
Abstract
Cushing’s disease (CD) is a severe endocrine condition caused by an adrenocorticotropin (ACTH)-producing pituitary adenoma that chronically stimulates adrenocortical cortisol production and with potentially serious complications if not or inadequately treated. Active CD may produce a fourfold increase in mortality and is associated with significant morbidities. Moreover, excess mortality risk may persist even after CD treatment. Although predictors of risk in treated CD are not fully understood, the importance of early recognition and adequate treatment is well established. Surgery with resection of a pituitary adenoma is still the first line therapy, being successful in about 60–70 % of patients; however, recurrence within 2–4 years may often occur. When surgery fails, medical treatment can reduce cortisol production and ameliorate clinical manifestations while more definitive therapy becomes effective. Compounds that target hypothalamic–pituitary axis, glucocorticoid synthesis or adrenocortical function are currently used to control the deleterious effects of chronic glucocorticoid excess. In this review we describe and analyze the molecular basis of the drugs targeting the disease at central level, suppressing ACTH secretion, as well as at peripheral level, acting as adrenal inhibitors, or glucocorticoid receptor antagonists. Understanding of the underlying molecular mechanisms in CD and of glucocorticoid biology should promote the development of new targeted and more successful therapies in the future. Indeed, most of the drugs discussed have been tested in limited clinical trials, but there is potential therapeutic benefit in compounds with better specificity for the class of receptors expressed by ACTH-secreting tumors. However, long-term follow-up with management of persistent comorbidities is needed even after successful treatment of CD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pituitary adrenocorticotropin (ACTH)-dependent hypercortisolism, caused by micro or macroadenomas originating from the corticotroph cells of the pituitary gland, is also known as Cushing’s disease (CD) [1]. CD is a rare but serious condition accounting for about 70 % of cases of endogenous hypercortisolism. The standard treatment of choice for CD is transsphenoidal adenomectomy [1–3]. Patients with residual or recurrent disease might benefit of a second surgery or radiotherapy, however for patients in which surgical resection is contraindicated, medical therapies represent a treatment option in order to ameliorate clinical manifestations of hypercortisolism even though some drugs display important limitations due to the severe toxicity [2]. Medications currently used in patients with CD include neuromodulatory agents, targeted therapy agents, steroidogenesis inhibitors, and glucocorticoid receptor (GR) antagonist.
This review focuses on current data about medical therapies acting at the hypothalamus pituitary levels, as well as on peripherally acting agents, and highlights the molecular mechanisms underlying their action. Compounds with neuromodulatory properties, including serotonin antagonists, gamma-aminobutyric acid (GABA) agonists, dopamine agonists and somatostatin (SST) analogs, drugs targeting membrane and nuclear hormone receptor or specific intracellular pathways, and finally agents inhibiting adrenal steroidogenesis and GR antagonists will be addressed with regard to clinical applications in CD.
Central targeting agents
Hypothalamic neuromodulators
Corticotrophin releasing hormone (CRH) neurons of the paraventricular nucleus receive significant innervations from many hypothalamic and extrahypothalamic brain regions that indirectly modulate the activity of the hypothalamic–pituitary–adrenal (HPA) axis. Indeed, HPA axis activity is regulated mainly by the hypothalamic CRH and vasopressin (AVP) which are, in turn, under the influence of several neurotransmitters and neuropeptides.
Along the last three decades many of these neurotransmitters and neuropeptides influencing the HPA axis have been investigated for a potential role in the treatment of CD. However, conflicting, and often unclear conclusions, have been drawn due to the difficulty of translating preclinical data to the clinical setting. Among these molecules serotonin (5-HT), which generally stimulates hypothalamic CRH release, and GABA, which mediates inhibitory inputs have been more largely explored, generating small clinical investigation studies [4, 5].
A stimulatory role of 5-HT on the HPA axis is well established in human. Indeed, the administration of 5-HT precursors, releasers, uptake inhibitors, or receptor agonists increases plasma ACTH, β-endorphin, and glucocorticoid levels [6]. Clearly, 5-HT receptor antagonists may counteract this effect [4]. This direct stimulatory effect on CRH is antagonized by metergoline, a 5-HT1 and 5-HT2 receptor antagonist and, with similar potency, by ketanserin and ritanserin, two rather selective 5-HT2 receptor antagonists, suggesting a main role of the 5-HT2 receptor in mediating the serotonin action at this level. Initially, a potential CRH-independent mechanism, mediated by a cholinergic interneuron, has been hypothesized, however more recent evidences exclude this possibility [4].
Considering this background, one of the first attempts in vivo for the treatment of CD has been explored with the use of cyproheptadine, an antiserotoninergic and antihistaminergic drug, which displays a poor selectivity and may also have anticholinergic activity. However, analyzing the few reported small clinical trials and single cases, a rather low percentage of partial response rates (<10 %) or only short temporary effects are recorded compared with the more selective serotonin antagonists, such as ritanserin and ketanserin, which resulted apparently effective in about 20 % of cases [7–11]. Based on the faint evidence that the majority of positive results were observed in microadenomas or in those patients with negative imaging (undetectable microadenomas, pituitary corticotroph cells hyperplasia, or hypothalamic CD), it was again hypothesized that the effects were exerted via the serotoninergic central nervous system stimuli on hypothetic hypothalamic factors promoting ACTH secretion, or alternatively via a potent direct inhibitory activity on CRH and AVP producing neurons [11]. Lately, other authors have also proposed a potential direct effect on signaling pathways in the corticotroph cells, therefore at pituitary level, without affecting or influencing neurotransmitters regulating HPA axis [11, 12]. However, besides the still unknown exact mechanism of action of this class of drugs, and the variability in the clinical outcome, the sedative side effects, as well as the orexigenic properties, strongly limited the use of anti-serotonergic drugs in the clinical practice [9, 13].
The neuroanatomical evidence for a possible role of GABA in the regulation of CRH release and of the HPA axis is the presence of GABA-synthesizing enzyme glutamate decarboxylase-positive nerve fibers in the hypothalamus and GABA terminals in the median eminence. Two types of receptors (GABA-A and GABA-B) are involved, being GABA-A a macromolecular complex with separate binding sites for GABA, benzodiazepines, and barbiturate-like substances [4]. Activation of the GABA-A receptor results in the opening of the chloride channel linked to this receptor, leading to an influx of chloride ions and hyperpolarization of the cell firing [4]. Conversely, the GABA-B does not have binding sites for benzodiazepines and is not linked to the chloride channel. Activation of the GABA-B receptor increases the influx of extracellular calcium ions [4, 14]. GABA inhibits HPA axis function in vivo, and this effect appears to be exerted mainly at central level. Indeed, subcutaneous injection of gamma-aminohydroxybutyric acid, a GABA derivative which does not cross the blood brain barrier, does not have any effect on HPA axis function [15], whereas GABA injected intracerebroventricularly causes a reduction of CRH levels in hypophysial-portal blood [16].
Sodium valproate (valproic acid) inhibits GABA aminotransferase, and in patients with epilepsy was shown to decrease ACTH levels, possibly inhibiting CRH and hence ACTH release [17, 18]. Based on this evidence placebo-controlled studies have been carried out in CD, however, the results did not support the use of sodium valproate as monotherapy, although its combination with inhibitors of cortisol secretion appeared to be more beneficial [19, 20].
Peroxisome proliferator-activated receptor-gamma agonists
Peroxisome proliferator-activated receptor gamma (PPARγ) belongs to the family of nuclear hormone receptor transcription factors, whose ligands, thiazolidinediones (TZD), are well-known insulin sensitizers and have been demonstrated to have anticancer properties as well [21]. The evidence of PPARγ expression in normal ACTH-secreting human anterior pituitary cells and in human ACTH-secreting pituitary adenomas prompted several studies to understand whether PPARγ agonists may be useful as medical tools for treating CD. In both murine and human ACTH-secreting pituitary adenoma cells, PPARγ activators induced G0/G1 cell-cycle arrest and apoptosis and suppressed ACTH secretion. Similarly, rosiglitazone, a clinically available TZD, prevented the development of corticotroph tumors in mice injected with the ACTH-secreting AtT20 cells; in addition, in these mice ACTH and corticosterone secretion was suppressed [22]. These data were confirmed in other experimental settings, where rosiglitazone determined a 20 % decrease in cell viability of a human ACTH-secreting pituitary tumor in vitro, without correlation between PPARγ expression and the anti-tumoral effect of the drug [23]. However, these encouraging results were not confirmed by further studies, demonstrating the efficacy of rosiglitazone in reducing ACTH secretion only in two out of eight primary cultures [24]. In addition, clinical studies showed variable but generally unsatisfactory results in terms of disease control in humans [21], probably due to differences in treatment duration and doses used. As a consequence, TZD did not find a clinical application in the medical treatment of CD.
Vasopressin antagonists
Another potential class of drugs for suppressing ACTH secretion is represented by AVP antagonists, also used in the diagnosis of CD. Indeed, desmopressin, a long-acting analog stimulate ACTH secretion from pituitary adenomas. This effect is due to the expression of V2 and V3 receptors in the neoplastic corticotroph cells. AVP antagonists, called vaptans, have been developed to antagonize both the pressor and antidiuretic effects of AVP [25]. There are three subtypes of receptors, V1a, V1b, and V2, belonging to the large rhodopsin-like G-protein-coupled receptor family. However, the recent cloning and molecular characterization of AVP/oxytocin receptor subtypes call for the revision of their nomenclature, V1a receptor is also called V1 vascular receptor, the V2 receptor, V2 renal receptor, and the V1b is the V3 pituitary receptor. Indeed, this latter receptor is located mainly in the anterior pituitary gland and plays a role in ACTH release. Both V1a and V1b receptors act through the intracellular phosphoinositol signaling pathway, Ca(++) being the second messenger [25]. V2 receptors work through cAMP generation, which promotes aquaporin 2 (AQP2) trafficking and allows water to enter the cell. The most important vaptans are selective V2 antagonists and they mainly influence free water elimination without affecting electrolyte excretion. There are several studies on the effects of these drugs in hypervolemic hyponatremia (heart failure, hepatic cirrhosis) as well as in normovolemic hyponatremia (SIADH) [26]. Development of receptor-specific AVP antagonists could lead to agents that suppress ACTH-secreting adenomas and reverse hypercortisolemia. The antagonists now available to bind preferentially to V2 and V1a receptors and their effect is mainly on water reabsorption by the renal tubule, and their influence on ACTH secretion is still unclear. Antagonists specific for V3 (V1b) receptors are more likely to be effective in CD [27].
Retinoic acid
Retinoic acid (RA) inhibits the transcriptional activity of AP-1 and the orphan receptors Nur77, and Nurr1 in ACTH-secreting tumor cells. Preliminary experiments demonstrated that RA treatment reduces pro-opiomelanocortin (POMC) transcription and ACTH production in animal and human pituitary ACTH-secreting tumor, but not in normal cells [28]. RA also inhibited cell proliferation and after prolonged treatment, increased caspase-3 activity and induced cell death in ACTH-secreting cells. The in vitro effects of RA were confirmed in a in vivo model in nude mice [28]. Due to its additional peripheral activity at adrenal level, these initial findings indicated that RA could be a promising option to inhibit ACTH, cortisol, and tumor growth in CD. This possibility was further supported by the evidence that RA also induces bone morphogenetic protein-4 (BMP-4), a member of the TGF-β family that plays a central role during pituitary organogenesis, transcription, and expression. In addition, RA antiproliferative action is blocked in Smad-4dn- and noggin transfected AtT-20 cells that do not respond to BMP-4. Therefore, RA induces BMP-4, which participates in its antiproliferative effect [29]. These preliminary findings promoted a randomized treatment with RA versus ketoconazole in dogs with CD [30]. A significant reduction in circulating hormones as well as pituitary adenoma size was recorded at the end the study, and RA treatment in animals also led to a resolution of the clinical phenotype [30]. Despite these promising outcomes, only one pilot study on a small number of patients (seven) is available so far [31].
Somatostatin analogs
The presence of SST receptors (SSTRs) in corticotroph cells provides the basis to explain SST analogs’ influence on these cells, both in normal and pathological conditions. SST has been demonstrated to influence normal corticotroph cells, since SST knock-out mice display elevated plasma corticosterone and increased POMC mRNA levels. In addition, SST administration in mice reduces pituitary POMC mRNA expression, ACTH synthesis and release, supporting the hypothesis that endogenous SST has an inhibitory action on corticotroph cells [32]. These effects are mediated by SSTRs, which expression has been demonstrated in mouse pituitary adenoma cell lines and in rat pituitary, where SSTR2 and SSTR5 transduce their inhibitory effects on CRF- and forskolin-induced ACTH secretion [33, 34]. In human pituitary adenoma corticotroph cells SSTR5 seems to be the most expressed subtype [35]. The biological activity of these receptors is witnessed by the evidence that in a mouse ACTH-secreting pituitary adenoma line, the AtT20 cells, SST reduces ACTH secretion, mainly interacting with SSTR2, which rapidly internalizes [36]. Conversely, SSTR5 persists at the membrane level, also after interacting with the ligand, indicating a functional difference between SSTR2 and SSTR5 [37]. Moreover, there is evidence indicating that SSTR5 regulates SSTR2 action, by attenuating the potency of SSTR2 agonists and retaining SSTR2 within the plasma membrane, preventing its internalization and desensitization. These data provide support to the hypothesis that SSTR5 activation is required to warrant efficacy of SST analogs in the treatment of patients with ACTH-secreting adenomas [36].
Further evidence for SST influence on corticotroph cells has been provided by a study in which, a SSTR pan-agonist, pasireotide, as well as octreotide and a selective SSTR5 agonist (BIM-23268), were demonstrated to reduce CRF-induced ACTH release in AtT-20 cells [38]. However, only compounds having greater affinity for SSTR5 (pasireotide and BIM-23268) showed an enhanced inhibitory effect on ACTH secretion in the presence of dexamethasone, indicating that SSTR5 is resistant to the negative control exerted by glucocorticoids. Indeed, there is evidence that dexamethasone down-regulates SSTR2 but not SSTR5 expression in AtT-20 cells [38, 39]. Pasireotide has been shown to inhibit ACTH release even after prolonged treatment, but did not influence cell proliferation or apoptosis, on the contrary of what demonstrated in non-functioning pituitary adenomas [40]. In addition, no evidence for inhibition of POMC synthesis was found, suggesting that pasireotide might act by inducing ACTH break-down [39]. These in vitro effects have been confirmed in rats, where pasireotide demonstrated a greater inhibitory effect on ACTH, as well as on corticosterone secretion, as compared to octreotide [41]. The effects of pasireotide and octreotide on mitotic activity, apoptosis and ACTH labeling index have also been explored in rat pituitaries before and after adrenalectomy. These studies showed that these analogs do not affect baseline pituitary cell turnover and apoptosis, while they block the expected rise in corticotroph mitotic index after adrenalectomy. Similarly, no effect was observed at baseline concerning ACTH secretion, which rise after adrenalectomy was completely blunted by treatment with pasireotide or octreotide, suggesting that SSTR2 and SSTR5 activation might mediate corticotroph responses to adrenalectomy [42]. In addition, these results indicate that ACTH secretion in normal rat corticotroph may be reduced by SST only when glucocorticoids fail to exert their physiological feedback on ACTH release [43]. Therefore, the available evidence demonstrates that SST inhibits CRH-induced ACTH secretion by rat pituitary cells in vitro in the absence of glucocorticoids [44].
Human corticotroph seem to respond differently to SST and its analogs, since SST infusion in healthy adults may result in an activation of the HPA axis, suggesting that changes in HPA axis might be mediated acutely by SST-induced reduction in GH plasma levels and complex interplay at central level [45]. Other studies, however, demonstrated that SST or octreotide administration do not influence ACTH secretion in normal healthy individuals [46, 47], but can repress ACTH secretion in patients with adrenal insufficiency [48] and in patients with Nelson’s syndrome [49]. This evidence suggests that SST and its analogs do not affect normal corticotroph function, but may influence it in pathological conditions. It has been shown, indeed, that octreotide can reduce CRF-induced ACTH release by corticotroph tumors in vitro, an effect that was abolished by hydrocortisone treatment [50]. Further studies demonstrated that in vivo octreotide has no effect on basal or CRF-stimulated ACTH secretion in patients with CD, likely due to the negative influence of high circulating levels of glucocorticoids [51–53]. In vitro studies demonstrated that glucocorticoids inhibit SSTR2 expression by corticotroph adenoma cells, but do not affect SSTR5 expression [38], providing an explanation for the lack of efficacy of SST analogs having greater affinity for SSTR2 in patients with CD. The negative effect of glucocorticoids on SSTR function was confirmed by another study, which demonstrated that this inhibitory effect is not restricted to corticotroph but is rather a generalized phenomenon, also affecting other tissues [54].
SST analogs are capable of inhibiting ACTH secretion in human corticotroph adenomas in the absence of glucocorticoid negative influence: pasireotide, better than octreotide, reduces ACTH secretion apparently through its interaction with SSTR5, the mostly expressed subtype in corticotroph tumors [39], where, besides SSTR2, also SSTR1 expression has been reported [55, 56], especially in silent corticotropic adenomas [57]. These results have been further confirmed and extended by a study [58], demonstrating that pasireotide is capable of reducing corticotroph cell proliferation, an effect likely mediated by a reduction in the MAPK pathway [59] promoted by SSTR5 activation [60]. Indeed, the latter receptor mediates both the short- and long-term enhanced action of pasireotide in corticotroph tumor cells, while pasireotide action on SSTR2 seems negligible [58]. In addition, pasireotide promotes SSTR5 internalization in a limited fashion, while it is clearly more potent than octreotide in inducing SSTR5-mediated signaling and endocytosis [61].
These studies, therefore, underline the differential effects promoted by the activation of the SSTR subtypes, as also previously reported in other tissues [62]. In addition these data provide the basis for the use of pasireotide in patients with CD [63], that may be effective especially when glucocorticoid levels are lowered by simultaneous treatment with other compounds [64].
Dopaminergic drugs
Dopaminergic pathways play a crucial role in the central nervous system, and dopamine is involved in the control or hormone production in endocrine glands as well. Indeed, dopamine receptors (DRs) are differentially expressed in pituitary cells. Among the five different G-protein coupled DR subtypes, codenamed D1–5, and subdivided in D1-like (D1, D5) and D2-like (D2, D3, D4) groups, the subtype 2 (D2) exists in two different isoforms, the long (D2long) and short (D2short) isoforms [65]. In rats, D2s are predominantly expressed in lactotrophs, however also the other pituitary cell subsets may display a variable presence of this subtype. In particular, D2 expression in corticotroph cells, initially suggested by the demonstration of D2 expression in the cell line AtT20, displays a rather complicated distribution [66]. In humans, D2 is expressed in more than 75 % of cells of the anterior pituitary gland, confirming a complex spectrum of expression extended to non-lactotroph cells. In fact, D2s have been clearly demonstrated in the “intermediate lobe” of the pituitary. With respect to the isoforms, several studies on experimental models have demonstrated that both D2long and D2short variants are expressed in lactotroph and melanotroph cells, although D2long seems the predominant one. Finally, D2 is frequently accompanied by the expression of D4, in particular its D4.4 variant, although its role in the physiology of the pituitary gland is not completely understood [65].
D2 is involved in the negative regulation of POMC gene expression in melanotroph cells. The principal products of the POMC gene are β-endorphin and α-MSH; in the corticotrophs POMC is principally processed into ACTH. D2-deficient mice display an increased POMC expression, intermediate lobe hypertrophy, and unexpected elevated ACTH levels with a corresponding increase of corticosteroids and consequent hypertrophy of the adrenal gland, resembling the human CD, suggesting the crucial role for dopamine in the control of POMC-derived peptides [67].
In the recent years, the increasing knowledge, as well as the availability of sophisticated techniques, together with the newer dopaminergic compounds entered in the clinical practice, increased the evidence on the pathophysiological role of dopamine and DRs in corticotroph tumors [68, 69]. Early in vitro studies with bromocriptine showed conflicting results, however the observed differences were probably related to the different study designs and to the lack of receptor characterization [70, 71]. More recently, Pivonello et al. [72] on the basis of previous studies showing the effectiveness of dopamine agonists in inducing shrinkage of silent ACTH-secreting pituitary tumors and of adenomas in Nelson’s syndrome, demonstrated by immunohistochemistry, receptor-ligand binding, RT-PCR as well as by dynamic experiments, the expression of functional D2 in around 80 % of ACTH-secreting pituitary tumors, and showed the effectiveness of treatment with cabergoline in normalizing ACTH and cortisol secretion in almost 40 % of these patients, suggesting the potential therapeutic use of dopaminergic drugs in the management of persistent and/or recurrent CD. In this series D4 receptor was found in 20 % of cases, whereas D1, D3, and D5 were undetectable [72].
Finally, based on the fact that many corticotroph cells may co-express both SSTRs and D2 receptors [69], and that these receptors may work synergistically, probably via membrane interaction or dimerization [73], new chimeric molecules, containing both SST and dopamine structural elements, have been tested. By binding two different receptors these molecules may draw the receptors together, in a spatial manner, leading to a higher potency of the chimeric drugs, compared to the separate activation of SSTRs and DRs [74]. Studies carried on different tumor models, including pituitary adenomas, revealed a higher potency of chimeric molecules, such as BIM-23A779, BIM-23A760, and BIM-23A781, in controlling tumor cell functions [75, 76]. While more convincing preclinical data are available for pituitary GH-secreting tumors, the efficacy of hybrid compounds on CD is, however, still under investigation. The key question is whether or not the exposure of such multiple ligands could increase their efficacy, by comparison to the effects of the single ligands, in controlling the different pituitary adenoma cells.
Interestingly, in different neuroendocrine cell line models, glucocorticoids selectively down-regulate SSTR2, whereas both D2 and SSTR5 appear more resistant to this effect which can be responsible for the low expression of SSTR2 in corticotroph adenomas and justify the current interest in SSTR5 and D2 as possible therapeutic targets for medical treatment of CD [54]. The co-expression of these receptors in corticotroph tumors open interesting areas of exploration for the development of new medical approaches in CD [56, 77].
The complete characterization and definition of the molecular basis of the signaling pathways involving SSTRs and DRs are strongly warranted since in experimental settings the new analogs may result in prolonged pharmacological properties, or could activate alternative intracellular signaling pathways involved in the control of cell growth, as well as cell secretion [68].
Preliminary data strongly support the possibility to use a combined treatment involving at least both pathways (SSTRs/DRs) and eventually with drugs inhibiting cortisol production at the adrenal level, such as ketoconazole. Feelders et al. [64] evaluated the sequential addition of pasireotide, cabergoline, and ketoconazole in patients with CD when they did not respond to initial and/or subsequent treatment. In this study, 17 patients initially received pasireotide 100 μg three times daily, with an increase to 250 μg/dose if the urinary free cortisol (UFC) did not normalize in 2 weeks. Cabergoline (0.5–.5 mg every other day) was added if there was no response at day 28, and ketoconazole at low doses (200 mg three times daily) was added to non-responders at day 60. Normal UFC was achieved in five patients with pasireotide, four with pasireotide and cabergoline, and six taking all three medications, for an biochemical control in nearly 90 % of patients. In a recent study the combined treatment with cabergoline (0.5–3 mg/week) and ketoconazole (200–600 mg/day) led to UFC normalization in 79 % of patients [78].
Temozolomide
Temozolomide is an orally administered second-generation alkylating chemotherapeutic agent, capable of crossing the blood–brain barrier [79]. Its derivative, methyl-triazeno-imidazole-carboxamide, methylates DNA at the O6 position of guanine [80], causing a mismatch with thymine during the next cycle of DNA replication, eventually leading to apoptosis, independently of the cell cycle phase. As a consequence, temozolomide can be effectively used also in relatively slow-growing pituitary tumors [81]. The efficacy of the drug is limited by the activity of the enzyme O6-methylguanine-DNA methyltransferase (MGMT), a DNA repairing protein, that reverses alkylation at the O6 position of guanine [82]. Low MGMT tumor expression, most likely due to epigenetic silencing of the MGMT gene, has been associated with enhanced sensitivity to temozolomide treatment [83]. However, the molecular mechanisms regulating MGMT expression have not been fully clarified yet. Among pituitary adenomas, ACTH-producing tumors frequently show low MGMT expression [84], also in the settings of invasive Crooke cell adenomas [85], with no correlation with patient age, sex, tumor size, invasiveness, or recurrence [86], suggesting that these tumors more likely respond to temozolomide treatment. Indeed, MGMT expression, but not MGMT gene promoter methylation, has been shown to have a good positive predictive value (nearly 80 %) in ACTH-secreting pituitary adenomas as concerns response to temozolomide treatment, defined as a 20 % decrease in maximal tumor size and as a 50 % decrease in hormone secretion [87]. At the tissue level, it has been reported that treatment with temozolomide leads to tumor softening and friability, facilitating surgical removal [88]. In addition, pituitary tumors from patients treated with temozolomide displayed better differentiation, fewer mitoses, lower Ki-67 labeling index, tissue hemorrhage, necrosis, focal fibrosis, and neuronal transformation also were observed [89]. These data suggest that temozolomide has a direct effect on pituitary adenoma cell proliferation. Sheehan et al. [90] provided further support to this hypothesis, since they showed that temozolomide exerts a direct antiproliferative effect in the mouse ACTH-secreting pituitary adenoma cell line, AtT20, where 50 μM temozolomide significantly inhibited cell viability and induced apoptosis. These data support the clinical efficacy recorded in several (but not all) cases of aggressive CD treated with temozolomide [84].
Tyrosine kinase and mTOR inhibitors
Growth factors regulate pituitary transdifferentiation processes and contribute to mutated pituitary cell transformation into a pituitary adenoma, mostly in an autocrine–paracrine fashion, since most of them are indeed produced by the pituitary itself [91]. Among the various pituitary growth factors, vascular endothelial growth factor (VEGF) is known to promote vasculogenesis during embryonic life and angiogenesis after birth, therefore regulating oxygen supply, formation of collateral circulation, regeneration after injury, as well as cancer blood supply. Microvessel density is increased in ACTH-secreting pituitary adenomas, but this characteristic is not associated with greater aggressiveness. On the contrary VEGF-R2 expression is associated with extrasellar extension [92], as well as with pituitary tumor transforming gene (PTTG) and fibroblast growth factor (FGF) expression [93], all characteristics associated with a greater pituitary adenoma proliferative potential. These data may provide the basis for the treatment of ACTH-secreting pituitary tumors, as recently attempted in a case of an invasive silent corticotroph adenoma, treated with bevacizumab, a monoclonal antibody that recognizes and blocks VEGF receptors. This drug stopped tumor progression and caused severe cell injury, vascular abnormalities and fibrosis, inducing a long-lasting disease stabilization, indicating that anti-angiogenic therapy may represent a new option in the treatment of aggressive pituitary tumors [94].
Epidermal growth factor (EGF) and its receptors are expressed both in normal and adenomatous pituitary cells [95, 96], where they participate in the control of POMC expression and in both basal and CRH-induced ACTH release [97]. EGF receptor (EGFR) expression has been reported to correlate with tumor invasiveness [98] and its activation in AtT20 cells promotes cell proliferation [99]. The importance of EGF blockade as a possible tool for medical treatment of CD has been highlighted by the evidence that anti-EGFR molecules suppress POMC expression in human and canine corticotroph cultured tumors [100]. In addition, in AtT20 cells transfected with EGFR, gefitinib, an EGFR tyrosine kinase inhibitor was capable of blocking the stimulatory effects of EGF on POMC expression, ACTH secretion and cell proliferation by interacting with the MAPK pathway and by inducing apoptosis. These powerful effects were also confirmed in vivo in a mouse model, further supporting the concept that inhibiting EGFR signaling may represent a new medical approach for treating CD [100].
The phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) cascade is a key signaling pathway that regulates cell homeostasis, energy balance, as well as normal and neoplastic growth. It has been recently demonstrated that the PI3K/Akt/mTOR pathway is upregulated in pituitary adenomas [99] and that its blockade in both pituitary cell lines and in pituitary adenoma primary cultures inhibits cell proliferation, promoting apoptosis [101, 102], suggesting that mTOR inhibition may be a promising therapeutic option for pituitary adenomas resistant and unresponsive to other treatments. Human ACTH-secreting pituitary tumors express mTOR and its active phosphorylated isoform at similar levels to those observed in the normal pituitary [103]. These results were confirmed by further studies demonstrating a slight activation of the PI3/AKT/mTOR signaling pathway in 18 ACTH-secreting pituitary adenomas, as evaluated by transcriptomic analysis [104], which mirrors the little efficacy of rapamycin in reducing cell viability in AtT-20 cells [105]. This lack of efficacy has been attributed to the positive feed-back loop on AKT activation by mTOR inhibitors, that confers resistance to these compounds [106]. In the AtT-20 cells, mTOR resistance can be effectively overcome by octreotide co-treatment, which induced G1 cell cycle arrest, with a concomitant increase in p27/Kip1 levels and a consequent reduction in E2F transcriptional activity and cyclin E levels [106]. These data suggest that adjuvant treatment with a SST analog may sensitize pituitary tumor cells to the antiproliferative effects of mTOR inhibitors, also in the setting of CD.
Peripheral targeting agents
Steroidogenesis inhibitors
Among the available medical therapies, agents that inhibit steroidogenesis are the most commonly used. Ketoconazole, metyrapone, mitotane, LCI699, aminoglutethimide, trilostane, and etomidate decrease cortisol production by direct inhibition of steroidogenesis at one or more enzymatic steps. Steroidogenesis inhibitors are highly effective to suppress glucocorticoid synthesis, but do not affect the underlying pituitary tumor associated with CD [27, 107].
Among the currently available drugs, ketoconazole appears to be the most effective [2, 106–110]. Ketoconazole is an imidazole derivative developed as an oral antifungal agent that, in high doses, reduces adrenal steroid production via inhibition of multiple steroidogenic enzymes, such as cholesterol side-chain cleavage, 17,20-lyase, 11-β-hydroxylase, and 17-α-hydroxylase [110, 111]. In addition to adrenal blocking effects, ketoconazole may also have a direct effect on corticotrope tumor cells in patients with CD. Ketoconazole inhibits in vitro both basal and CRF-stimulated ACTH release from rat anterior pituitary cells [112]. When administered to five women with CD for 3 months both urine cortisol excretion and plasma ACTH levels decreased, supporting the hypothesis that ketoconazole may have an additional site of inhibition at the pituitary level [113]. However, in other studies ACTH response to CRH was enhanced [114] or unchanged [115] during ketoconazole treatment in patients with CD. Because of its potent inhibitory effects on P450-dependent enzymes modulating adrenal steroidogenesis, several side effects occur during therapy with ketoconazole: hepatotoxicity, adrenal insufficiency, and gastrointestinal complaints, as well as hypogonadism and gynecomastia in men. A meta-analysis of 82 patients with CD showed that single agent treatment with ketoconazole reduces plasma cortisol levels in ~70 % of cases (range 25–93 %) [116]. Another advantage of this compound is its effect on lipid metabolism. Ketoconazole reduces total cholesterol and LDL cholesterol [117]. This beneficial effect is secondary to the inhibition of cholesterol synthesis. Interestingly, ketoconazole, owing to its imidazole group, inhibits LDL oxidation by acting as an iron chelator and/or inhibitor of prooxidant forms of heme-containing enzymes [118].
Metyrapone is the most potent, short-acting inhibitor of cortisol synthesis with a rapid onset of action. It inhibits 11-β-hydroxylase, consequently blocking the last step of adrenal steroid biosynthesis in which the biologically inactive 11-deoxycortisol is converted to cortisol [119]. Nausea, vomiting, and dizziness are potential side-effects related to sudden cortisol reduction and adrenal insufficiency [28, 107]. Other side-effects, particularly when administered to patients with CD, are hirsutism and acne, which can be clearly problematic in women. These events are due to the increase in androgenic precursors secondary to the inhibition of 11-β-hydroxylase coupled with high ACTH levels. Adverse effects due to increased 11-deoxycorticosterone levels (hypokalemia, edema, and hypertension) are less frequent, presumably because of the benefits of lower circulating cortisol levels [111, 120]. These side effects are less commonly observed in association with mitotane, presumably due to its inhibitory activity on other enzymes involved in adrenal androgen and mineralocorticoid synthesis [121]. Metyrapone therapy can be effective in CD, with rapid reductions in cortisol levels. In a 16-week study of 53 patients, normalization of mean daily plasma cortisol levels has been observed in 75 % of cases treated with metyrapone [122]. However; in several countries metyrapone is currently not commercially available. In addition, a major hurdle of metyrapone and ketoconazole is the fact that both agents tend to lose their beneficial effects, as a result of increased secretion of ACTH by the pituitary tumor in response to decreased feedback inhibition, secondary to the reduction of cortisol levels. Increasing doses of the drug are often required to maintain adequate clinical remission, but this raises problems of compliance and tolerability.
Mitotane or o,p′-DDD (1-(o-chlorophenyl)-1-(p-chlorophenyl)-2,2-dichloroethane), is an isomer of DDD, an insecticide analog of DDT. Mitotane is used in the treatment of CD since it blocks cortisol synthesis, causing alterations in the mitochondrial function and altering the extra-adrenal metabolism of cortisol and androgens. Mitotane reduces cortisol production by blocking cholesterol side-chain cleavage and 11-β-hydroxylase and at doses greater than 4 g/day it has adrenolytic action. Mitotane is hydroxylated at the β-carbon and transformed to an acyl-chloride by a mitochondrial P-450 enzyme. The acyl-chloride covalently binds to specific bionucleophiles in adrenal cells, leading to cellular necrosis [111, 120, 123, 124]. This effect underscores its primary use in patients with adrenocortical carcinoma [125]. It also appears to alter the extra-adrenal metabolism of steroids leading to a reduction in measurable 17-hydroxy corticosteroids, even though plasma corticosteroid concentrations do not fall and increase the formation of 6-β-hydroxy cortisol. Interestingly, the cortisol response to mitotane therapy should be estimated by measuring UFC. Serum cortisol levels can be elevated even when circulating free cortisol is not elevated, since mitotane increases binding of cortisol to corticosteroid binding globulin [27]. In addition, it has been recently demonstrated that mitotane reduces cell viability and reduces ACTH secretion in AtT20 cells and in primary cultures. In AtT20 cells mitotane reduces POMC expression and blocks the stimulatory effects of CRH on cell viability, ACTH secretion, and POMC expression. These effects were apparent at mitotane doses greater than those usually necessary for reducing cortisol secretion in Cushing’s syndrome, but still in the therapeutic window for adrenocortical carcinoma treatment. These data indicate that mitotane could have direct pituitary effects on corticotroph cells [126].
Mitotane, particularly at doses above 6 g daily, has serious gastrointestinal, neurological, pituitary [127], and hepatic side effects and it can induce adrenal insufficiency. Side effects of this medication and the delayed onset of mitotane action (weeks or months) may limit its use. Because of its adrenolytic action, mitotane seems to be effective in the long-term management of CD, even after treatment withdrawal, because the drug has a long half-life and is stored in adipose tissue [124]. Contrary to other steroidogenesis inhibitors, its mechanism of action prevents the escape phenomenon. Mitotane alone can achieve remission in up to 83 % of cases with a sustained remission after discontinuation of treatment in about a third of subjects [128]. More often mitotane is used with radiation therapy for the treatment of CD [129]. The concomitant pituitary radiotherapy in patients who undergo mitotane treatment could be useful to reduce the risk of developing Nelson syndrome.
LCI699 is a drug belonging to the category of the steroidogenesis inhibitors and its structure is similar to FAD286 (R-enantiomer of fadrozole). It is a potent inhibitor of human recombinant aldosterone synthase, the enzyme that catalyzes the generation of aldosterone from the hydroxycorticosterone, and of 11-β-hydroxylase, that catalyzes both the hydroxylation of deoxycorticosterone and of deoxycortisol in order to obtain corticosterone and cortisol, respectively [130]. LCI699 ability of inhibiting aldosterone synthase and 11-β-hydroxylase is justified by the amino acid sequences of these two enzymes that are 95 % identical in the coding regions [130]. The inhibition of CYP11B1 induces an increase in 11-deoxycorticosterone, 11-deoxycortisol, and ACTH plasma levels. Aldosterone is the final component of the renin–angiotensin–aldosterone system (RAAS) and RAAS is a key regulator of blood pressure. The reduced plasma aldosterone levels and blood pressure as effects of LCI699 have been already investigated in patients with primary hyperaldosteronism [131] and in a double-blind randomized trial in patients with primary hypertension [132]. Currently, LCI699 is under investigation as medical agent for the management of CD [133]. Preliminary results of a multicenter proof-of-concept study conducted on adult patients with mild to severe CD have demonstrated the ability of LCI699 in normalizing UFC in 100 % of investigated patients during treatment, but after treatment discontinuation UFC increased in 66.6 % of patients [134].
Aminoglutethimide is an anticonvulsant agent that also suppresses adrenal steroid production by inhibition of the side-chain cleavage of cholesterol to pregnenolone and additionally inhibits 11-β-hydroxylase, 18-hydroxylase, and aromatase activity [27, 111]. Aminoglutethimide has been used to decrease cortisol production in CD in the past, although with an unfavorable adverse event profile. The manufacturer stopped producing the drug in 2007 and it is no longer available [27].
Trilostane is a competitive inhibitor of the steroidogenic enzyme 3-β-hydroxysteroid dehydrogenase, which is an essential enzyme in the synthesis of cortisol, aldosterone, and androstenedione. It is an effective inhibitor of steroid synthesis in vitro but in human the results have been disappointing [135, 136]. Currently, trilostane is used in veterinary practice as it is very effective in controlling CD in dogs [120].
Etomidate, a commonly used short-acting intravenous anesthetic, is a potent inhibitor of side chain cleavage enzyme 11-β-hydroxylase, aldosterone synthase, as well as an inhibitor of 17-α-hydroxylase [27, 120, 124, 137]. There have been a number of case reports on the successful use of etomidate in controlling hypercortisolemia in seriously ill patients with CD even in the pediatric population [123, 138]. Etomidate’s use is limited by the need to be given intravenously. However, in situations where rapid control of cortisol levels is required and oral therapy is problematic, etomidate therapy may be preferred [2]. Figure 1 illustrates the sites of action of major adrenolytic drugs on steroidogenesis.

Adrenolitic drugs acting on steroidogenesis and their sites of action
Combination therapy with several steroidogenesis inhibitors acting at different pathways of hypercortisolemia may be a promising procedure to suppress effectively cortisol secretion and to allow lower doses of drugs with serious adverse events. Kamenicky et al. [139] recently evaluated the efficacy and safety of combination therapy with mitotane, metyrapone, and ketoconazole in 11 patients with severe ACTH-dependent Cushing’s syndrome. This approach benefits from two fast-acting steroidogenesis inhibitors (metyrapone and ketoconazole), providing rapid clinical and biological control of severe hypercortisolism, thus covering the time lag before obtaining favorable effects of mitotane. Furthermore, the concomitant administration of ketoconazole may prevent metyrapone-induced excessive deoxycorticosterone and androgens production responsible of hypokalemia, hypertension, and hirsutism. In all patients a marked clinical improvement and a striking decrease of 24-h UFC have been observed. In seven patients, metyrapone and ketoconazole were discontinued and UFC excretion remained controlled by mitotane monotherapy. Five patients were able to undergo etiological surgery. Adverse effects were tolerable, consisting mainly of gastrointestinal discomfort. This combination may provide an alternative to emergency high-risk bilateral adrenalectomy.
Glucocorticoid receptor antagonists
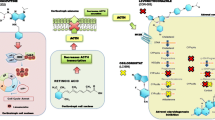
Mifepristone, designed as 11β-[p-(dimethylamino)phenil]-17β-hydroxy-17-(1-propynyl)estra-4,9-dien-3-one, is a GRs and progesterone receptors (PR) antagonist. It has been mainly used as an “abortion” pill given for its antiprogestin activity [140]. The GR, expressed widely in human body, belong to the superfamily of nuclear receptors and show a characteristic three-domain structure constituted by a ligand-binding domain (LBD), at C-terminal part; a DNA-binding domain (DBD), located in the center, and a transactivation domain in N-terminal part [141]. GR also contain sequences necessary to bind some heat shock proteins (HSPs), necessary to have high-affinity steroid-binding activity, for the translocation from the cytoplasm to the nucleus and interaction with some transcription factors and transcription intermediary factors [141]. When GR are not bound to the ligand, they are located mainly in the cytosol but in some cells types they are localized constitutively in the nucleus, as a heteromeric complex consisting of the receptor and several HSPs: HSP90, HSP70, HSP56, and HSP23 [138] (Fig. 2). Upon the binding of a specific agonist, i.e., cortisol, one of the mechanisms believed to undergo the GR transcriptional regulation is the occurrence of GR transformation followed by the conformational change of the molecule that leads to the dissociation from HSPs complex. GR homodimerization, aided by LBD, allows the receptors to translocate into the nucleus binding the glucocorticoids responsive elements (GREs). After the interaction with chromatin and other transcription factors, such as c-Jun, the transcription machinery (trans-activation) is activated [142–144]. In presence of an antagonist, such as mifepristone (this exhibits an affinity 18 times higher than cortisol and 4 times higher than dexamethasone), GR are bound strongly to the HSP complex leading to a non-functional nuclear translocation with no alteration changes in chromatin and loss of transcription activation function (trans-repression) (Fig. 2), as demonstrated also in AtT20 cell line [141–143, 145–147].

Human GR are present in cytoplasm as multimeric complexes consisting of the receptor, several HSP, and TPR protein. The 4 main receptor associated TPR proteins are: FKBP52, FKBP51, Cyp40, and PP5. Hormone and antagonists binding leads to receptor conformational change, dissociation of cytoplasmic complex, GR dimerization, and traslocation into the nucleus (a, b). Mifepristone binding stabilizes the multimeric complex but fails to trigger the transcription machinery (b). GR glucocorticoid receptors, HSP heat shock protein, TPR tetratricopeptide repeat, GRE glucocorticoid response element, nGRE negative glucocorticoid response element
Mifepristone is orally administered, rapidly absorbed in gastric milieu, primarily metabolized in the liver by the enzyme CYP3A4, and after demethylation and hydroxylation, three major metabolites are formed. These may contribute to the overall effects of mifepristone given their high concentrations in serum, although their affinity for the receptors and their potency are less than those of mifepristone [148]. The use of mifepristone in hypercortisolism has been investigated in some cases, in retrospective studies and in one recent large prospective multicenter trial [149–154]. In CD as in malignant Cushing’s syndrome, mifepristone induced an improvement of clinical signs of hypercortisolism. During the first month of mifepristone treatment, metabolic and blood pressure benefits have been also observed. In patients with severe psychosis, rapid improvements of psychiatric signs have been reported [152–154]. However, a long-term administration of mifepristone produces persistent elevation of ACTH and cortisol circulating levels due to alterations in negative feedback loop. Indeed, unlike the adrenolytic agents, mifepristone is not able to decrease cortisol levels as a result of receptor-blocking strategy. The cortisol excess may not be regulated by 11-β-hydroxysteroid dehydrogenase and since mineralocorticoid receptors (MR) are not blocked, the high levels of cortisol could lead to the MR activation [155, 156] and consequently to hypokalemia status [149–154]. Therefore, the treatment with mifepristone should require at the same time the administration of MR antagonists, i.e., spironolactone and eplerenone [27]. Despite these issues, in February 2012, the Food and Drug Administration (FDA) has approved mifepristone (Korlym) to control hyperglycemia in adults with endogenous Cushing’s syndrome, who have type II diabetes or glucose intolerance, and remained unresponsive to previous surgery or were not eligible candidates for surgery [157].
Medical management of aggressive and rare malignant tumors
Aggressive corticotroph adenomas and carcinomas are rare pituitary tumors with poor prognosis; carcinomas are characterized by the presence of distant metastases to the extracranial region or the cerebrospinal space [158]. Medical therapy of such malignant tumors is clinically very challenging, since limited information is available, mainly represented by case reports. The effects of treatment with DA agonists, SST analogs, tamoxifen, cyproheptadine, mitotane, and systemic chemotherapy have been reported as variable or palliative [159]. In addition, these approaches do not seem to influence long-term survival [160, 161], which might be improved by surgical excision of metastases [162].
The first goal of medical therapy, even in the settings of an aggressive pituitary tumor is to normalize or at least reduce ACTH and cortisol levels, in order to reduce CD morbidity and mortality. In these settings, pasireotide has proven to be effective in CD patients not eligible for pituitary surgery, most of which had severe disease, since it was capable of reducing UFC in 15–26 % of patients after 6 months and in ~65 % of patients after 12 months [63]. It has been reported that this drug was also effective in achieving short-term control of hypercortisolism [163], and to induce stable disease in an ACTH-secreting pituitary carcinoma previously treated with temozolomide [164].
Medical treatment with temozolomide has been suggested as a possible effective means to try and control aggressive CD disease. Several reports document a significant tumor shrinkage associated with an improvement in clinical conditions, not invariably correlated with a decrease in ACTH levels, in patients with pituitary ACTH-secreting pituitary carcinoma [81, 85, 165–169]. However, clinical validation studies are still lacking.
Treatment with the GR antagonist, mifepristone, has been reported to be effective in controlling symptoms in the settings of aggressive CD [150], even for a very long time (95 months) [170]. A multi-center, open-label study showed that this drug indeed improves CD clinical manifestations with an acceptable benefit-risk profile [154]. However, data on long-term effects on tumor bulk are not available, so far.
In emergency conditions, when any other medical treatment is precluded (i.e., acute psychosis), intravenous etomidate infusion can be effective in controlling hypercortisolemia, allowing to perform bilateral adrenalectomy [171]. However, this therapeutic approach should be undertaken in intensive care settings, with tight serum cortisol monitoring to assess the need for replacement hydrocortisone infusion.
Current guidelines recommend repeated surgery, whenever feasible, followed by radiation therapy, medical therapy, or bilateral adrenalectomy performed in specialized centers with the collaboration of a multidisciplinary neuroendocrine team in order to properly pursue every treatment modality available [2]. Figure 3 depicts a possible algorithm, which may be useful in case of aggressive and/or malignant tumors.

Algorithm for aggressive and/or malignant tumors
Conclusions
The treatment of CD patients involves surgical resection of the pituitary tumor. However, pharmacological therapy may have an important role in the management of this disease. There is a long “negative” history in searching for medical therapy of CD, however, the emerging knowledge on the inter- and intracellular mechanisms in pituitary cells, including the growth factors circuitries and the potential gene regulation, and their interactions with other signaling pathways will help in elucidating what exactly happens in the formation of a benign corticotroph adenoma in order to set up novel forms of pharmacological tumor control. We would expect that over the next 5–10 years new potential central targets will be discovered, opening a new exciting scenario in the treatment of pituitary tumors including CD. At moment the new SST analogs, pasireotide, has been approved as the first drug targeting the corticotroph tumors. Moreover, at peripheral level, mifepristone and LCI699 represent the latest available therapeutic tools. Mifepristone has been recently approved by the United States FDA for use in Cushing’s syndrome, in patients who are not surgical candidates or have not achieved remission from surgery, while LCI699 is still under investigation. Understanding of the underlying molecular mechanisms in CD and of glucocorticoid biology will promote the development of new targeted and more successful therapies in the future. In addition, the efficacy of novel combination therapies with acceptable tolerability, particularly in patients with more severe hypercortisolism, needs to be better explored. Finally, long-term follow-up with management of persistent comorbidities is needed even after successful surgical or medical treatment of CD.
References
R. Pivonello, M.C. De Martino, M. De Leo, G. Lombardi, A. Colao, Cushing’s syndrome. Endocrinol. Metab. Clin. N. Am. 37, 135–149 (2008)
B.M. Biller, A.B. Grossman, P.M. Stewart, S. Melmed, X. Bertagna, J. Bertherat, M. Buchfelder, A. Colao, A.R. Hermus, L.J. Hofland, A. Klibanski, A. Lacroix, J.R. Lindsay, J. Newell-Price, L.K. Nieman, S. Petersenn, N. Sonino, G.K. Stalla, B. Swearingen, M.L. Vance, J.A. Wass, M. Boscaro, Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J. Clin. Endocrinol. Metab. 93, 2454–2462 (2008)
J.S. Lim, S.K. Lee, S.H. Kim, E.J. Lee, S.H. Kim, Intraoperative multiple-staged resection and tumor tissue identification using frozen sections provide the best result for the accurate localization and complete resection of tumors in Cushing’s disease. Endocrine 40, 452–461 (2011)
A.E. Calogero, Neurotransmitter regulation of the hypothalamic corticotropin-releasing hormone neuron. Ann. N. Y. Acad. Sci. 771, 31–40 (1995)
R. Giordano, M. Pellegrino, A. Picu, L. Bonelli, M. Balbo, R. Berardelli, F. Lanfranco, E. Ghigo, E. Arvat, Neuroregulation of the hypothalamus–pituitary–adrenal (HPA) axis in humans: effects of GABA-, mineralocorticoid-, and GH-secretagogue-receptor modulation. Sci. World J. 6, 1–11 (2006)
R.W. Fuller, The involvement of serotonin in regulation of pituitary–adrenocortical function. Front. Neuroendocrinol. 13, 250–270 (1992)
B. Ambrosi, M. Gaggini, F. Secchi, G. Faglia, Lack of effect of antiserotoninergic and/or dopaminergic treatment in patients with pituitary-dependent Cushing’s syndrome. Horm. Metab. Res. 11, 318–319 (1979)
F. Cavagnini, U. Raggi, P. Micossi, A. Di Landro, C. Invitti, Effect of an antiserotoninergic drug, metergoline, on the ACTH and cortisol response to insulin hypoglycemia and lysine-vasopressin in man. J. Clin. Endocrinol. Metab. 43, 306–312 (1976)
N. Sonino, G.A. Fava, F. Fallo, A. Franceschetto, P. Belluardo, M. Boscaro, Effect of the serotonin antagonists ritanserin and ketanserin in Cushing’s disease. Pituitary 3, 55–59 (2000)
R. Tanakol, F. Alagöl, H. Azizlerli, O. Sandalci, T. Terzioğlu, F. Berker, Cyproheptadine treatment in Cushing’s disease. J. Endocrinol. Invest. 19, 242–247 (1996)
K.I. Alexandraki, A.B. Grossman, Pituitary-targeted medical therapy of Cushing’s disease. Expert Opin. Investig. Drugs 17, 669–677 (2008)
T. Suda, F. Tozawa, T. Mouri, A. Sasaki, T. Shibasaki, H. Demura, K. Shizume, Effects of cyproheptadine, reserpine, and synthetic corticotropin-releasing factor on pituitary glands from patients with Cushing’s disease. J. Clin. Endocrinol. Metab. 56, 1094–1099 (1983)
D.T. Krieger, L. Amorosa, F. Linick, Cyproheptadine-induced remission of Cushing’s disease. N. Engl. J. Med. 293, 893–896 (1975)
N.G. Bowery, D.R. Hill, A.L. Hudson, G.W. Price, W.J. Turnbull, G.P. Wilson, Heterogeneity of mammalian GABA receptors, in Actions and interactions of GABA and benzodiazepine, ed. by N.G. Bowery (Raven Press, New York, 1984), pp. 81–108
J. Takahara, S. Yumoki, W. Yakushji, J. Yamauchi, H. Hosogi, T. Ofuji, Stimulatory effects of gamma-aminohydroxybutyric acid (GABOB) on growth hormone, prolactin and cortisol release in man. Horm. Metab. Res. 12, 31–34 (1980)
P.M. Plotsky, S. Otto, S. Sutton, Neurotransmitter modulation of corticotropin releasing factor secretion into the hypophysial-portal circulation. Life Sci. 41, 1311–1317 (1987)
H.P. Koppeschaar, R.J. Croughs, J.H. Thijssen, F. Schwarz, Sodium valproate and cyproheptadine may independently induce a remission in the same patient with Cushing’s disease. Acta Endocrinol. (Copenh). 104, 160–163 (1983)
A. Beckers, A. Stevenaert, G. Pirens, P. Flandroy, J. Sulon, G. Hennen, Cyclical Cushing’s disease and its successful control under sodium valproate. J. Endocrinol. Invest. 13, 923–929 (1990)
A. Colao, R. Pivonello, F.S. Tripodi, F. Orio Jr, D. Ferone, G. Cerbone, C. Di Somma, B. Merola, G. Lombardi, Failure of long-term therapy with sodium valproate in Cushing’s disease. J. Endocrinol. Invest. 20, 387–392 (1997)
S.S. Nussey, P. Price, J.S. Jenkins, A.R. Altaher, B. Gillham, M.T. Jones, The combined use of sodium valproate and metyrapone in the treatment of Cushing’s syndrome. Clin. Endocrinol. (Oxf). 28, 373–380 (1988)
M. Mannelli, G. Cantini, G. Poli, M. Mangoni, G. Nesi, L. Canu, E. Rapizzi, E. Borgogni, T. Ercolino, V. Piccini, M. Luconi, Role of the PPAR-γ system in normal and tumoral pituitary corticotropic cells and adrenal cells. Neuroendocrinology 92(Suppl 1), 23–27 (2010)
A.P. Heaney, M. Fernando, W.H. Yong, S. Melmed, Functional PPAR-gamma receptor is a novel therapeutic target for ACTH-secreting pituitary adenomas. Nat. Med. 8, 1281–1287 (2002)
K. Winczyk, J. Kunert-Radek, A. Gruszka, M. Radek, H. Ławnicka, M. Pawlikowski, Effects of rosiglitazone-peroxisome proliferators-activated receptor gamma (PPARgamma) agonist on cell viability of human pituitary adenomas in vitro. Neuro. Endocrinol. Lett. 30, 107–110 (2009)
L. Kreutzer, I. Jeske, B. Hofmann, I. Blumcke, R. Fahlbusch, M. Buchfelder, R. Buslei, No effect of the PPAR-gamma agonist rosiglitazone on ACTH or cortisol secretion in Nelson’s syndrome and Cushing’s disease in vitro and in vivo. Clin. Neuropathol. 28, 430–439 (2009)
M. Manning, A. Misicka, A. Olma, K. Bankowski, S. Stoev, B. Chini, T. Durroux, B. Mouillac, M. Corbani, G. Guillon, Oxytocin and vasopressin agonists and antagonists as research tools and potential therapeutics. J. Neuroendocrinol. 24, 609–628 (2012)
G. Narayen, S.N. Mandal, Vasopressin receptor antagonists and their role in clinical medicine. Indian J. Endocrinol. Metab. 16, 183–191 (2012)
D.E. Schteingart, Drugs in the medical treatment of Cushing’s syndrome. Expert Opin. Emerg. Drugs 14, 661–671 (2009)
M. Páez-Pereda, D. Kovalovsky, U. Hopfner, M. Theodoropoulou, U. Pagotto, E. Uhl, M. Losa, J. Stalla, Y. Grübler, C. Missale, E. Arzt, G.K. Stalla, Retinoic acid prevents experimental Cushing syndrome. J. Clin. Invest. 108, 1123–1131 (2001)
D. Giacomini, M. Páez-Pereda, M. Theodoropoulou, M. Labeur, D. Refojo, J. Gerez, A. Chervin, S. Berner, M. Losa, M. Buchfelder, U. Renner, G.K. Stalla, E. Arzt, Bone morphogenetic protein-4 inhibits corticotroph tumor cells: involvement in the retinoic acid inhibitory action. Endocrinology 147, 247–256 (2006)
V. Castillo, D. Giacomini, M. Páez-Pereda, J. Stalla, M. Labeur, M. Theodoropoulou, F. Holsboer, A.B. Grossman, G.K. Stalla, E. Arzt, Retinoic acid as a novel medical therapy for Cushing’s disease in dogs. Endocrinology 147, 4438–4444 (2006)
F. Pecori Giraldi, A.G. Ambrogio, M. Andrioli, F. Sanguin, I. Karamouzis, S.M. Corsello, C. Scaroni, E. Arvat, A. Pontecorvi, F. Cavagnini, Potential role for retinoic acid in patients with Cushing’s disease. J. Clin. Endocrinol. Metab. 97, 3577–3583 (2012)
R.M. Luque, M.D. Gahete, U. Hochgeschwender, R.D. Kineman, Evidence that endogenous SST inhibits ACTH and ghrelin expression by independent pathways. Am. J. Physiol. Endocrinol. Metab. 291, E395–E403 (2006)
D. Cervia, D. Fehlmann, D. Hoyer, Native somatostatin sst2 and sst5 receptors functionally coupled to Gi/o-protein, but not to the serum response element in AtT-20 mouse tumour corticotrophs. Naunyn Schmiedebergs Arch. Pharmacol. 367, 387–578 (2003)
M.Z. Strowski, M.P. Dashkevicz, R.M. Parmar, H. Wilkinson, M. Kohler, J.M. Schaeffer, A.D. Blake, Somatostatin receptor subtypes 2 and 5 inhibit corticotropin-releasing hormone-stimulated adrenocorticotropin secretion from AtT-20 cells. Neuroendocrinology 75, 339–346 (2002)
S. Nielsen, S. Mellemkjaer, L.M. Rasmussen, T. Ledet, N. Olsen, M. Bojsen-Møller, J. Astrup, J. Weeke, J.O. Jørgensen, Expression of somatostatin receptors on human pituitary adenomas in vivo and ex vivo. J. Endocrinol. Invest. 24, 430–437 (2001)
U.I. Richardson, A. Schonbrunn, Inhibition of adrenocorticotropin secretion by somatostatin in pituitary cells in culture. Endocrinology 108, 281–290 (1981)
A. Ben-Shlomo, K.A. Wawrowsky, I. Proekt, N.M. Wolkenfeld, S.R. Ren, J. Taylor, M.D. Culler, S. Melmed, Somatostatin receptor type 5 modulates somatostatin receptor type 2 regulation of adrenocorticotropin secretion. J. Biol. Chem. 280, 24011–24021 (2005)
J. van der Hoek, M. Waaijers, P.M. van Koetsveld, D. Sprij-Mooij, R.A. Feelders, H.A. Schmid, P. Schoeffter, D. Hoyer, D. Cervia, J.E. Taylor, M.D. Culler, S.W. Lamberts, L.J. Hofland, Distinct functional properties of native somatostatin receptor subtype 5 compared with subtype 2 in the regulation of ACTH release by corticotroph tumor cells. Am. J. Physiol. Endocrinol. Metab. 289, E278–E287 (2005)
L.J. Hofland, J. van der Hoek, R. Feelders, M.O. van Aken, P.M. van Koetsveld, M. Waaijers, D. Sprij-Mooij, C. Bruns, G. Weckbecker, W.W. de Herder, A. Beckers, S.W. Lamberts, The multi-ligand somatostatin analogue SOM230 inhibits ACTH secretion by cultured human corticotroph adenomas via somatostatin receptor type 5. Eur. J. Endocrinol. 152, 645–654 (2005)
M.C. Zatelli, D. Piccin, C. Vignali, F. Tagliati, M.R. Ambrosio, M. Bondanelli, V. Cimino, A. Bianchi, H.A. Schmid, M. Scanarini, A. Pontecorvi, L. De Marinis, G. Maira, E.C. degli Uberti, Pasireotide, a multiple somatostatin receptor subtypes ligand, reduces cell viability in non-functioning pituitary adenomas by inhibiting vascular endothelial growth factor secretion. Endocr. Relat. Cancer 14, 91–102 (2007)
A.P. Silva, P. Schoeffter, G. Weckbecker, C. Bruns, H.A. Schmid, Regulation of CRH-induced secretion of ACTH and corticosterone by SOM230 in rats. Eur. J. Endocrinol. 153, R7–R10 (2005)
L.A. Nolan, H.A. Schmid, A. Levy, Octreotide and the novel multi-receptor ligand somatostatin receptor agonist pasireotide (SOM230), block the adrenalectomy-induced increase in mitotic activity in male rat anterior pituitary. Endocrinology 148, 2821–2827 (2007)
J. van der Hoek, S.W. Lamberts, L.J. Hofland, The role of somatostatin analogs in Cushing’s disease. Pituitary 7, 257–264 (2004)
L.J. Hofland, S.W.J. Lamberts, R.A. Feelders, Role of somatostatin receptors in normal and tumoral pituitary corticotropic cells. Neuroendocrinology 92(suppl 1), 11–16 (2010)
M.R. Ambrosio, M. Campo, M.C. Zatelli, S.G. Cella, G. Trasforini, A. Margutti, A.E. Rigamonti, E.E. Müller, E.C. degli Uberti, Unexpected activation of pituitary-adrenal axis in healthy young and elderly subjects during somatostatin infusion. Neuroendocrinology 68, 123–128 (1998)
C. Invitti, F. Pecori Giraldi, A. Dubini, M. Piolini, F. Cavagnini, Effect of Sandostatin on CRF-stimulated secretion of ACTH, beta-lipotropin and beta-endorphin. Horm. Metab. Res. 23, 233–235 (1991)
P.J. Stafford, P.J. Kopelman, K. Davidson, L. McLoughlin, A. White, L.H. Rees, G.M. Besser, D.H. Coy, A. Grossman, The pituitary-adrenal response to CRF-41 is unaltered by intravenous somatostatin in normal subjects. Clin. Endocrinol. (Oxf). 30, 661–666 (1989)
H.L. Fehm, K.H. Voigt, R. Lang, K.E. Beinert, S. Raptis, E.F. Pfeiffer, Somatostatin: a potent inhibitor of ACTH-hypersecretion in adrenal insufficiency. Klin Wochenschr. 54, 173–175 (1976)
G. Benker, K. Hackenberg, B. Hamburger, D. Reinwein, Effects of growth hormone release-inhibiting hormone and bromocryptine (CB 154) in states of abnormal pituitary-adrenal function. Clin. Endocrinol. (Oxf). 5, 187–190 (1976)
G.K. Stalla, S.J. Brockmeier, U. Renner, C. Newton, M. Buchfelder, J. Stalla, O.A. Müller, Octreotide exerts different effects in vivo and in vitro in Cushing’s disease. Eur. J. Endocrinol. 130, 125–131 (1994)
B. Ambrosi, D. Bochicchio, C. Fadin, P. Colombo, G. Faglia, Failure of somatostatin and octreotide to acutely affect the hypothalamic–pituitary–adrenal function in patients with corticotropin hypersecretion. J. Endocrinol. Invest. 13, 257–261 (1990)
S.W. Lamberts, J. Zuyderwijk, F. den Holder, P. van Koetsveld, L. Hofland, Studies on the conditions determining the inhibitory effect of somatostatin on adrenocorticotropin, prolactin and thyrotropin release by cultured rat pituitary cells. Neuroendocrinology 50, 44–50 (1989)
J. Julesz, F. Laczi, T. Janáky, F. László, Effects of somatostatin and bromocryptine on the plasma ACTH level in bilaterally adrenalectomized patients with Cushing’s disease. Endokrinologie 76, 68–72 (1980)
C. de Bruin, R.A. Feelders, A.M. Waaijers, P.M. van Koetsveld, D.M. Sprij-Mooij, S.W.J. Lamberts, L.J. Hofland, Differential regulation of human dopamine D2 and somatostatin receptor subtype expression by glucocorticoids in vitro. J. Mol. Endocrinol. 42, 47–56 (2009)
A. Saveanu, P. Jaquet, Somatostatin-dopamine ligands in the treatment of pituitary adenomas. Rev. Endocr. Metab. Disord. 10, 83–90 (2009)
C. de Bruin, A.M. Pereira, R.A. Feelders, J.A. Romijn, F. Roelfsema, D.M. Sprij-Mooij, M.O. van Aken, A.J. van der Lelij, W.W. de Herder, S.W. Lamberts, L.J. Hofland, Coexpression of dopamine and somatostatin receptor subtypes in corticotroph adenomas. J. Clin. Endocrinol. Metab. 94, 1118–1124 (2009)
T. Tateno, M. Kato, Y. Tani, K. Oyama, S. Yamada, Y. Hirata, Differential expression of somatostatin and dopamine receptor subtype genes in adrenocorticotropin (ACTH)-secreting pituitary tumors and silent corticotroph adenomas. Endocr. J. 56, 579–584 (2009)
D.L. Batista, X. Zhang, R. Gejman, P.J. Ansell, Y. Zhou, S.A. Johnson, B. Swearingen, E.T. Hedley-Whyte, C.A. Stratakis, A. Klibanski, The effects of SOM230 on cell proliferation and adrenocorticotropin secretion in human corticotroph pituitary adenomas. J. Clin. Endocrinol. Metab. 91, 4482–4488 (2006)
E. Hubina, A.M. Nanzer, M.R. Hanson, E. Ciccarelli, M. Losa, D. Gaia, M. Papotti, M.R. Terreni, S. Khalaf, S. Jordan, S. Czirják, Z. Hanzély, G.M. Nagy, M.I. Góth, A.B. Grossman, M. Korbonits, Somatostatin analogues stimulate p27 expression and inhibit the MAP kinase pathway in pituitary tumours. Eur. J. Endocrinol. 155, 371–379 (2006)
A. Ben-Shlomo, H. Schmid, K. Wawrowsky, O. Pichurin, E. Hubina, V. Chesnokova, N.A. Liu, M. Culler, S. Melmed, Differential ligand-mediated pituitary somatostatin receptor subtype signaling: implications for corticotroph tumor therapy. J. Clin. Endocrinol. Metab. 94, 4342–4350 (2006)
S. Lesche, D. Lehmann, F. Nagel, H.A. Schmid, S. Schulz, Differential effects of octreotide and pasireotide on somatostatin receptor internalization and trafficking in vitro. J. Clin. Endocrinol. Metab. 94, 654–661 (2009)
M.C. Zatelli, F. Tagliati, J.E. Taylor, D. Piccin, M.D. Culler, E.C. degli Uberti, Somatostatin, but not somatostatin receptor subtypes 2 and 5 selective agonists, inhibits calcitonin secretion and gene expression in the human medullary thyroid carcinoma cell line, TT. Horm. Metab. Res. 34, 229–233 (2002)
A. Colao, S. Petersenn, J. Newell-Price, J.W. Findling, F. Gu, M. Maldonado, U. Schoenherr, D. Mills, L.R. Salgado, B.M. Biller, Pasireotide B2305 Study Group, A 12-month phase 3 study of pasireotide in Cushing’s disease. N. Engl. J. Med. 366, 914–924 (2012)
R.A. Feelders, C. de Bruin, A.M. Pereira, J.A. Romijn, R.T. Netea-Maier, A.R. Hermus, P.M. Zelissen, F.H. de Jong, A.J. van der Lely, W.W. de Herder, L.J. Hofland, S.W.J. Lamberts, Stepwise medical treatment of Cushing’s disease with pasireotide mono- or combination therapy with cabergoline and low-dose ketoconazole. N. Engl. J. Med. 362, 1846–1848 (2010)
R. Pivonello, D. Ferone, G. Lombardi, A. Colao, S.W. Lamberts, L.J. Hofland, Novel insights in dopamine receptor physiology. Eur. J. Endocrinol. 156(Suppl 1), S13–S21 (2007)
M. Boschetti, F. Gatto, M. Arvigo, D. Esposito, A. Rebora, M. Talco, M. Albertelli, E. Nazzari, U. Goglia, F. Minuto, D. Ferone, Role of dopamine receptors in normal and tumoral pituitary corticotropic cells and adrenal cells. Neuroendocrinology 92(Suppl 1), 17–22 (2010)
A. Saiardi, E. Borrelli, Absence of dopaminergic control on melanotrophs leads to Cushing’s-like syndrome in mice. Mol. Endocrinol. 12, 1133–1139 (1998)
D. Ferone, R. Pivonello, E. Resmini, M. Boschetti, A. Rebora, M. Albertelli, V. Albanese, A. Colao, M.D. Culler, F. Minuto, Preclinical and clinical experiences with the role of dopamine receptors in the treatment of pituitary adenomas. Eur. J. Endocrinol. 156(Suppl 1), S37–S43 (2007)
D. Ferone, F. Gatto, M. Arvigo, E. Resmini, M. Boschetti, C. Teti, D. Esposito, F. Minuto, The clinical-molecular interface of somatostatin, dopamine and their receptors in pituitary pathophysiology. J. Mol. Endocrinol. 42, 361–370 (2009)
W.E. Farrell, A.J. Clark, M.F. Stewart, S.R. Crosby, A. White, Bromocriptine inhibits pro-opiomelanocortin mRNA and ACTH precursor secretion in small cell lung cancer cell lines. J Clin Invest. 90, 705–710 (1992)
D. Yin, S. Kondo, J. Takeuchi, T. Morimura, Induction of apoptosis in murine ACTH-secreting pituitary adenoma cells by bromocriptine. FEBS Lett. 339, 73–75 (1994)
R. Pivonello, D. Ferone, W.W. de Herder, J.M. Kros, M.L. De Caro, M. Arvigo, L. Annunziato, G. Lombardi, A. Colao, L.J. Hofland, S.W. Lamberts, Dopamine receptor expression and function in corticotroph pituitary tumors. J. Clin. Endocrinol. Metab. 89, 2452–2462 (2004)
M. Rocheville, D.C. Lange, U. Kumar, S.C. Patel, R.C. Patel, Y.C. Patel, Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science 288, 154–157 (2000)
D. Ferone, A. Saveanu, M.D. Culler, M. Arvigo, A. Rebora, F. Gatto, F. Minuto, P. Jaquet, Novel chimeric somatostatin analogs: facts and perspectives. Eur. J. Endocrinol. 156(Suppl 1), S23–S28 (2007)
A. Saveanu, G. Gunz, S. Guillen, H. Dufour, M.D. Culler, P. Jaquet, Somatostatin and dopamine-somatostatin multiple ligands directed towards somatostatin and dopamine receptors in pituitary adenomas. Neuroendocrinology 83, 258–263 (2006)
D. Ferone, M. Arvigo, C. Semino, P. Jaquet, A. Saveanu, J.E. Taylor, J.P. Moreau, M.D. Culler, M. Albertelli, F. Minuto, A. Barreca, Somatostatin and dopamine receptor expression in lung carcinoma cells and effects of chimeric somatostatin-dopamine molecules on cell proliferation. Am. J. Physiol. Endocrinol. Metab. 289, E1044–E1050 (2005)
C. De Bruin, R.A. Feelders, S.W. Lamberts, L.J. Hofland, Somatostatin and dopamine receptors as targets for medical treatment of Cushing’s syndrome. Rev. Endocr. Metab. Disord. 10, 91–102 (2009)
M. Barbot, N. Albiger, F. Ceccato, M. Zilio, A.C. Frigo, L. Denaro, F. Mantero, C. Scaroni, Combination therapy for Cushing’s disease: effectiveness of two schedules of treatment. Should we start with cabergoline or ketoconazole? Pituitary (2013). doi:10.1007/s11102-013-0475-3
E.S. Newlands, G.R.P. Blackledge, J.A. Slack, G.J.S. Rustin, D.B. Smith, N.S.A. Stuart, C.P. Quarterman, R. Hoffman, M.F.G. Stevens, M.H. Brampton, A.C. Gibson, Phase I trial of temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br. J. Cancer 65, 287–291 (1992)
S. Neidle, D.E. Thurston, Chemical approaches to the discovery and development of cancer therapies. Nat. Rev. Cancer 5, 285–296 (2005)
S. Lim, H. Shahinian, M.M. Maya, W. Yong, A.P. Heaney, Temozolomide: a novel treatment for pituitary carcinoma. Lancet Oncol. 7, 518–520 (2006)
F.J. Rodriguez, S.N. Thibodeau, R.B. Jenkins, K.V. Schowalter, B.L. Caron, B.P. O’neill, C.D. James, S. Passe, J. Slezak, C. Giannini, MGMT immunohistochemical expression and promoter methylation in human glioblastoma. Appl. Immunohistochem. Mol. Morphol. 16, 59–65 (2008)
S. Sharma, F. Salehi, B.W. Scheithauer, F. Rotondo, L.V. Syro, K. Kovacs, Role of MGMT in tumor development, progression, diagnosis, treatment and prognosis. Anticancer Res. 29, 3759–3768 (2009)
L.V. Syro, L.D. Ortiz, B.W. Scheithauer, R. Lloyd, Q. Lau, R. Gonzalez, H. Uribe, M. Cusimano, K. Kovacs, E. Horvath, Treatment of pituitary neoplasms with temozolomide: a review. Cancer 117, 454–462 (2011)
A. Takeshita, N. Inoshita, M. Taguchi, C. Okuda, N. Fukuhara, K. Oyama, K. Ohashi, T. Sano, Y. Takeuchi, S. Yamada, High incidence of low O(6)-methylguanine DNA methyltransferase expression in invasive macroadenomas of Cushing’s disease. Eur. J. Endocrinol. 161, 553–559 (2009)
F. Salehi, B.W. Scheithauer, K. Kovacs, E. Horvath, L.V. Syro, S. Sharma, B. Manoranjan, M. Cusimano, O-6-methylguanine-DNA methyltransferase (MGMT) immunohistochemical expression in pituitary corticotroph adenomas. Neurosurgery 70, 491–496 (2012)
G. Raverot, F. Castinetti, E. Jouanneau, I. Morange, D. Figarella-Branger, H. Dufour, J. Trouillas, T. Brue, Pituitary carcinomas and aggressive pituitary tumours: merits and pitfalls of temozolomide treatment. Clin. Endocrinol. 76, 769–775 (2012)
K. Kovacs, B.W. Scheithauer, M. Lombardero, R.E. McLendon, L.V. Syro, H. Uribe, L.D. Ortiz, L.C. Penagos, MGMT immunoexpression predicts responsiveness of pituitary tumors to temozolomide therapy. Acta Neuropathol. 115, 261–262 (2008)
S.S. Agarwala, J.M. Kirkwood, Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist 5, 144–151 (2000)
J. Sheehan, J. Rainey, J. Nguyen, R. Grimsdale, S. Han, Temozolomide-induced inhibition of pituitary adenoma cells. J. Neurosurg. 114, 354–358 (2011)
A. Spada, Growth factors and human pituitary adenomas. Eur. J. Endocrinol. 138, 255–257 (1998)
M. Niveiro, F.I. Aranda, G. Peiró, C. Alenda, A. Picó, Immunohistochemical analysis of tumor angiogenic factors in human pituitary adenomas. Hum. Pathol. 36, 1090–1095 (2005)
C.J. McCabe, K. Boelaert, L.A. Tannahill, A.P. Heaney, A.L. Stratford, J.S. Khaira, S. Hussain, M.C. Sheppard, J.A. Franklyn, N.J. Gittoes, Vascular endothelial growth factor, its receptor KDR/Flk-1, and pituitary tumor transforming gene in pituitary tumors. J. Clin. Endocrinol. Metab. 87, 4238–4244 (2002)
L.D. Ortiz, L.V. Syro, B.W. Scheithauer, A. Ersen, H. Uribe, C.E. Fadul, F. Rotondo, E. Horvath, K. Kovacs, Anti-VEGF therapy in pituitary carcinoma. Pituitary 15, 445–449 (2012)
S. Ezzat, L. Zheng, H.S. Smyth, S.L. Asa, The c-erbB-2/neu proto-oncogene in human pituitary tumours. Clin. Endocrinol. (Oxf). 46, 599–606 (1997)
G. Kontogeorgos, L. Stefaneanu, K. Kovacs, Z. Cheng, Localization of epidermal growth factor (EGF) and epidermal growth factor receptor (EGFr) in human pituitary adenomas and nontumorous pituitaries: an immunocytochemical study. Endocr. Pathol. 7, 63–70 (1996)
O. Cooper, G. Vlotides, H. Fukuoka, M.I. Greene, S. Melmed, Expression and function of ErbB receptors and ligands in the pituitary. Endocr. Relat. Cancer 18, R197–R211 (2011)
D. Lubke, W. Saeger, D.K. Ludecke, Proliferation markers and EGF in ACTH-secreting adenomas and carcinomas of the pituitary. Endocr. Pathol. 6, 45–55 (1995)
P.A. van Wijk, J.W. van Neck, A. Rijnberk, R.J. Croughs, J.A. Mol, Proliferation of the murine corticotropic tumour cell line AtT20 is affected by hypophysiotrophic hormones, growth factors and glucocorticoids. Mol. Cell. Endocrinol. 111, 13–19 (1995)
H. Fukuoka, O. Cooper, A. Ben-Shlomo, A. Mamelak, S.G. Ren, D. Bruyette, S. Melmed, EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J. Clin. Invest. 121, 4712–4721 (2011)
A. Gorshtein, H. Rubinfeld, E. Kendler, M. Theodoropoulou, V. Cerovac, G.K. Stalla, Z.R. Cohen, M. Hadani, I. Shimon, Mammalian target of rapamycin inhibitors rapamycin and RAD001 (everolimus) induce anti-proliferative effects in GHsecreting pituitary tumor cells in vitro. Endocr. Relat. Cancer 16, 1017–1027 (2009)
M.C. Zatelli, M. Minoia, C. Filieri, F. Tagliati, M. Buratto, M.R. Ambrosio, M. Lapparelli, M. Scanarini, E.C. degli Uberti, Effect of everolimus on cell viability in nonfunctioning pituitary adenomas. J. Clin. Endocrinol. Metab. 95, 968–976 (2010)
D. Dworakowska, E. Wlodek, C.A. Leontiou, S. Igreja, M. Cakir, M. Teng, N. Prodromou, M.I. Goth, S. Grozinsky-Glasberg, M. Gueorguiev, B. Kola, M. Korbonits, A.B. Grossman, Activation of RAF/MEK/ERK and PI3 K/AKT/mTOR pathways in pituitary adenomas and their effects on downstream effectors. Endocr. Relat. Cancer 16, 1329–1338 (2009)
E. Jouanneau, A. Wierinckx, F. Ducray, V. Favrel, F. Borson-Chazot, J. Honnorat, J. Trouillas, G. Raverot, New targeted therapies in pituitary carcinoma resistant to temozolomide. Pituitary 15, 37–43 (2012)
V. Cerovac, J. Monteserin-Garcia, H. Rubinfeld, M. Buchfelder, M. Losa, T. Florio, M. Paez-Pereda, G.K. Stalla, M. Theodoropoulou, The somatostatin analogue octreotide confers sensitivity to rapamycin treatment on pituitary tumor cells. Cancer Res. 70, 666–674 (2010)
K.E. O’Reilly, F. Rojo, Q.B. She, D. Solit, G.B. Mills, D. Smith, H. Lane, F. Hofmann, D.J. Hicklin, D.L. Ludwig, J. Baselga, N. Rosen, mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508 (2006)
P.M. Stewart, S. Petersenn, Rationale for treatment and therapeutic options in Cushing’s disease. Best Pract. Res. Clin. Endocrinol. Metab. 23(Suppl 1), S15–S22 (2009)
M. Boscaro, N. Sonino, A. Rampazzo, F. Mantero, Response of pituitary-adrenal axis to corticotrophin releasing hormone in patients with Cushing’s disease before and after ketoconazole treatment. Clin. Endocrinol. (Oxf). 27, 461–467 (1987)
P. Loli, M.E. Berselli, M. Tagliaferri, Use of ketoconazole in the treatment of Cushing’s syndrome. J. Clin. Endocrinol. Metab. 63, 1365–1371 (1986)
N. Sonino, The use of ketoconazole as an inhibitor of steroid production. N. Engl. J. Med. 317, 812–818 (1987)
R.A. Feelders, L.J. Hofland, W.W. de Herder, Medical treatment of Cushing’s syndrome: adrenal-blocking drugs and ketaconazole. Neuroendocrinology 92(Suppl 1), 111–115 (2010)
G.K. Stalla, J. Stalla, M. Huber, J.P. Loeffler, V. Hollt, K. von Werder, O.A. Muller, Ketoconazole inhibits corticotropic cell function in vitro. Endocrinology 122, 618–623 (1988)
A. Angeli, R. Frairia, Ketoconazole therapy in Cushing’s disease. Lancet 1, 821 (1985)
M. Boscaro, N. Sonino, A. Rampazzo, F. Mantero, Response of pituitary-adrenal axis to corticotrophin releasing hormone in patients with Cushing’s disease before and after ketoconazole treatment. Clin. Endocrinol. (Oxf). 27, 461–467 (1987)
P. Loli, M.E. Berselli, M. Tagliaferri, Use of ketoconazole in the treatment of Cushing’s syndrome. J. Clin. Endocrinol. Metab. 63, 1365–1371 (1986)
D. Engelhardt, M.M. Weber, Therapy of Cushing’s syndrome with steroid biosynthesis inhibitors. J. Steroid Biochem. Mol. Biol. 49, 261–267 (1994)
T.A. Miettinen, Cholesterol metabolism during ketoconazole treatment in man. J. Lipid Res. 29, 43–51 (1988)
M.I. Dushkin, N.K. Zenkov, E.B. Menshikova, E.N. Pivovarova, G. Lyubimov, N.N. Volsky, Ketoconazole inhibits oxidative modification of low density lipoprotein. Atherosclerosis 114, 9–18 (1995)
X. Bertagna, L. Guignat, L. Groussin, J. Bertherat, Cushing’s disease. Best Pract. Res. Clin. Endocrinol. Metab. 23, 607–623 (2009)
C.N. Dang, O. Trainer, Pharmacological management of Cushing’s syndrome: an update. Arq. Bras. Endocrinol. Metabol. 51, 1339–1348 (2007)
B.A. Gross, S.A. Mindea, A.J. Pick, J.P. Chandler, H.H. Batjer, Medical management of Cushing disease. Neurosurg. Focus 23, E10 (2007)
J.A. Verhelst, P.J. Trainer, T.A. Howlett, L. Perry, L.H. Rees, A.B. Grossman, J.A. Wass, G.M. Besser, Short and long-term responses to metyrapone in the medical management of 91 patients with Cushing’s syndrome. Clin. Endocrinol. (Oxf). 35, 169–178 (1991)
T. Mancini, T. Porcelli, A. Giustina, Treatment of Cushing disease: overview and recent findings. Ther. Clin. Risk Manag. 6, 505–516 (2010)
L.K. Nieman, Medical therapy of Cushing’s disease. Pituitary 5, 77–82 (2002)
M. Terzolo, A. Angeli, M. Fassnacht, F. Daffara, L. Tauchmanova, P.A. Conton, R. Rossetto, L. Buci, P. Sperone, E. Grossrubatscher, G. Reimondo, E. Bollito, M. Papotti, W. Saeger, S. Hahner, A.C. Koschker, E. Arvat, B. Ambrosi, P. Loli, G. Lombardi, M. Mannelli, P. Bruzzi, F. Mantero, B. Allolio, L. Dogliotti, A. Berruti, Adjuvant mitotane treatment for adrenocortical carcinoma. N. Engl. J. Med. 356, 2372–2380 (2007)
E. Gentilin, F. Tagliati, M. Terzolo, M. Zoli, M. Lapparelli, M. Minoia, M.R. Ambrosio, E.C. degli Uberti, M.C. Zatelli, Mitotane reduces human and mouse ACTH-secreting pituitary cells viability and function. J. Endocrinol. 218, 275–285 (2013)
M.C. Zatelli, E. Gentilin, F. Daffara, F. Tagliati, G. Reimondo, G. Carandina, M.R. Ambrosio, M. Terzolo, E.C. degli Uberti, Therapeutic concentrations of mitotane (o,p′-DDD) inhibit thyrotroph cell viability and TSH expression and secretion in a mouse cell line model. Endocrinology 151, 2453–2461 (2010)
J.P. Luton, J.A. Mahoudeau, P. Bouchard, P. Thieblot, M. Hautecouverture, D. Simon, M.H. Laudat, Y. Touitou, H. Bricaire, Treatment of Cushing’s disease by o,p′DDD. Survey of 62 cases. N. Engl. J. Med. 300, 459–464 (1979)
D.E. Schteingart, H.S. Tsao, C.I. Taylor, A. McKenzie, R. Victoria, B.A. Therrien, Sustained remission of Cushing’s disease with mitotane and pituitary irradiation. Ann. Intern. Med. 92, 613–619 (1980)
D. LaSala, Y. Shibanaka, A.Y. Jeng, Coexpression of CYP11B2 or CYP11B1 with adrenodoxin and adrenodoxin reductase for assessing the potency and selectivity of aldosterone synthase inhibitors. Anal. Biochem. 394, 56–61 (2009)
L. Amar, M. Azizi, J. Menard, S. Peyrard, C. Watson, P.F. Plouin, Aldosterone synthase inhibition with LCI699: a proof-of-concept study in patients with primary aldosteronism. Hypertension 56, 831–838 (2010)
D.A. Calhoun, W.B. White, H. Krum, W. Guo, G. Bermann, A. Trapani, M.P. Lefkowitz, J. Menard, Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: results of a randomized, double-blind, placebo- and active-controlled phase 2 trial. Circulation 124, 1945–1955 (2011)
N.A. Tritos, B.M. Biller, Advances in medical therapies for Cushing’s syndrome. Discov. Med. 13, 171–179 (2011)
X. Bertagna, R. Pivonello, M. Fleseriu, J. Zhang, P. Robinson, A. Taylor, C. Watson, M. Maldonado, A. Hamraian, M. Boscaro, Patients with Cushing’s disease achieve normal urinary cortisol with LCI699, a potent 11B-hydroxylase inhibitor: preliminary results from a multicenter, proof-of-concept study, in 15th International and 14th European Congress of Endocrinology (ICE/ECE 2012), Florence, Italy (abstract OC1.2) (2012)
P. Dewis, D.C. Anderson, D.E. Bu’lock, R. Earnshaw, W.F. Kelly, Experience with trilostane in the treatment of Cushing’s syndrome. Clin. Endocrinol. (Oxf). 18, 533–540 (1983)
P. Komanicky, R.F. Spark, J.C. Melby, Treatment of Cushing’s syndrome with trilostane (WIN 24,540), an inhibitor of adrenal steroid biosynthesis. J. Clin. Endocrinol. Metab. 47, 1042–1051 (1978)
S. Hahner, A. Stürmer, M. Fassnacht, R.W. Hartmann, K. Schewe, S. Cochran, M. Zink, A. Schirbel, B. Allolio, Etomidate unmasks intraadrenal regulation of steroidogenesis and proliferation in adrenal cortical cell lines. Horm. Metab. Res. 42, 528–534 (2010)
N. Mettauer, J. Brierley, A novel use of etomidate for intentional adrenal suppression to control severe hypercortisolemia in childhood. Pediatr. Crit. Care Med. 10, e37–e40 (2009)
P. Kamenicky, C. Droumaguet, S. Salenave, A. Blanchard, C. Jublanc, J.F. Gautier, S. Brailly-Tabard, S. Leboulleux, M. Schlumberger, E. Baudin, P. Chanson, J. Young, Mitotane, metyrapone, and ketoconazole combination therapy as an alternative to rescue adrenalectomy for severe ACTH-dependent Cushing’s syndrome. J. Clin. Endocrinol. Metab. 96, 2796–2804 (2011)
S. Johanssen, B. Allolio, Mifepristone (RU 486) in Cushing’s syndrome. Eur. J. Endocrinol. 157, 561–569 (2007)
M.C. Lebeau, E.E. Baulieu, Steroid antagonists and receptor-associated proteins. Hum. Reprod. 9, 437–444 (1994)
C.M. Bamberger, G.P. Chrousos, The glucocorticoid receptor and RU 486 in man. Ann. N. Y. Acad. Sci. 761, 296–310 (1995)
M.D. Heitzer, I.M. Wolf, E.R. Sanchez, S.F. Witchel, D.B. DeFranco, Glucocorticoid receptor physiology. Rev. Endocr. Metab. Disord. 8, 321–330 (2007)
H.F. Yang-Yen, J.C. Chambard, Y.L. Sun, T. Smeal, T.J. Schmidt, J. Drouin, M. Karin, Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein–protein interaction. Cell 62, 1205–1215 (1990)
C.M. Jewell, J.C. Webster, K.L. Burnstein, M. Sar, J.E. Bodwell, J.A. Cidlowski, Immunocytochemical analysis of hormone mediated nuclear translocation of wild type and mutant glucocorticoid receptors. J. Steroid Biochem. Mol. Biol. 55, 135–146 (1995)
B.W. Peeters, G.S. Ruigt, M. Craighead, P. Kitchener, Differential effects of the new glucocorticoid receptor antagonist ORG 34517 and RU486 (mifepristone) on glucocorticoid receptor nuclear translocation in the AtT20 cell line. Ann. N. Y. Acad. Sci. 1148, 536–541 (2008)
F. Spiga, D.M. Knight, S.K. Droste, B. Conway-Campbell, Y. Kershaw, C.P. MacSweeney, F.J. Thomson, M. Craighead, B.W. Peeters, S.L. Lightman, Differential effect of glucocorticoid receptor antagonists on glucocorticoid receptor nuclear translocation and DNA binding. J. Psychopharmacol. 25, 211–221 (2011)
R. Sitruk-Ware, I.M. Spitz, Pharmacological properties of mifepristone: toxicology and safety in animal and human studies. Contraception 68, 409–420 (2003)
X. Bertagna, C. Bertagna, M.H. Laudat, J.M. Husson, F. Girard, J.P. Luton, Pituitary-adrenal response to the antiglucocorticoid action of RU 486 in Cushing’s syndrome. J. Clin. Endocrinol. Metab. 63, 639–643 (1986)
J.W. Chu, D.F. Matthias, J. Belanoff, A. Schatzberg, A.R. Hoffman, D. Feldman, Successful long-term treatment of refractory Cushing’s disease with high-dose mifepristone (RU 486). J. Clin. Endocrinol. Metab. 86, 3568–3573 (2001)
A.M. Isidori, G.A. Kaltsas, C. Pozza, V. Frajese, J. Newell-Price, R.H. Reznek, P.J. Jenkins, J.P. Monson, A.B. Grossman, G.M. Besser, The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J. Clin. Endocrinol. Metab. 91, 371–377 (2006)
F. Castinetti, M. Fassnacht, S. Johanssen, M. Terzolo, P. Bouchard, P. Chanson, C. Do Cao, I. Morange, A. Pico, S. Ouzounian, J. Young, S. Hahner, T. Brue, B. Allolio, B. Conte-Devolx, Merits and pitfalls of mifepristone in Cushing’s syndrome. Eur. J. Endocrinol. 160, 1003–1010 (2009)
F. Castinetti, B. Conte-Devolx, T. Brue, Medical treatment of Cushing’s syndrome: glucocorticoid receptor antagonists and mifepristone. Neuroendocrinology 92(Suppl 1), 125–130 (2010)
M. Fleseriu, B.M. Biller, J.W. Findling, M.E. Molitch, D.E. Schteingart, C. Gross, Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing’s syndrome. J. Clin. Endocrinol. Metab. 97, 2039–2049 (2012)
P.M. Stewart, B.R. Walker, G. Holder, D. O’Halloran, C.H. Shackleton, 11 beta-Hydroxysteroid dehydrogenase activity in Cushing’s syndrome: explaining the mineralocorticoid excess state of the ectopic adrenocorticotropin syndrome. J. Clin. Endocrinol. Metab. 80, 3617–3620 (1995)
S.H. van Uum, J.W. Lenders, A.R. Hermus, Cortisol, 11beta-hydroxysteroid dehydrogenases, and hypertension. Semin. Vasc. Med. 4, 121–128 (2004)
J.D. Carmichael, M. Fleseriu, Mifepristone: is there a place in the treatment of Cushing’s disease? Endocrine 44, 20–32 (2013)
G.A. Kaltsas, P. Nomikos, G. Kontogeorgos, M. Buchfelder, A.B. Grossman, Clinical review: diagnosis and management of pituitary carcinomas. J. Clin. Endocrinol. Metab. 90, 3089–3099 (2005)
B.T. Ragel, W.T. Couldwell, Pituitary carcinoma: a review of the literature. Neurosurg. Focus 16, 1–9 (2004)
R.E. Landman, M. Horwith, R.E. Peterson, A.G. Khandji, S.L. Wardlaw, Long-term survival with ACTH-secreting carcinoma of the pituitary: a case report and review of the literature. J. Clin. Endocrinol. Metab. 87, 3084–3089 (2002)
G.A. Kaltsas, J.J. Mukherjee, The role of cytotoxic chemotherapy in the management of aggressive and malignant pituitary tumors. J. Clin. Endocrinol. Metab. 83, 4233–4238 (1998)
A.A. van der Klaauw, T. Kienitz, C.J. Strasburger, J.W. Smit, J.A. Romijn, Malignant pituitary corticotroph adenomas: report of two cases and a comprehensive review of the literature. Pituitary 12, 57–69 (2009)
K. Cukier, R. Tewari, F. Kurth, H.A. Schmid, C. Lai, D.J. Torpy, Significant response to pasireotide (SOM230) in the treatment of a patient with persistent, refractory Cushing’s disease. Clin. Endocrinol. (Oxf). 71, 305–307 (2009)
H. Bode, M. Seiz, A. Lammert, M.A. Brockmann, W. Back, H.P. Hammes, C. Thomé, SOM230 (pasireotide) and temozolomide achieve sustained control of tumour progression and ACTH secretion in pituitary carcinoma with widespread metastases. Exp. Clin. Endocrinol. Diabetes 118, 760–763 (2010)
G. Raverot, N. Sturm, F. de Fraipont, M. Muller, S. Salenave, P. Caron, O. Chabre, P. Chanson, C. Cortet-Rudelli, R. Assaker, H. Dufour, S. Gaillard, P. François, E. Jouanneau, J.G. Passagia, M. Bernier, A. Cornélius, D. Figarella-Branger, J. Trouillas, F. Borson-Chazot, T. Brue, Temozolomide treatment in aggressive pituitary tumors and pituitary carcinomas: a French multicenter experience. J. Clin. Endocrinol. Metab. 95, 4592–4599 (2010)
C.E. Fadul, A.L. Kominsky, L.P. Meyer, L.S. Kingman, W.B. Kinlaw, C.H. Rhodes, C.J. Eskey, N.E. Simmons, Long-term response of pituitary carcinoma to temozolamide. Report of two cases. J. Neurosurg. 105, 621–626 (2006)
C. Hagen, H.D. Schroder, S. Hansen, C. Hagen, M. Andersen, Temozolamide treatment of a pituitary carcinoma and two pituitary macroadenomas resistant to conventional therapy. Eur. J. Endocrinol. 161, 631–637 (2009)
A.I. McCormack, K.L. McDonald, A.J. Gill, S.J. Clark, M.G. Burt, K.A. Campbell, W.J. Braund, N.S. Little, R.J. Cook, A.B. Grossman, B.G. Robinson, R.J. Clifton-Bligh, Low O6-methylguanine-DNA methyltransferase (MGMT) expression and response to temozolomide in aggressive pituitary tumours. Clin. Endocrinol. 71, 226–233 (2009)
M. Thearle, P. Freda, J. Bruce, S. Isaacson, Y. Lee, R. Fine, Temozolomide (Temodar®) and capecitabine (Xeloda®) treatment of an aggressive corticotroph pituitary tumor. Pituitary 14, 418–424 (2011)
M. Basina, H. Liu, A.R. Hoffman, D. Feldman, Successful long-term treatment of Cushing disease with mifepristone (RU486). Endocr. Pract. 18, e114–e120 (2012)
L.F. Chan, M. Vaidya, B. Westphal, J. Allgrove, L. Martin, F. Afshar, P.C. Hindmarsh, M.O. Savage, A.B. Grossman, H.L. Storr, Use of intravenous etomidate to control acute psychosis induced by the hypercortisolaemia in severe paediatric Cushing’s disease. Horm. Res. Paediatr. 75, 441–446 (2011)
Acknowledgments
This study was supported by grants from the Italian Ministry of Education, Research and University (FIRB RBAP11884M; RBAP1153LS; PRIN 2008LFK7J5_004), Fondazione Cassa di Risparmio di Ferrara, and Associazione Italiana per la Ricerca sul Cancro (AIRC) in collaboration with Laboratorio in rete del Tecnopolo “Tecnologie delle terapie avanzate” (LTTA) of the University of Ferrara; CARIGE Foundation, Genova.
Disclosure
D. Ferone, A. Colao, R. Pivonello report serving as an ad hoc consultant to Novartis, and receiving research grant support. The remaining authors do not have any relationships to disclose.
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was conducted on behalf of ABC (Altogether to Beat Cushing’s syndrome) Group.
The members of the ABC Group are listed in Appendix.
D. Ferone, C. Pivonello, G. Vitale and M. C. Zatelli have contributed equally to this work.
Appendix
Appendix
ABC (Altogether to Beat Cushing’s syndrome) 2012 Group: N. Albiger, A. Ambrogio, G. Arnaldi, E. Arvat, R. Baldelli, R. Berardelli, M. Boscaro, S. Cannavò, F. Cavagnini, A. Colao, S.M. Corsello, A. Cozzolino, A. De Bartolomeis, M. De Leo, G. Di Minno, C. Di Somma, K. Esposito, G. Fabbrocini, D. Ferone, C. Foresta, M. Galderisi, C. Giordano, D. Giugliano, A. Giustina, F. Grimaldi, A.M. Isidori, E. Jannini, F. Lombardo, L. Manetti, M. Mannelli, F. Mantero, G. Marone, G. Mazziotti, S. Moretti, E. Nazzari, R.M. Paragliola, R. Pasquali, S. Pecorelli, F. Pecori Giraldi, C. Pivonello, R. Pivonello, G. Reimondo, C. Scaroni, A. Scillitani, C. Simeoli, A. Stigliano, V. Toscano, L. Trementino, G. Vitale, M.C. Zatelli.
Rights and permissions
About this article
Cite this article
Ferone, D., Pivonello, C., Vitale, G. et al. Molecular basis of pharmacological therapy in Cushing’s disease. Endocrine 46, 181–198 (2014). https://doi.org/10.1007/s12020-013-0098-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-013-0098-5