
Abstract
The aim of this study was to identify mutations in three different genes, the arginine-vasopressin-neurophysin II (AVP-NPII) gene, the arginine-vasopressin receptor 2 (AVPR2) gene, and the vasopressin-sensitive water channel aquaporin-2 (AQP2) gene in Turkish patients affected by central diabetes insipidus or nephrogenic diabetes insipidus. This study included 15 patients from unrelated families. Prospective clinical data were collected for all patients including the patients underwent a water deprivation–desmopressin test. The coding regions of the AVPR2, AQP2, and AVP-NPII genes were amplified by polymerase chain reaction and submitted to direct sequence analysis. Of the 15 patients with diabetes insipidus referred to Gulhane Military Medical Academy, Department of Endocrinology and Metabolism, eight patients have AVPR2 mutations, five patients have AQP2 mutations and two patients have AVP-NPII mutations. Of the patients, which have AVPR2 mutations, one is compound heterozygous for AVPR2 gene. Seven of these mutations are novel. Comparison of the clinical outcomes of these mutations may facilitate in understanding the functions of AVP-NPII, AQP2, and AVPR2 genes in future studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes insipidus (DI) is a metabolic disease caused by inadequate secretion of antidiuretic hormone (ADH)—also known as arginine vasopressin (AVP)—from the hypothalamus or inadequate response of the kidney to ADH [1]. Disease is characterized by polyuria, hypo-osmolar urine, and hypernatremia. DI has different types, each with a different cause. Central diabetes insipidus (CDI) results from insufficient production of vasopressin while nephrogenic diabetes insipidus (NDI) results from inability of the kidney to respond to vasopressin [2]. CDI can be caused by damage to the hypothalamus or pituitary gland as a result of hypothalamic tumors, surgery, trauma, congenital malformations or autoimmune, infiltrative, metastatic, inflammatory, infectious processes or mutations in the AVP-NPII gene [3]. AVP-NPII gene is located on chromosome 20p13, consists of three exons [4]. Prepro-vasopressin has 164 amino acids and is encoded by 2.5 kb AVP gene. NDI may occur as an inherited disorder or may be acquired by iatrogenic (lithium or demeclocycline), hypokalemia, hypercalcemia, various types of renal disease, and sickle cell anemia [1, 5]. The genetic grounds are X-linked and caused by mutations in AVPR2 gene or autosomal and caused by the mutations in the AQP2 water channel gene which have a much lower prevalence [6, 7]. The AVPR2 gene, belongs to a large superfamily of G-protein coupled receptors (GPCRs), have 371 amino acids with seven transmembrane, four extracellular and four cytoplasmic domains. AVPR2 is located on chromosome Xq28 and consists of three exons. About 90 % of NDI patients were reported as male with X-linked recessive NDI and have mutations in the AVPR2 gene, whereas 10 % of the NDI patients were reported as having autosomal mode of inheritance and is mainly due to mutations in the AQP2 gene [8]. However, it is reported that in some females who had classic features of the disease by the result of AVPR2 mutations, had too many mutant cells because of skewed X inactivation, the most severe had the greatest amount of skewing. The cellular mosaicism resulting from X inactivation in female patients is mostly protecting carriers of X-linked mutations from the severe clinical manifestations seen in males [9]. AQP2 gene which codes the vasopressin-regulated water channel of the apical membrane of the kidney collecting tubule has 271 amino acids and is located on chromosome 12q13 and has four exons and three introns [4].
AVP is a hormone produced in the supraoptic and paraventricular nuclei regions of hypothalamus and stored in the posterior pituitary. In healthy subjects, after an increase of sodium chloride in the extracellular concentration or a decrease in the blood pleasure, AVP is released into the bloodstream and binds to V2 GPCRs within the distal convoluted tubules. This binding increases the volume of cyclic AMP (cAMP) and activates protein kinase A (PKA). PKA acts to promote the water channels aquoporin 2 (AQP2) and phosphorylation of AQP2 allowing the reabsorption of free water into the collecting duct cells. In the absence of AVP, AQP2 is localized in vesicles in the subapical region of the cell. AQP2 relocate into the apical membrane by binding of AVP to its V2 receptor and supply high water permeability to apical membrane [3, 4, 10–12].
Here we report 12 mutations on AVPR2, AVP-NPII, and AQP2 genes, which belong to 15 Turkish patients. Seven of these mutations are novel. Two of the novel mutations, which were detected in AVPR2 gene, constitute a compound heterozygosity in a patient.
Materials and methods
Patients
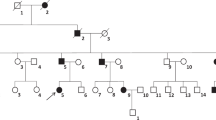
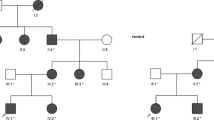
Fifteen patients with DI were included in this study among patients with polyuria or/and polydipsia who had referred to Gulhane Military Medical Academy, Department of Endocrinology and Metabolism. Nine of the patients have been diagnosed as NDI, while 6 of the patients have been diagnosed as CDI according to water deprivation test. The patients enrolled in this study were unrelated to each other and were all affected males. Patients had taken neither lithium nor demeclocycline. Patients with a history of psychiatric and other diseases including psychogenic polydipsia, cardiac diseases, neurologic diseases, renal diseases, diabetes mellitus, or any other metabolic or endocrinologic disorders, smoking habits, and the takers of medications that are used other than in DI treatment were excluded from the study. Other exclusion criteria were the presence of any kind of pituitary tumor or any other hypothalamic-pituitary disorders. Patients with any cranial surgery were also excluded. The clinical features of the patients are given in Table 1. The patients were referred to the Gulhane Military Medical Academy, Department of Endocrinology and Metabolism because of polyuria and polydipsia between the years of 2005 and 2011. Because this hospital is the biggest, tertiary military medical center in Turkey, almost all young male patients who have signs, symptoms of DI were referred to our center for evaluation regarding eligibility for military service. All subjects have undergone routine laboratory tests, such as glycemia, natrium, potassium, BUN, creatinine, calcium, urine analysis, and complete blood count. Plasma osmolality, a mean of 24-h urine osmolality, and 24-h urine volume have also been measured. All patients have undergone a water deprivation–desmopressin test. Diagnosis of DI was made according to water deprivation test. At the beginning of the test, all patients were weighted and serum concentrations of natrium were measured. When two sequential, hourly urine osmolality measurements has differed <10 % and weight loss <2 %, samples have been collected again for natrium plasma osmolality and vasopressin. Patients have been given parenteral 2-µg desmopressin, and urine output, and osmolality have been measured for at least two more hours. If the patients have lost 3 % or more of body weight or natrium got above the upper normal range at any time, test has been stopped. Patients with primary polydipsia were also differentiated from DI with maximum urine concentration often above 700 mOsm/kg, and without further response to desmopressin. These patients had also lower natrium and uric acid values. CDI was diagnosed at the end of the dehydration if there was minimal urine concentration and after vasopressin at least 50 % increase in urine osmolality. NDI was diagnosed if there was again minimal increase in urine concentration despite dehydration, and no further increase after vasopressin. Diagnoses of patients have also been cross checked with the treatment. The study protocol was in adherence to the tenets of the declaration of Helsinki. Informed consent was obtained from all patients after giving the explanation on the nature and possible consequences of the study.
Genomic DNA extraction, amplification, and sequencing
Genomic DNA was purified from peripheral blood using the phenol–chloroform extraction method. After the extraction, genomic DNA was stored at −20 °C. From the genomic DNA the entire AVPR2, AVP-NPII, and AQP2 genes were amplified using the polymerase chain reaction (PCR). The primer sets, which were used to amplify the entire coding sequence of three genes, are given in detail in Table 2. After PCR amplification, PCR products were electrophoresed on 1.5 % agarose gel (Sigma-Aldrich, Steinheim, Germany) and visualized under ultraviolet light after ethidium bromide staining. PCR products of expected sizes were purified with enzymatic purification method before sequencing. For each purification reaction 4 μl PCR product, 0.4 μl Exonuclease I and 0.4 μl Shrimp alkaline phosphatase were used. Purification carried out at 37 °C for 20 min, 80 °C for 20 min, and 94 °C for 2 min. PCR fragments sequenced directly on an automated DNA sequencer (310 Genetic Analyzer; ABI Prism) using the BigDye kit (Applied Biosystems, USA). The PCR primers were also used as sequencing primers. Sequencing reaction was carried out at 96 °C for 10 s, 50 °C for 5 s, and 60 °C for 4 min at 25 cycles. For optimal results sequencing products were purified using ethanol/sodium acetate precipitation method.
Results
This study included 15 patients with DI according to the results of the water deprivation test. Six patients diagnosed as CDI, whereas nine patients diagnosed NDI according to vasopressin test. In 15 patients with DI, we have detected 12 different mutations. We have collected male patients who had referred to Gulhane Military Medical Academy, Department of Endocrinology and Metabolism in the course of military medical examination and screened them to identify these 12 different mutations. Families of some male patients were also screened and we identified mutations also in their families. Seven of the mutations were detected in the AVPR2 gene. The six of these mutations are missense mutations (G107W, R106C, T273M, R68W, V162A, G12E) and one of the mutations is in-frame deletion (271del9). 271del9 and G107W mutations were identified in the same NDI patient. Two missense mutations (L137P, A147T) and one nonsense mutation (R85X) were detected in the AQP2 gene and two missense mutations were detected in the AVP-NPII gene (G88V, C104F). Five of the AVPR2 mutations (271del9, G107W, T273M, R68W, V162A), one of the AQP2 mutations (L137P) and one of the AVP-NPII mutations (G88V) are novel mutations, whereas two of the AVPR2 mutations (R106C [13] and G12E [14]), two of the AQP2 mutations (A147T [10] and R85X [15]) and one of the AVP-NPII mutations (C104F [16]) had already been reported in the literature. Except the patients 13 (R85X), 14 (G88V), and 15 (C104F) all the other patients were found to be homozygous for the mutations. All the patients and mutations are shown in Table 3 in detail. Patient 1, whose clinic manifestation is more serious with polydipsia, severe fatigue, hyposthenuria, and more than 10 l voiding, harbors the G107W mutation and also 271del9 mutation that is a 9-bp in-frame deletion. Clinic of patient 9 is the most serious one with severe fatigue, polydipsia, prominent hyposthenuria, and more than 20 l voiding.
Discussion
In this study, mutation profiles of AVPR2, AVP-NPII, and AQP2 genes of 15 Turkish patients with NDI or CDI referred to the Gulhane Military Medical Academy, Department of Endocrinology and Metabolism have been identified. Mutations were evaluated in accordance with the manifestations of the patients. Molecular analysis of 15 DI patients revealed seven novel and five previously reported mutations.
AVPR2 gene mutations have been classified in three types according to the function and subcellular localization of the mutant protein. Type 1 mutant receptors reach the cell surface but display impaired ligand binding and are unable to induce normal cAMP production. Type 2 mutant receptors have defective intracellular transport as a result they do not reach the cell surface and accumulate in a pre-Golgi compartment. Type 3 mutant receptors are transcribed inefficiently; consequently, lead to rapidly degraded unstable mRNA [4, 12]. To date, 200 disease-causing mutations, which were reported as 50 % missense mutations, 27 % small deletions/insertions, 9 % nonsense mutations, and 8 % large or complex deletions [17], have been published in NDI families. The first patient in our study harbors two novel mutations, G107W mutation and 271del9 mutation, which is a 9-bp in-frame deletion. 271del9 frameshift mutation results from the deletion of codons 67-68-69-70 (Arg-Arg-Gly-His). These amino acids are located in the middle of the first and the second transmembrane domains or in the first intracellular loop. Missing amino acids may result in impaired ligand binding, misfolding of the receptor, inefficient respond and functional defects. G107W mutation is located in the first extracellular loop. G107, which is highly conserved, is important for folding and trafficking of AVPR2. Mutations in this location may affect the ligand binding capacity, specificity and cell surface expression [17]. These two mutations together could lead to this severe clinical picture. Patient’s mother and sister were heterozygous for this mutation and his father was healthy. The second patient has R106C mutation in V2 receptor gene that was reported previously by Bichet et al. [13]. The mutation leads to the substitution of arginine by cysteine is located in the first extracellular loop and might cause a change in the region of disulfide bridge joining extracellular loops I and II. This alteration may affect the binding affinity of the V2 receptor to AVP and decrease the stimulation of adenylate cyclase [18]. It was reported that R106C mutation might present urological complications [19]. Patient 3 has T273M mutation that is placed in a transmembrane domain of V2 receptor gene. Because of the location of the mutation, it may disrupt proper assembly of the V2 receptor. R68W mutation, which was detected for patient 4, is located in the first intracellular loop. V162A mutation, which leads to the substitution of apolar valine by apolar alanine were found in patient 5. This mutation is placed in the fourth transmembrane domain and may affect the binding affinity of receptor and AVP. Three patients have G12E mutation, which has previously reported [14], is located in the N-terminal extracellular tail and may influence ligand binding.
So far, several AQP2 mutations have been reported and functional analysis of these mutations showed disruption in protein synthesis [20]. It was reported that endoplasmic reticulum retardation of the misfolded protein leads to degradation by cytosolic proteasomes [21]. In this study, three missense mutations in the AQP2 gene of five patients, coding for L137P substituted (patient 9), A147T substituted (patients 10 and 11) and R85X substituted (patients 12 and 13) AQP2 proteins, were reported. L137P mutation was not reported before. Both L137P and A147T mutations are located in a transmembrane domain. Misfolding may cause the exposure of hydrophobic regions on the surface of the molecule since the mutations are in a transmembrane domain [10]. We think that, mutation L137P is especially important for the severity of clinic. Because this patient’s clinic was the worst we had examined. Mulders et al. [10] reported that A147T mutant protein less stable than wild-type AQP2 and functional as a water channel. They predicted that major cause underlying autosomal recessive NDI resulting in A147T mutation is the misrouting of AQP2 mutant proteins. Vargas-Poussou et al. [15] previously reported the R85X mutation in exon 2 of AQP2 gene in a Turkish family. This mutation is located in the second intracellular loop of AQP2 and conserved in the major intrinsic protein family.
Two hypotheses for the role of AVP-NPII gene for DI pathogenesis have been reported so far, and these hypotheses have been supported by clinical and experimental observations [22, 23]. First hypothesis is retaining of mutant precursor hormones in endoplasmic reticulum protein quality machinery and consequently, cytotoxic accumulation and aggregation of mutant precursor protein in the neurons [24, 25]. According to second hypothesis; cytosolic proteolytic system degrades the mutant AVP prohormone, because of the impaired function of mutant NPII molecules, which function in protecting AVP from proteolytic degradation and assisting AVP in its axonal transport [22]. Numerous mutations in AVP have been reported. These mutations might be in the neurophysin II-coding region which may be located in the AVP-binding site or which may lead to misfolding by disrupting the intrachain disulfide bonds within neurophysin II, might be in the AVP-coding region itself which may disrupt AVP-binding to a “pocket” formed by several residues in the amino-terminal domain of neurophysin or might affect the signal peptide and disrupt signal protein processing [23]. G88V mutation is a novel mutation and detected in AQP2 gene of the patient 14. This mutation might be responsible for AVP deficiency in this patient. According to Ito et al. [26] and computer-assisted prediction of the secondary structure of NPII molecule, this substitution could influence AVP-binding or may lead to a conformational change of the NPII molecule since Gly88 is a conserved amino acid. C104F missense mutation was reported before [16]. It is predicted that the ability to create the disulfide bridge between Cys104 and Cys92 is disrupted because of the C104F missense mutation [16].
In conclusion, we have presented the results of mutation profiles and have investigated their possible involvement with the disease phenotype and severity in a group of Turkish subjects. We report five novel and two recurrent mutations of the AVPR2 gene, a novel and two recurrent mutations of the AQP2 gene, and a novel and a recurrent mutation of the AVP-NPII gene. For the hereditary diseases, molecular studies are important for diagnosis, prognosis and pharmacologic studies. Juul [27] suggested that potential clinical and pharmacologic implications of a better phylogenetic understanding of the biological systems are important for body fluid homeostasis relates to especially deficiencies of any part of the AVP, AVPR2, and AQP2 axes causing CDI or NDI. Concerning the hereditary metabolic diseases, establishment of the relationship between the genotype and the phenotype is extremely important in understanding the effects of the location and the type of mutation on the type of activity failure. So far, molecular genetic studies based on DNA sequencing, comprise a wide mutation database in terms of the mutation profiles of the genes. Our study will contribute to these mutations’ database and with the future functional analyses studies these mutations data will shed light on the approaches for treatment of the disease and the literature.
References
A.N. Makaryus, S.I. McFarlane, Diabetes insipidus: diagnosis and treatment of a complex disease. Clevel. Clin. J. Med. 73(1), 65–71 (2006)
J.A. Majzoub, A. Srivatsa, Diabetes insipidus: clinical and basic aspects. Pediatr. Endocrinol. Rev. 4(Suppl 1), 60–65 (2006)
S.A. Ranadive, B. Ersoy, H. Favre, C.C. Cheung, S.M. Rosenthal, W.L. Miller, C. Vaisse, Identification, characterization and rescue of a novel vasopressin-2 receptor mutation causing nephrogenic diabetes insipidus. Clin. Endocrinol. (Oxf) 71(3), 388–393 (2009)
T.M. Fujiwara, D.G. Bichet, Molecular biology of hereditary diabetes insipidus. J. Am. Soc. Nephrol. 16, 2836–2846 (2005)
P.W. Oakley, I.M. Whyte, G.L. Carter, Lithium toxicity: an iatrogenic problem in susceptible individuals. Aust. N. Z. J. Psychiatry 35(6), 833–840 (2001)
N.V.A.M. Knoers, P.M.T. Deen, Molecular and cellular defects in nephrogenic diabetes insipidus. Pediatr. Nephrol. 16, 1146–1152 (2001)
H. Mizuno, Y. Sugiyama, Y. Ohro, H. Imamine, M. Kobayashi, S. Sasaki, S. Uchida, H. Togari, Clinical characteristics of eight patients with congenital nephrogenic diabetes insipidus. Endocrine 24(1), 55–59 (2004)
D.G. Bichet, V2R mutations and nephrogenic diabetes insipidus. Prog. Mol. Biol. Transl. Sci. 89, 15–29 (2009)
B.R. Migeon, X inactivation, female mosaicism, and sex differences in renal diseases. J. Am. Soc. Nephrol. 19(11), 2052–2059 (2008)
S.M. Mulders, N.V. Knoers, A.F. Van Lieburg, L.A. Monnens, E. Leumann, E. Wühl, E. Schober, J.P. Rijss, C.H. Van Os, P.M. Deen, New mutations in the AQP2 gene in nephrogenic diabetes insipidus resulting in functional but misrouted water channels. J. Am. Soc. Nephrol. 8(2), 242–248 (1997)
P. Saborio, G.A. Tipton, J.C. Chan, Diabetes insipidus. Pediatr. Rev. 21(4), 122–129 (2000). quiz 129
E. Spanakis, E. Milord, C. Gragnoli, AVPR2 variants and mutations in nephrogenic diabetes insipidus: review and missense mutation significance. J. Cell Physiol. 217(3), 605–617 (2008)
D.G. Bichet, M. Birnbaumer, M. Lonergan, M.F. Arthus, W. Rosenthal, P. Goodyer, H. Nivet, S. Benoit, P. Giampietro, S. Simonetti et al., Nature and recurrence of AVPR2 mutations in X-linked nephrogenic diabetes insipidus. Am. J. Hum. Genet. 55(2), 278–286 (1994)
D. Wenkert, J.J. Merendino Jr, A. Shenker, N. Thambi, G.L. Robertson, A.M. Moses, A.M. Spiegel, Novel mutations in the V2 vasopressin receptor gene of patients with X-linked nephrogenic diabetes insipidus. Hum. Mol. Genet. 3(8), 1429–1430 (1994)
R. Vargas-Poussou, L. Forestier, M.D. Dautzenberg, P. Niaudet, M. Déchaux, C. Antignac, Mutations in the vasopressin V2 receptor and aquaporin-2 genes in 12 families with congenital nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 8(12), 1855–1862 (1997)
J. Santiprabhob, J. Browning, D. Repaske, A missense mutation encoding Cys73Phe in neurophysin II is associated with autosomal dominant neurohypophyseal diabetes insipidus. Mol. Genet. Metab. 77(1–2), 112–118 (2002)
I. Böselt, D. Tramma, S. Kalamitsou, T. Niemeyer, P. Nykänen, K.J. Gräf, H. Krude, K.S. Marenzi, S. Di Candia, T. Schöneberg, A. Schulz, Functional characterization of novel loss-of-function mutations in the vasopressin type 2 receptor gene causing nephrogenic diabetes insipidus. Nephrol. Dial. Transplant. 27(4), 1521–1528 (2011)
C.H. Chen, W.Y. Chen, H.L. Liu, T.T. Liu, A.P. Tsou, C.Y. Lin, T. Chao, Y. Qi, K.J. Hsiao, Identification of mutations in the arginine vasopressin receptor 2 gene causing nephrogenic diabetes insipidus in Chinese patients. J. Hum. Genet. 47(2), 66–73 (2002)
M. Fysekidis, J.J. Boffa, L. Baud, J.P. Haymann, The R106C mutation of the V2 vasopressor receptor gene (AVPR2) causing X linked congenital nephrogenic diabetes insipidus is responsive to short term desmopressin challenge. Endocr. Abstr. 20, P291 (2009)
C. Guyon, Y. Lussier, P. Bissonnette, A. Leduc-Nadeau, M. Lonergan, M.F. Arthus, R.B. Perez, A. Tiulpakov, J.Y. Lapointe, D.G. Bichet, Characterization of D150E and G196D aquaporin-2 mutations responsible for nephrogenic diabetes insipidus: importance of a mild phenotype. Am. J. Physiol. Renal. Physiol. 297(2), F489–F498 (2009)
E.J. Kamsteeg, P.M. Deen, C.H. van Os, Defective processing and trafficking of water channels in nephrogenic diabetes insipidus. Exp. Nephrol. 8, 326–331 (2000)
J. Davies, D. Murphy, Autophagy in hypothalamic neurones of rats expressing a familial neurohypophysial diabetes insipidus transgene. J. Neuroendocrinol. 14, 629–637 (2002)
C.M. Hedrich, A.Z. Buczynska, A. Gawlil, S. Russ, G. Hahn, K. Koehler, E.M. Tendera, A. Huebner, Autosomal neurohypophyseal diabetes insipidus in two families. Horm. Res. 71, 111–119 (2009)
S. Rittig, C. Siggaard, M. Ozata, I. Yetkin, N. Gregersen, E.B. Pedersen, G.L. Robertson, Autosomal dominant neurohypophyseal diabetes insipidus due to substitution of histidine for tyrosine-2 in the vasopressin moiety of the hormone precursor. J. Clin. Endocrinol. Metab. 87, 3351–3355 (2002)
J.H. Christensen, C. Siggaard, S. Rittig, Autosomal dominant familial neurohypophyseal diabetes insipidus. APMIS 109, 92–95 (2003)
M. Ito, Y. Mori, Y. Oiso, H. Saito, A single base substitution in the coding region for neurophysin II associated with familial central diabetes insipidus. J Clin. Invest. 87(2), 725–728 (1991)
K.V. Juul, The evolutionary origin of the vasopressin/V2-type receptor/aquaporin axis and the urine-concentrating mechanism. Endocrine. (2012). doi:10.1007/s12020-012-9634-y
Conflict of interest
The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Duzenli, D., Saglar, E., Deniz, F. et al. Mutations in the AVPR2, AVP-NPII, and AQP2 genes in Turkish patients with diabetes insipidus. Endocrine 42, 664–669 (2012). https://doi.org/10.1007/s12020-012-9704-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-012-9704-1