cushing’s disease. J. Clin. Endocrinol. Metab. 82, 3196–3202 (1997)
C. Di Somma, R. Pivonello, S. Loche, A. Faggiano, M. Klain, M. Salvatore, G. Lombardi, A. Colao, Effect of 2 years of cortisol normalization on the impaired bone mass and turnover in adolescent and adult patients with Cushing’s disease: a prospective study. Clin. Endocrinol. 58, 302–308 (2003)
I. Jeong, M. Oh, J.H. Kim, J.H. Cho, J.H. Choi, H.W. Yoo, Long-term follow-up on Cushing disease patient after transsphenoidal surgery. Ann. Pediatr. Endocrinol. Metab. 19, 164–168 (2014)
S. Scommegna, J.P. Greening, H.L. Storr, K.M. Davies, N.J. Shaw, J.P. Monson, A.B. Grossman, M.O. Savage, Bone mineral density at diagnosis and following successful treatment of pediatric Cushing’s disease. J. Endocrinol. Invest. 28, 231–235 (2005)
G.M. Leong, L.B. Mercado-Asis, J.C. Reynolds, S.C. Hill, E.H. Oldfield, G.P. Chrousos, : The effect of Cushing’s disease on bone mineral density, body composition, growth, and puberty: a report of an identical adolescent twin pair. J. Clin. Endocrinol. Metab. 81, 1905–1911 (1996)
G.M. Leong, V. Abad, E. Charmandari, J.C. Reynolds, S. Hill, G.P. Chrousos, L.K. Nieman, Effects of child- and adolescent-onset endogenous cushing syndrome on bone mass, body composition, and growth: a 7-year prospective study into young adulthood. J. Bone Miner. Res. 22, 110–118 (2007)
M.B. Lodish, H.P. Hsiao, A. Serbis, N. Sinaii, A. Rothenbuhler, M.F. Keil, S.A. Boikos, J.C. Reynolds, C.A. Stratakis, Effects of Cushing disease on bone mineral density in a pediatric population. J. Pediatr. 156, 1001–1005 (2010)
Y. Zhang, D.Y.M. Leung, E. Goleva, Vitamin D enhances glucocorticoid action in human monocytes. J. Biol. Chem. 288, 14544–14553 (2013)
S.M. Tse, H.W. Kelly, A.A. Litonjua, M.L. Van Natta, S.T. Weiss, K.G. Tantisira; Childhood Asthma Management Program Research Group, Corticosteroid use and bone mineral accretion in children with asthma: effect modification by vitamin D. J. Allergy Clin. Immunol. 130, 53–60 (2012)
Z.E. Davidson, K.Z. Walker, H. Truby, Do glucocorticoids alter vitamin D status? A systematic review with meta-analyses of observational studies. J. Clin. Endocrinol. Metab. 97, 738–744 (2012)
E. Romagnoli, J. Pepe, S. Piemonte, C. Cipriani, S. Minisola, Management of endocrine disease: value and limitations of assessing vitamin D nutritional status and advised levels of vitamin D supplementation. Eur. J. Endocrinol. 169, R59–R69 (2013)
C. Cipriani, J. Pepe, S. Piemonte, L. Colangelo, M. Cilli, S. Minisola, Vitamin d and its relationship with obesity and muscle. Int. J. Endocrinol. 2014, 841248 (2014)
S. Huybers, T.H. Naber, R.J. Bindels, J.G. Hoenderop, Prednisolone-induced Ca2+ malabsorption is caused by diminished expression of the epithelial Ca2+ channel TRPV6. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G92–G97 (2007)
J. Pepe, E. Romagnoli, I. Nofroni, M.T. Pacitti, S. De Geronimo, C. Letizia, G.,G. Tonnarini, A. Scarpiello, E. D’Erasmo, S. Minisola, Vitamin D status as the major factor determining the circulating levels of parathyroid hormone: a study in normal subjects. Osteoporos. Int. 16, 805–812 (2005)
E. Paz-Pacheco, G. El-Hajj Fuleihan, M. Leboff, Intact parathyroid hormone levels are not elevated in glucocorticoid-treated subjects. J. Bone Miner. Res. 10, 1713–1718 (1995)
S. Bonadonna, A. Burattin, M. Nuzzo, G. Bugari, E. Agabiti-Rosei, D. Valle, N. Iori, J.P. Bilezikian, J.D. Veldhuis, A. Giustina, Chronic glucocorticoid treatment alters spontaneous pulsatile parathyroid hormone secretory dynamics in human subjects. Eur. J. Endocrinol. 152, 199–205 (2005)
K.E. Hansen, H.A. Wilson, C. Zapalowski, H.A. Fink, S. Minisola, R.A. Adler, Uncertainties in the prevention and treatment of glucocorticoid-induced osteoporosis. J. Bone Miner. Res. 26, 1989–96 (2011)
J.M. Grossman, R. Gordon, V.K. Ranganath, C. Deal, L. Caplan, W. Chen, J.R. Curtis, D.E. Furst, M. McMahon, N.M. Patkar, E. Volkmann, K.G. Saag, American college of rheumatology recommendations for the prevention and treatment of glucocorticoid induced osteoporosis. Arthritis Care Res. 62, 1515–1526 (2010)
S. Lekamwasam, J.D. Adachi, D. Agnusdei, J.P. Bilezikian, S. Boonen, F. Borgström, C. Cooper, A. Diez Perez, R. Eastell, L.C. Hofbauer, J.A. Kanis, B.L. Langdahl, O. Lesnyak, R. Lorenc, E. McCloskey, O.D. Messina, N. Napoli, B. Obermayer-Pietsch, S.H. Ralston, P.N. Sambrook, S. Silverman, M. Sosa, J. Stepan, G. Suppan, D.A. Wahl, J.E. Compston; Joint IOF-ECTS GIO Guidelines Working Group, A framework for the development of guidelines for the management of glucocorticoid-induced osteoporosis. Osteoporosis Int. 23, 2257–2276 (2012)
A.C. Ross, J.E. Manson, S.A. Abrams, J.F. Aloia, P.M. Brannon, S.K. Clinton, R.A. Durazo-Arvizu, J.C. Gallagher, R.L. Gallo, G. Jones, C.S. Kovacs, S.T. Mayne, C.J. Rosen, S.A. Shapses, The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J. Clin. Endocrinol. Metab. 96, 53–58 (2011)
R.P. Heaney, M.F. Holick, Why the IOM recommendations for vitamin D are deficient. J. Bone Miner. Res. 26, 455–457 (2011)
K.G. Saag, R. Emkey, T.J. Schnitzer, J.P. Brown, F. Hawkins, S. Goemaere, G. Thamsborg, U.A. Liberman, P.D. Delmas, M.P. Malice, M. Czachur, A.G. Daifotis, Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced osteoporosis intervention study group. N. Engl. J. Med. 339, 292–299 (1998)
J.D. Adachi, K.G. Saag, P.D. Delmas, U.A. Liberman, R.D. Emkey, E. Seeman, N.E. Lane, J.M. Kaufman, P.E. Poubelle, F. Hawkins, R. Correa-Rotter, C.J. Menkes, J.A. Rodriguez-Portales, T.J. Schnitzer, J.A. Block, J. Wing, H.H. McIlwain, R. Westhovens, J. Brown, J.A. Melo-Gomes, B.L. Gruber, M.J. Yanover, M.O. Leite, K.G. Siminoski, M.C. Nevitt, J.T. Sharp, M.P. Malice, T. Dumortier, M. Czachur, W. Carofano, A. Daifotis, Two-year effects of alendronate on bone mineral density and vertebral fracture in patients receiving glucocorticoids: a randomized, double-blind, placebo-controlled extension trial. Arthritis Rheum. 44, 202–211 (2001)
S. Wallach, S. Cohen, D.M. Reid, R.A. Hughes, D.J. Hosking, R.F. Laan, S.M. Doherty, M. Maricic, C. Rosen, J. Brown, I. Barton, A.A. Chines, Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif. Tissue Int. 67, 277–285 (2000)
D.M. Reid, J.P. Devogelaer, K.G. Saag, C. Roux, C.S. Lau, J.Y. Reginster, P. Papanastasiou, A. Ferreira, F. Hartl, T. Fashola, P. Mesenbrink, P.N. Sambrook; HORIZON investigators, Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 373, 1253–1263 (2009)
T. Petranova, I. Sheytanov, S. Monov, R. Nestorova, R. Rashkov, Denosumab improves bone mineral density and microarchitecture and reduces bone pain in women with osteoporosis with and without glucocorticoid treatment. Biotechnol. Biotechnol. Equip. 28, 1127–1137 (2014)
C.C. Mok, L.Y. Ho, K.M. Ma, Switching of oral bisphosphonates to denosumab in chronic glucocorticoid users: a 12-month randomized controlled trial. Bone 75, 222–228 (2015)
T. Takeuchi, Y. Tanaka, N. Ishiguro, H. Yamanaka, T. Yoneda, T. Ohira, N. Okubo, H.K. Genant, D. van der Heijde, Effect of denosumab on Japanese patients with rheumatoid arthritis: a dose-response study of AMG 162 (Denosumab) in patients with rheumatoId arthritis on methotrexate to validate inhibitory effect on bone Erosion (DRIVE)-a 12-month, multicentre, randomised, double-blind, placebo-controlled, phase II clinical trial. Ann. Rheum. Dis. 75, 983–990 (2016)
L. Gennari, J.P. Bilezikian, Glucocorticoid-induced osteoporosis: hope on the HORIZON. Lancet 373, 1225–1226 (2009)
K.G. Saag, E. Shane, S. Boonen, F. Marín, D.W. Donley, K.A. Taylor, G.P. Dalsky, R. Marcus, Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N. Eng. J. Med. 357, 2028–2039 (2007)
K.G. Saag, J.R. Zanchetta, J.P. Devogelaer, R.A. Adler, R. Eastell, K. See, J.H. Krege, K. Krohn, M.R. Warner, Effects of teriparatide versus alendronate for treating glucocorticoid-induced osteoporosis: thirty-six-month results of a randomized, double-blind, controlled trial. Arthritis Rheum. 60, 3346–3355 (2009)
J.P. Devogelaer, R.A. Adler, C. Recknor, K. See, M.R. Warner, M. Wong, K. Krohn, Baseline glucocorticoid dose and bone mineral density response with teriparatide or alendronate therapy in patients with glucocorticoid-induced osteoporosis. J. Rheumatol. 37, 141–148 (2010)
B.L. Langdahl, F. Marin, E. Shane, H. Dobnig, J.R. Zanchetta, M. Maricic, K. Krohn, K. See, M.R. Warner, Teriparatide versus alendronate for treating glucocorticoid-induced osteoporosis: an analysis by gender and menopausal status. Osteoporosis Int. 12, 2095–2104 (2009)
C.C. Glüer, F. Marin, J.D. Ringe, F. Hawkins, R. Möricke, N. Papaioannu, P. Farahmand, S. Minisola, G. Martínez, J.M. Nolla, C. Niedhart, N. Guañabens, R. Nuti, E. Martín-Mola, F. Thomasius, G. Kapetanos, J. Peña, C. Graeff, H. Petto, B. Sanz, A. Reisinger, P.K. Zysset, Comparative effects of teriparatide and risedronate in glucocorticoid-induced osteoporosis in men: 18-month results of the EuroGIOPs trial. J. Bone Miner. Res. 28, 1355–1368 (2013)
G. Mazziotti, F. Maffezzoni, M. Doga, L.C. Hofbauer, R.A. Adler, A. Giustina, Outcome of glucose homeostasis in patients with glucocorticoid-induced osteoporosis undergoing treatment with bone active-drugs. Bone 67, 175–180 (2014)
K. Briot, B. Cortet, C. Roux, L. Fardet, V. Abitbol, J. Bacchetta, D. Buchon, F. Debiais, P. Guggenbuhl, M. Laroche, E. Legrand, E. Lespessalilles, C. Marcelli, G. Weryha, T. Thomas, Bone section of the french society for rheumatology (SFR) and osteoporosis research and information group (GRIO). Joint Bone Spine 81, 493–501 (2014)
H.O. Tory, D.H. Solomon, S.P. Desai, Analysis of quality improvement efforts in preventing glucocorticoid-induced osteoporosis. Sem. Arthritis Rheum. 44, 483–488 (2015)
A.H. Warriner, R.C. Outman, J.J. Allison, J.R. Curtis, N.J. Markward, D.T. Redden, M.M. Safford, E.J. Stanek, A.R. Steinkellner, K.G. Saag, An internet-based controlled trial aimed to improve osteoporosis prevention among chronic glucocorticoid users. J. Rheumatol. 42, 1478–1483 (2015)
J.R. Curtis, A.O. Westfall, J. Allison, Challenges in improving the quality of osteoporosis care for long-term glucocorticoid users: a prospective randomized trial. Arch. Intern. Med. 167, 591–596 (2007)
D.H. Solomon, J.N. Katz, J.P. Jacobs, A.M. La Tourette, J. Coblyn, Multifaceted intervention to improve rheumatologists’ management of glucocorticoid-induced osteoporosis: a randomized controlled trial. Arthritis Rheum. 51, 383–387 (2004)
M.J. Kohler, M. Amezaga, J. Drozd, S.T. Crowley, B. Gulanski, D.R. Anderson, L. Fraenkel, Use of a computerized order set to increase prescription of calcium and vitamin D supplementation in patients receiving glucocorticoids. J. Gen. Intern. Med. 28, 825–829 (2013)
E.D. Newman, C.K. Matzko, T.P. Olenginski, J.L. Perruquet, T.M. Harrington, G. Maloney-Saxon, T. Culp, G.C. Wood, Glucocorticoid-induced osteoporosis program (GIOP): a novel, comprehensive, and highly successful care program with improved outcomes at1 year. Osteoporos. Int. 17, 1428–1434 (2006)
Funding
This study was not funded by any grant
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
G.M. received lecture fee from Abiogen Pharma SpA, J.P.B. was consultant for Merck, Amgen, Radius and received Research Grant from Shire Pharmaceuticals, A.Gr. received lecture fees and Advisory Board payments from Novartis, Ipsen and HRA Pharma, S.M. received lecture fees from Abiogen Pharma SpA, Amgen, Diasorin, Eli Lilly, Italfarmaco, Fujii, Merck Sharp & Dohme, Takeda and Advisory Board payments from Amgen and Eli Lilly, A.Gi. was consultant for Abiogen Pharma SpA. A.M.F., R.A.A. and E.S. declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Mazziotti, G., Formenti, A.M., Adler, R.A. et al. Glucocorticoid-induced osteoporosis: pathophysiological role of GH/IGF-I and PTH/VITAMIN D axes, treatment options and guidelines. Endocrine 54, 603–611 (2016). https://doi.org/10.1007/s12020-016-1146-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-016-1146-8