
Abstract
Osteoimmunology is new field of research dedicated to the study of the interactions between the immune system and bone. Among the cells of the immune system that regulate bone and hemopoietic cells are T lymphocytes. These cells secrete osteoclastogenic cytokines such as RANKL and TNF, as well as factors that stimulate bone formation and hemopoietic cells, one of which is Wnt10b. This article will review the evidence that T cells are implicated in the mechanism of action of parathyroid hormone (PTH) in bone and on the hemopoietic system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Effects of PTH on bone and the hemopoietic niche
PTH plays a critical regulatory role in calcium metabolism. Secreted in response to small decrements in serum ionized calcium, this hormone defends against hypocalcemia, in part by stimulating bone resorption and thereby the release of calcium from the skeleton. In addition to its role in regulating the level of serum calcium, sustained overproduction, or in vivo continuous infusion of PTH (cPTH), is a cause of bone disease. Secondary hyperparathyroidism has been implicated in the pathogenesis of senile osteoporosis [1], and primary hyperparathyroidism, is associated with accelerated bone loss and osteopenia [2]. However, when injected daily in humans and animals at low dose, a regimen known as intermittent PTH (iPTH) treatment, the hormone stimulates trabecular and cortical bone formation, leading to marked increases in bone volume and strength [3]. Attesting to potency, iPTH, has been shown to decrease the risk of fractures in humans, and is an FDA approved treatment modality for postmenopausal women and men with osteoporosis [4].
PTH exerts its anabolic activity by binding to the PPR receptor (also known as PTH-1R), which is expressed on osteoblasts (OBs), osteocytes, and stromal cells (SCs) [5]. Mice lacking PPR have decreased trabecular bone and increased thickness of cortical bone during fetal development [6]. Conversely, osteoblastic expression of the constitutively active PPR display a substantial increase in trabecular bone volume and a decrease in cortical bone thickness of the long bones [7]. Transgenic mice expressing a constitutively active PTH receptor exclusively in osteocytes also exhibit increased bone mass and bone remodeling, as well as reduced expression of the osteocyte-derived Wnt antagonist sclerostin, increased Wnt signaling, increased osteoclast (OC) and OB number, and decreased OB apoptosis [8]. PTH receptor signaling in osteocytes has been shown to increase bone mass and the rate of bone remodeling through LRP5 dependent and independent mechanisms, respectively [9].
PTH promotes bone formation by increasing the number of OBs [10, 11] through multiple effects, including activation of quiescent lining cells [12], increased OB proliferation [13] and differentiation [14], attenuation of pre-OB and OB apoptosis [15, 16], and signaling in osteocytes [8]. However, the specific contribution of each of these effects to the overall anabolic activity of PTH remains controversial.
One of the major effects of PTH is to activate Wnt signaling in osteoblastic cells. Activation of Wnt signaling induces OB proliferation [17] and differentiation [18], prevents pre-OB and OB apoptosis [19], and augments OB production of OPG [20]. Wnt proteins initiate a canonical signaling cascade by binding to receptors of the Frizzled family together with coreceptors, members of the low-density lipoprotein receptor-related protein (LRP) family, LRP5 and LRP6, which results in the stabilization of cytosolic β-catenin. A nuclear complex of beta-catenin and the T cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors then interacts with DNA to regulate the transcription of Wnt target genes [21]. Wnt proteins also signal through non-canonical pathways that involve the Src/ERK and Pi3K/Akt cascades [19].
PTH is a canonical Wnt signaling agonist that increases β-catenin levels in osteoblastic cells [22]. PTH, once bound to PPR, is also capable of forming a complex with LRP6 which results in LRP6 signaling and β-catenin activation [23]. Thus, PTH activates Wnt signaling in osteoblastic cells through both Wnt ligands dependent and Wnt ligands independent mechanisms. Moreover, PTH down regulates the production of sclerostin, an osteocyte-derived Wnt antagonist that blocks Wnt signaling by binding to LRP5 and LRP6 [24, 25]. Recently, convincing evidence has emerged that PTH receptor signaling in osteocytes and the resulting direct regulation of sclerostin production play a particularly relevant role in the anabolic activity of PTH [8]. PTH also regulates Dickkopf-1, a soluble LRP5 and LRP6 signaling inhibitor [22], and Sfrp-4, a factor that binds Wnt proteins thus antagonizing both canonical and non-canonical Wnt signaling [26]. Uncertainty remains with regard to the identity and the source of Wnt ligands which activate Wnt signaling in response to PTH treatment are not completely understood.
In addition, PTH has important effects on the hemopoietic system. PTH expands the hemopoietic stem cells (HSCs) pool and regulates the activity of the HSC niche [27], the specialized microenvironment that maintains HSPs. Accordingly, patients with primary hyperparathyroidism have an increased number of circulating bone marrow derived HSCs in the peripheral blood [28]. Moreover, some PTH regimens increase the population that can later be mobilized by G-CSF [29], while others induce mobilization of progenitor cells from the BM into the peripheral circulation [30], thus mimicking the effects of G-CSF. Because of these properties, PTH has been investigated as a potential therapeutic agent to enhance HSCs mobilization [31].
A pivotal effect of PTH is that of increasing the HSC pool through regulatory actions on the HSC niche. The HSC niche comprises a variety of cells including SCs and OBs [32]. Early studies had linked this activity of PTH to its capacity to increase the osteoblastic expression of the Notch ligand, Jagged1, leading to the activation of Notch signaling in HSCs in vivo [27].
T cells and PTH induced bone loss
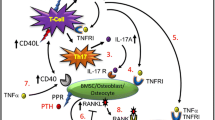
T lymphocytes, expresses functional PPR [33], responds to PTH [34], and stimulates OB differentiation [35]. Hory et al. [36] were the first to suggest a link between T cells and PTH as they reported that transplantation of human parathyroid gland fragments from patients with primary hyperparathyroidism into nude mice fails to stimulate OC formation and bone resorption. These observations prompted more in depth investigations on the role of T cells as mediators of the pro-resorptive effect of cPTH treatment. We found that an infusion of cPTH that mimics hyperparathyroidism fails to induce OC formation, bone resorption, and cortical bone loss in mice lacking T cells [37]. By contrast, cPTH equally stimulated bone formation in T cell replete and T cell deficient mice. These studies further revealed the existence of a cross-talk between T cells and SCs mediated by the CD40L/CD40 signaling system (Fig. 1). T cells sensitize SCs to PTH through CD40L, a surface molecule of activated T cells that induces CD40 signaling in SCs. Attesting to the relevance of CD40L, mice lacking T cells or T cell expressed CD40L possess SCs that produce lower amount of RANKL and have an even smaller suppression of OPG secretion in response to PTH. Therefore, SCs from T cell deficient mice have a lower capacity to support OC formation in vivo and in vitro [37]. Moreover, cPTH stimulates T cell production of TNF [38]. TNF further increases the SC production of RANKL, and upregulates the expression of CD40 in SCs, thus increasing their response to T cell expressed CD40L. Attesting to the relevance of T cell produced TNF, cPTH fails to induce bone loss and stimulate bone resorption in mice specifically lacking T cell TNF production [38]. We have also shown that conditional silencing of the PTH receptor PPR in T cells blunts the stimulation of bone resorption induced by cPTH without affecting bone formation, thus blocking cortical bone loss and converting the effects of cPTH in trabecular bone from catabolic to anabolic [38]. These findings demonstrate the critical relevance of direct PPR signaling in T cells.

Schematic representation of the role of T cells in the mechanism by which continuous PTH induces bone loss. PTH binding to PPR in T cells stimulates the production of TNF. This cytokine increases CD40 expression by SCs. Binding of CD40 by T cell expressed CD40L increases SC sensitivity to PTH resulting in enhanced SC production of RANKL and diminished secretion of OPG in response to PTH. T cell produced TNF further stimulates OC formation through its direct effects on maturing OC precursors. The red arrows represent the main modifications induced by activation of PPR signaling in T cells. Reproduced with permission from Tawfeek et al. [38]
It is now known that the survival of naive T cells and some memory T cells requires continuous contact with a diverse repertoire of self Ags bound to MHC molecules [39, 40].
Consistent with a requirement for T cell survival, we found that cPTH did not induce bone loss in mice lacking class I MHC or class II MHC-TCR interactions [41]. Additional studies revealed that cPTH does not increase MHC-TCR interactions, thus suggesting that cPTH activity requires tonic interactions of T cells and MHC complexes bound to self-peptides. We also found that cPTH does not increase T cell activation. Production of TNF in the absence of enhanced expression of activation markers is a feature of T cells undergoing “bystander activation,” a process mediated by cytokines produced by other cells [42]. Thus, T cell production of TNF may require not only direct PPR signaling in T cells but also T cell stimulation by cytokines produced by other cells [42].
A second set of signals required for T cell survival and function is provided by the interaction of the costimulatory molecules on antigen presenting cells with the T cell expressed counter receptors such as CD28. A pharmacological approach to test the role of costimulation is provided by Abatacept, an agent approved for the treatment of Rheumatoid Arthritis. We found that PTH induced bone loss is prevented using this inhibitor of costimulation [41]. These findings provide further evidence of a novel regulatory link between the immune system and the mechanism of action of PTH.
Role of T cells in the anabolic activity of intermittent PTH treatment
The hypothesis that T cells may play a role in the anabolic response to iPTH was first proposed by Pettway et al. [43], who investigated the effects of daily injections of PTH for up to 7 weeks on the growth of ectopic “ossicles” implanted in nude mice. This investigation disclosed that iPTH increased the bone content of the implanted WT ossicles, a structure that contains normal BM, but failed to induce vertebral bone growth in host nude mice, a strain devoid of T cells [43].
These observations were followed by our investigation on whether T cells contribute to the anabolic response to iPTH. Studies conducted in T cell deficient mice revealed that mice lacking T cells, exhibit a blunted increase in bone formation and trabecular bone volume in response to iPTH [33]. Furthermore, adoptive transfer of T cells into T cell deficient mice restored a normal response to iPTH. T cells were found to augment the capacity of iPTH to improve architecture in trabecular but not in cortical bone. Although the reason of this selectivity is unknown, a lack of access of T cells to cortical surfaces is not a likely explanation, as T cells reach endosteal and periosteal bone surface through blood vessels and recirculate in and out of the BM [44].
The effects of iPTH on bone volume and the indices of bone strength measurable by μCT are blunted but not abolished in the absence of T cells. By contrast, direct measurements of bone strength by 4-point bending revealed that the capacity of iPTH to improve bone strength is completed dependent on the presence of T cells. Although the reason for this discrepancy is unknown, is possible that T cells might be required to improve the material property of bone. It should also be noted that while the stimulation of bone formation induced by iPTH was severely blunted in T cell deficient mice, T cells did not improve the capacity of cPTH to stimulate bone formation. The reason for this critical difference remains to be determined.
With regard of the mechanism by which T cells potentiate the bone anabolic activity of iPTH, studies have disclosed that in the absence of T cells iPTH is unable to increase the commitment of SCs to the osteoblastic lineage, induce OB proliferation and differentiation, and mitigate OB apoptosis. All of these action of PTH were found to hinge on the capacity of T cells to activate Wnt signaling in osteoblastic cells [33]. Although it is well established that Wnt activation is a key mechanism by which iPTH expands the osteoblastic pool, little information is available on the nature and the source of the Wnt ligand required to activate Wnt signaling in OBs. We have found that PTH stimulates BM CD8+ T cells to produce large amounts of Wnt10b [33], a Wnt protein which activates Wnt signaling in SCs and OBs, thus increasing OB proliferation, differentiation, and life-span. Treatment with iPTH also caused a small increase in the production of Wnt10b by BM CD4+ cells. The relevance of CD8+ cells was demonstrated by the inability of iPTH to promote bone anabolism in class I MHC−/− mice, a strain that lacks CD8+ cells [33]. Additional studies revealed that iPTH does not improve bone architecture in T cell deficient mice reconstituted with CD4+ cells, while it does so in mice adoptively transferred with CD8+ cells [33]. The pivotal role of T cell produced Wnt10b was revealed by the hampered effect of iPTH on bone volume in global Wnt10b null mice [45] and T cell null mice reconstituted with T cells from Wnt10b−/− mice [33].
Together the data indicate that CD8+ T cells potentiates the anabolic activity of PTH by providing Wnt10b, which is a critical Wnt ligand required for activating Wnt signaling in osteoblastic cells Therefore in the absence of CD8+ cells, stimulation of osteoblastic cells by PTH is not sufficient to elicit maximal Wnt activation due to the lack of a critical Wnt ligand (Fig. 2). The residual bone anabolic activity of PTH observed in T cell deficient mice is presumably due to ligand independent activation of LRP6 [23], and suppressed production of sclerostin [8, 24, 25].

Schematic representation of the role of T cells in the mechanism by which intermittent PTH treatment stimulates bone formation. PTH stimulates T cells to secrete Wnt10b, a Wnt ligand required to activate Wnt signaling in SCs and OBs. In the presence of T cell produced Wnt10b, stimulation of osteoblastic cells by PTH result in the activation of the Wnt signaling pathway. This event leads to increased commitment of mesenchymal stem cells to the osteoblastic lineage, increased osteoblast proliferation and differentiation, and decreased osteoblast apoptosis. Reproduced with permission from Bedi et al. [45]
Our earlier studies did not reveal whether direct activation of PPR receptors in T cells by PTH is required for iPTH treatment to exert its full anabolic activity. To address these issues we made use of PPRT cells−/− mice, a strain with a silent PPR in all T cells [38]. First, we treated PPRT cells−/− with iPTH for 4 weeks to determine whether the hormone induces T cell production of Wnt10b by directly targeting T cells. These studies disclosed that iPTH increased Wnt10b mRNA levels in T cells from control mice. By contrast PTH had no stimulatory effects in T cells from PPRT cells−/− mice [45]. Next we addressed the question of whether iPTH increases bone volume by directly activating the PPR receptor in T cells. We found that the capacity of iPTH to increase trabecular bone volume was decreased, but not completely abolished, in young PPRT cells−/− mice. By contrast, the anabolic activity of iPTH was completely blocked in mature PPRT cells−/− mice. These findings suggest that the contribution of T cells to the activity of iPTH increases with age. Analysis of histomorphometric and biochemical indices of bone turnover and cellular studies revealed that silencing of PPR signaling in T cells blunts the stimulation of osteoblastogenesis and bone turnover induced by iPTH. In summary the data demonstrate that T cells are direct targets of PTH that play a pivotal role in the osteoblastogenic response to iPTH (Fig. 3).

Schematic representation of the role of T cells in the mechanism by which intermittent PTH treatment stimulates HSPCs expansion. Direct PPR signaling in T cells stimulates T cells to secrete Wnt10b, a Wnt ligand required to activate Wnt signaling. In the presence of T cell produced Wnt10b, PTH activates Wnt signaling in stromal cells and HSPCs. Wnt signaling activation also upregulates the expression of the Notch ligand Jagged 1 in SCs. These events result in HSPCs expansion. Reproduced with permission from Li et al. [46]
Interestingly our data show that continuous activation of PPR in T cells induced by continuous PTH treatment is required for the hormone to stimulate bone resorption but not bone formation [38]. The changes in bone turnover induced by cPTH reflect the capacity of continuous activation of PPR in T cells to increase the production of TNF but not of Wnt10b [38]. By contrast, intermittent activation of T cell expressed PPR by iPTH is required for the hormone to fully stimulate bone formation and bone resorption. Moreover, intermittent activation of T cell expressed PPR induces T cell production of Wnt10b but not of TNF [45]. Thus, cell autonomous effects of PTH in T cells resulting from intermittent or continuous PPR signaling play pivotal roles in the overall mechanism of action of PTH in bone.
Role of T cells on the expansion of HSCs induced by PTH
Although it is well established that iPTH expands HSCs, the specific HSC population regulated by iPTH has remained unknown until recently. HSCs comprise at least two major populations. The first is the most primitive long-term reconstituting subset of HSPCs (LT-HSPCs). The second is the short-term reconstituting subset of HSPCs (ST-HSPCs), a population which arise from LT-HSPCs and possess limited self-renewal activity. Therefore, studies were designed to identify the population of HSPCs regulated by iPTH. Moreover, since iPTH regulates bone cells only in the presence of T cells [33, 45], additional studies were designed to determine whether T cells are required for PTH to expand HSPCs. Comparisons of T cell replete mice and T cell deficient mice treated with PTH for revealed that iPTH specifically expands ST-HSCs and does so only in mice with T cells [46].
To determine whether iPTH needs to directly targets T cells to expand HSCs, studies were conducted using PPRT cells−/− mice, a strain with a silent PPR in all T cells [38]. These studies disclosed that iPTH expands ST-HSPCs in control mice but not in PPRT cells−/− mice [46].
To determine whether the stimulatory effects of iPTH on HSPCs might be relevant in BM transplantation studies were conducted to determine whether iPTH increases survival of lethally irradiated mice subjected to BM transplantation. Thee studies revealed that iPTH treatment of either donor or recipient mice increased survival by approximately threefold, but only in the presence of T cells.
Activation of Wnt signaling in both HSPCs and BM stromal cells (SCs) is known to be required for the expansion of HSPCs. This consideration led to the hypothesis that increased production of Wnt10b by iPTH-stimulated T cells might lead to Wnt activation and HSCs expansion. Indeed studies revealed that iPTH activates Wnt signaling in SCs through PTH receptor signaling in T cells.
Activation of Wnt signaling has been linked to upregulation of Jagged1 expression in SCs. Increased expression of Jagged1 by SCs is a potential mechanism by which iPTH expands HSPCs [27]. Not surprisingly studies revealed that iPTH increases the expression of Jagged1 in SCs from control mice. By contrast iPTH did not increase Jagged1 expression in SCs from mice lacking T cells or the T cell production of Wnt10b. Together these findings demonstrate that T cells and their production of Wnt10b are required for iPTH to activate Wnt signaling and upregulate Jagged-expression in SCs.
Attesting to the relevance of Wnt10b, iPTH failed to expand ST-HSPCs in mice with T cell specific deletion of Wnt10b. Moreover, iPTH fails to promote HSCs engraftment after BM transplantation in Wnt10b null mice. Additional experiments disclosed that iPTH had no effect on the survival of mice transplanted with BM from Wnt10b null mice. Thus, T cells and Wnt10b are required for iPTH to activate Wnt signaling in HSPs and increase survival after BM transplantation. In summary, direct PTH receptor signaling in T cells and the resulting production of Wnt10b play a pivotal role in the mechanism by which iPTH expands ST-HSCs.
Conclusions
Remarkable progress has been made in understanding how T cells participate in the regulation of bone remodeling and the hemopoietic stem cell niche in health and disease. Progress has been made in recognizing that T cells play an unexpected role in the function of a major calciotropic hormone such as PTH, and therefore in common and clinically relevant forms of bone loss such primary hyperparathyroidism. Much remains to be done especially in translating observations accrued in experimental animals into studies in humans. Strategies to utilize T cells/bone cells interactions as the bases of new therapies also remain to be developed.
References
B.L. Riggs, L.J. Melton, Medical progress: involutional osteoporosis. N. Eng. J. Med. 314, 1676–1684 (1986)
J. Potts, Primary hyperparathyroidism, in Metabolic Bone Diseases, 3rd edn, vol. 1, ed. by L.V. Avioli, S. Krane (Academic Press, San Diego, 1998), pp. 411–442
G. Mazziotti, J. Bilezikian, E. Canalis, D. Cocchi, A. Giustina, New understanding and treatments for osteoporosis. Endocrine 41(1), 58–69 (2012)
E. Canalis, A. Giustina, J.P. Bilezikian, Mechanisms of anabolic therapies for osteoporosis. N. Engl. J. Med. 357(9), 905–916 (2007)
L. Qin, L.J. Raggatt, N.C. Partridge, Parathyroid hormone: a double-edged sword for bone metabolism. Trends Endocrinol. Metab. 15(2), 60–65 (2004)
B. Lanske, M. Amling, L. Neff, J. Guiducci, R. Baron, H.M. Kronenberg, Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leads to distinct abnormalities in bone development. J. Clin. Invest. 104(4), 399–407 (1999)
L.M. Calvi, N.A. Sims, J.L. Hunzelman, M.C. Knight, A. Giovannetti, J.M. Saxton, H.M. Kronenberg, R. Baron, E. Schipani, Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J. Clin. Invest. 107(3), 277–286 (2001)
C.A. O’Brien, L.I. Plotkin, C. Galli, J.J. Goellner, A.R. Gortazar, M.R. Allen, A.G. Robling, M. Bouxsein, E. Schipani, C.H. Turner, R.L. Jilka, R.S. Weinstein, S.C. Manolagas, T. Bellido, Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 3(8), e2942 (2008)
C. Galli, L.A. Zella, J.A. Fretz, Q. Fu, J.W. Pike, R.S. Weinstein, S.C. Manolagas, C.A. O’Brien, Targeted deletion of a distant transcriptional enhancer of the receptor activator of nuclear factor-kappaB ligand gene reduces bone remodeling and increases bone mass. Endocrinology 149(1), 146–153 (2008)
R. Lindsay, F. Cosman, H. Zhou, M.P. Bostrom, V.W. Shen, J.D. Cruz, J.W. Nieves, D.W. Dempster, A novel tetracycline labeling schedule for longitudinal evaluation of the short-term effects of anabolic therapy with a single iliac crest bone biopsy: early actions of teriparatide. J. Bone Miner. Res. 21(3), 366–373 (2006)
Y.L. Ma, Q. Zeng, D.W. Donley, L.G. Ste-Marie, J.C. Gallagher, G.P. Dalsky, R. Marcus, E.F. Eriksen, Teriparatide increases bone formation in modeling and remodeling osteons and enhances IGF-II immunoreactivity in postmenopausal women with osteoporosis. J. Bone Miner. Res. 21(6), 855–864 (2006)
H. Dobnig, R.T. Turner, Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology 136(8), 3632–3638 (1995)
G.J. Pettway, J.A. Meganck, A.J. Koh, E.T. Keller, S.A. Goldstein, L.K. McCauley, Parathyroid hormone mediates bone growth through the regulation of osteoblast proliferation and differentiation. Bone 42, 806–818 (2007)
X.W. Meng, X.G. Liang, R. Birchman, D.D. Wu, D.W. Dempster, R. Lindsay, V. Shen, Temporal expression of the anabolic action of PTH in cancellous bone of ovariectomized rats. J. Bone Miner. Res. 11(4), 421–429 (1996)
R.L. Jilka, R.S. Weinstein, T. Bellido, P. Roberson, A.M. Parfitt, S.C. Manolagas, Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Invest. 104(4), 439–446 (1999)
T. Bellido, A.A. Ali, L.I. Plotkin, Q. Fu, I. Gubrij, P.K. Roberson, R.S. Weinstein, C.A. O’Brien, S.C. Manolagas, R.L. Jilka, Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J. Biol. Chem. 278(50), 50259–50272 (2003)
M. Kato, M.S. Patel, R. Levasseur, I. Lobov, B.H. Chang, D.A. Glass 2nd, C. Hartmann, L. Li, T.H. Hwang, C.F. Brayton, R.A. Lang, G. Karsenty, L. Chan, Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 157(2), 303–314 (2002)
R.L. Jilka, Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 40(6), 1434–1446 (2007)
M. Almeida, L. Han, T. Bellido, S.C. Manolagas, S. Kousteni, Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by beta-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J. Biol. Chem. 280(50), 41342–41351 (2005)
D.A. Glass II, P. Bialek, J.D. Ahn, M. Starbuck, M.S. Patel, H. Clevers, M.M. Taketo, F. Long, A.P. McMahon, R.A. Lang, G. Karsenty, Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 8(5), 751–764 (2005)
J. Behrens, J.P. von Kries, M. Kuhl, L. Bruhn, D. Wedlich, R. Grosschedl, W. Birchmeier, Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 382(6592), 638–642 (1996)
N.H. Kulkarni, D.L. Halladay, R.R. Miles, L.M. Gilbert, C.A. Frolik, R.J. Galvin, T.J. Martin, M.T. Gillespie, J.E. Onyia, Effects of parathyroid hormone on Wnt signaling pathway in bone. J. Cell. Biochem. 95(6), 1178–1190 (2005)
M. Wan, C. Yang, J. Li, X. Wu, H. Yuan, H. Ma, X. He, S. Nie, C. Chang, X. Cao, Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 22(21), 2968–2979 (2008)
T. Bellido, A.A. Ali, I. Gubrij, L.I. Plotkin, Q. Fu, C.A. O’Brien, S.C. Manolagas, R.L. Jilka, Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146(11), 4577–4583 (2005)
H. Keller, M. Kneissel, SOST is a target gene for PTH in bone. Bone 37(2), 148–158 (2005)
L. Qin, P. Qiu, L. Wang, X. Li, J.T. Swarthout, P. Soteropoulos, P. Tolias, N.C. Partridge, Gene expression profiles and transcription factors involved in parathyroid hormone signaling in osteoblasts revealed by microarray and bioinformatics. J. Biol. Chem. 278(22), 19723–19731 (2003)
L.M. Calvi, G.B. Adams, K.W. Weibrecht, J.M. Weber, D.P. Olson, M.C. Knight, R.P. Martin, E. Schipani, P. Divieti, F.R. Bringhurst, L.A. Milner, H.M. Kronenberg, D.T. Scadden, Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425(6960), 841–846 (2003)
S. Brunner, H.D. Theiss, A. Murr, T. Negele, W.M. Franz, Primary hyperparathyroidism is associated with increased circulating bone marrow-derived progenitor cells. Am J Physiol Endocrinol Metab 293(6), E1670–E1675 (2007)
G.B. Adams, R.P. Martin, I.R. Alley, K.T. Chabner, K.S. Cohen, L.M. Calvi, H.M. Kronenberg, D.T. Scadden, Therapeutic targeting of a stem cell niche. Nat. Biotechnol. 25(2), 238–243 (2007)
S. Brunner, M.M. Zaruba, B. Huber, R. David, M. Vallaster, G. Assmann, J. Mueller-Hoecker, W.M. Franz, Parathyroid hormone effectively induces mobilization of progenitor cells without depletion of bone marrow. Exp. Hematol. 36(9), 1157–1166 (2008)
K.K. Ballen, E.J. Shpall, D. Avigan, B.Y. Yeap, D.C. Fisher, K. McDermott, B.R. Dey, E. Attar, S. McAfee, M. Konopleva, J.H. Antin, T.R. Spitzer, Phase I trial of parathyroid hormone to facilitate stem cell mobilization. Biol. Blood Marrow Transpl. 13(7), 838–843 (2007)
S. Mendez-Ferrer, T.V. Michurina, F. Ferraro, A.R. Mazloom, B.D. Macarthur, S.A. Lira, D.T. Scadden, A. Ma’ayan, G.N. Enikolopov, P.S. Frenette, Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466(7308), 829–834 (2010)
M. Terauchi, J.Y. Li, B. Bedi, K.H. Baek, H. Tawfeek, S. Galley, L. Gilbert, M.S. Nanes, M. Zayzafoon, R. Guldberg, D.L. Lamar, M.A. Singer, T.F. Lane, H.M. Kronenberg, M.N. Weitzmann, R. Pacifici, T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab. 10(3), 229–240 (2009)
O. Stojceva-Taneva, G.Z. Fadda, M. Smogorzewski, S.G. Massry, Parathyroid hormone increases cytosolic calcium of thymocytes. Nephron 64(4), 592–599 (1993)
L. Rifas, T-cell cytokine induction of BMP-2 regulates human mesenchymal stromal cell differentiation and mineralization. J. Cell. Biochem. 98(4), 706–714 (2006)
B.G. Hory, M.C. Roussanne, S. Rostand, A. Bourdeau, T.B. Drueke, J. Gogusev, Absence of response to human parathyroid hormone in athymic mice grafted with human parathyroid adenoma, hyperplasia or parathyroid cells maintained in culture. J. Endocrinol. Invest. 23(5), 273–279 (2000)
Y. Gao, X. Wu, M. Terauchi, J.Y. Li, F. Grassi, S. Galley, X. Yang, M.N. Weitzmann, R. Pacifici, T cells potentiate PTH-induced cortical bone loss through CD40L signaling. Cell Metab. 8(2), 132–145 (2008)
H. Tawfeek, B. Bedi, J.Y. Li, J. Adams, T. Kobayashi, M.N. Weitzmann, H.M. Kronenberg, R. Pacifici, Disruption of PTH receptor 1 in T cells protects against PTH-induced bone loss. PLoS ONE 5(8), e12290 (2010)
C.D. Surh, J. Sprent, Homeostasis of naive and memory T cells. Immunity 29(6), 848–862 (2008)
B. Ernst, D.S. Lee, J.M. Chang, J. Sprent, C.D. Surh, The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity 11(2), 173–181 (1999)
B. Bedi, J.Y. Li, F. Grassi, H. Tawfeek, M.N. Weitzmann, R. Pacifici, Inhibition of antigen presentation and T cell costimulation blocks PTH-induced bone loss. Ann. N. Y. Acad. Sci. 1192(1), 215–221 (2010)
N. Zhang, M.J. Bevan, CD8(+) T cells: foot soldiers of the immune system. Immunity 35(2), 161–168 (2011)
G.J. Pettway, A. Schneider, A.J. Koh, E. Widjaja, M.D. Morris, J.A. Meganck, S.A. Goldstein, L.K. McCauley, Anabolic actions of PTH (1–34): use of a novel tissue engineering model to investigate temporal effects on bone. Bone 36(6), 959–970 (2005)
F. Di Rosa, T-lymphocyte interaction with stromal, bone and hematopoietic cells in the bone marrow. Immunol. Cell Biol. 87, 20–29 (2008)
B. Bedi, J.Y. Li, H. Tawfeek, K.H. Baek, J. Adams, S.S. Vangara, M.K. Chang, M. Kneissel, M.N. Weitzmann, R. Pacifici, Silencing of parathyroid hormone (PTH) receptor 1 in T cells blunts the bone anabolic activity of PTH. Proc. Natl. Acad. Sci. U. S. A. 109, E725–E733 (2012)
J.Y. Li, J. Adams, L.M. Calvi, T.F. Lane, R. Dipaolo, M.N. Weitzmann, R. Pacifici, PTH expands short-term murine hemopoietic stem cells through T cells. Blood 120, 4352–4362 (2012)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pacifici, R. Role of T cells in the modulation of PTH action: physiological and clinical significance. Endocrine 44, 576–582 (2013). https://doi.org/10.1007/s12020-013-9960-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-013-9960-8