
Abstract
HIV-infected patients on highly active antiretroviral therapy (HAART) have increased prevalence of a number of chronic metabolic disorders of multifactorial but unclear etiology. These include disorders of lipid metabolism with or without lipodystrophy, insulin resistance, and an increased prevalence of impaired glucose tolerance, diabetes mellitus, and cardiometabolic syndrome. While much attention has been focused on the lipid and cardiovascular disorders, few investigations have attempted to characterize the prevalence, incidence, etiology, mechanisms, and management of glycemic disorders in HIV patients. In this review, we have focused specifically on a comprehensive assessment of dysglycemia in the context of HIV infection and HAART.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the introduction of highly active antiretroviral therapy (HAART) in the mid-1990s, the mortality and morbidity of HIV/AIDS has decreased substantially [1]. In fact, HIV/AIDS has become chronic illness for many patients on HAART. With the increased survival of these patients, there have emerged a number of unexpected consequences of chronic illness, especially in the form of metabolic disease. The chief manifestations of the metabolic defects are related to lipid metabolism, with or without an associated fat “redistribution” or lipodystrophy. Another manifestation—perhaps a consequence of the high prevalence of insulin resistance (IR) resulting from the defects in fat metabolism—is an increased frequency of glucose intolerance or frank diabetes mellitus (DM). The mechanisms underlying dysglycemia, and the relationship of the glucose metabolic disorders to those of lipid metabolism and fat redistribution, and to ethnicity, diet and specific HAART agents, are multifaceted, complex and, to some degree, enigmatic. This review will describe the current state of the epidemiology, mechanisms, screening, diagnosis, and treatment of dysglycemia in HIV patients.
Epidemiology
With the marked increase worldwide in the incidence and prevalence of different forms of diabetes, there has been great interest in determining the frequency and characteristics of diabetes, impaired glucose tolerance (IGT), metabolic syndrome, and cardiac events in HIV-infected patients. In addition to the effects of HAART, other etiologic factors are being investigated. These studies are difficult to perform, and it is especially difficult to separate the effects of HAART from those of HIV infection per se or of immune responses to the infection and treatment [1].
The epidemiology of DM has been studied to a limited extent in antiretroviral-naïve HIV patients. In a study which enrolled 419 racially diverse antiretroviral-naïve patients, the baseline prevalence of DM was 2.6% [2]. A similar group of patients had a higher baseline prevalence of DM when co-infected with hepatitis C virus (HIV/HCV co-infected group 5.9 vs. 3.3% HIV alone) [3]. In the Women’s Interagency HIV Study, HIV-infected women reporting no recent HAART showed a DM incidence rate of 1.53/100 person-years; those reporting HAART containing a protease inhibitor (PI) had a DM incidence of 2.50/100 person-years, and those reporting non-PI-containing HAART had a DM incidence of 2.89/100 person-years. The incidence of DM among HIV-uninfected women was 1.96/100 person-years [4].
Most epidemiological data regarding DM and IGT come from studies of patients on HAART [1]. An early study of HIV patients with lipodystrophy reported a 35% prevalence of IGT and a 7% prevalence of DM, compared to 5 and 0.5%, respectively, for matched non-HIV controls [5]. The Multicenter AIDS Cohort Study showed a 14% incidence of DM in HIV-infected men exposed to HAART, which was four times higher than among HIV-seronegative controls [6]. Other groups have reported 13.2% incidence of IR in HIV-infected children [7] and 13% incidence of IR after 1 year of HAART [8]. The prospective D:A:D study, composed primarily of patients on HAART, showed that the incidence of DM increased with cumulative exposure to HAART (particularly stavudine, a thymidine analog) with a relative risk of 1.11 after adjustment for other potential risk factors [9].
Several groups have studied the prevalence of metabolic syndrome (for which one possible criterion is impaired fasting glucose or IGT) in HIV-infected individuals. One group showed that the prevalence of metabolic syndrome was higher among HIV-infected patients on antiretroviral therapy (ART) than among non-HIV-infected healthy controls (15.8 vs. 3.2%) [10]. A Spanish study estimated the prevalence of metabolic syndrome to be 17% in HIV-infected patients receiving HAART [11]. In the Women’s Interagency HIV Study, metabolic syndrome was more prevalent in HIV-seropositive than HIV-seronegative women (33 vs. 22%, respectively) [12]. Samaras et al. who also studied HIV-infected individuals on HAART, found the prevalence of metabolic syndrome to be 14–18% (using International Diabetes Federation or the National Cholesterol Education Program’s Adult Treatment Protocol III criteria, respectively) [13]. However, Mondy et al. who studied a US population, found that the prevalence of metabolic syndrome was similar between HIV-infected patients (most of them on HAART) and a matched HIV-negative group (25.5 vs. 26.5%, respectively), and that traditional risk factors (anthropomorphic measurements, dyslipidemia, age, and glucose control) played a more significant role in the development of metabolic syndrome that HIV treatment associated factors [14]. Notably, a case–control study suggested that another component of the metabolic syndrome—hypertension—may be directly linked to increased IR in persons with HIV [15].
Data from relatively small studies and heterogeneous HIV populations suggest that metabolic (including glycemic) abnormalities among HIV patients may be affected by ethnicity [16–19]. HbA1c levels in HIV patients are associated with older age and ethnicity [20]. Among a Hispanic population of HIV patients, the prevalence of metabolic syndrome was 35%, which is high compared to the 24–26% prevalence reported in US adults living with HIV, but the same as for the general population of Puerto Rico [21]. To date, no studies have systematically addressed the effect of ethnicity and its interaction with glycemic parameters in HIV patients. To address this issue, we analyzed data collected in the Heart Positive study—a large, multiethnic study of hypertriglyceridemic but otherwise healthy patients with HIV infection on stable HAART, all of whom reported no history of diabetes and underwent oral glucose tolerance testing [22, 23]. African-Americans and Hispanics had significantly greater impairment of glycemic parameters than NHWs (Misra, Balasubramanyam, et al., unpublished data).
These findings are important because there is also an increased risk of myocardial infarction (MI) in HIV-infected patients, and both metabolic syndrome and dysglycemia are risk factors for MI. A higher MI incidence rate was seen in HIV-positive men exposed to PIs for 18 months or more [24]. In HIV-infected patients on HAART, the incidence of MI increased with longer exposure to therapy (adjusted relative rate per year of exposure, 1.26) [25]. An extensive search of hospital databases showed that the rate of acute MI was higher in an HIV-infected cohort over non-HIV patients, with a relative risk of 1.75, even after adjusting for age, gender, race, hypertension, DM, and dyslipidemia [26]. The D:A:D group also studied the effects of 13 anti-HIV drugs on the development of MI; of these, only indinavir, lopinavir–ritonavir, didanosine, and abacavir were associated with a significantly increased risk of MI [27].
Mechanisms
The mechanisms of glycemic dysregulation and associated defects in insulin action and secretion in HIV-infected patients are numerous and complex, with new information continually surfacing. The literature to date has focused primarily on defects in lipid metabolism and inflammation, indicating that lipotoxicity and inflammation lead to IR—one of the consequences of IR could be dysglycemia [28]. For the purpose of this review, we shall organize the extant data into three classes: studies that exclude the effects of HAART, studies done in the presence of HAART agents while elucidating non-antiretroviral mechanisms, and studies done in the presence of HAART agents that elucidate mechanisms directly related to HAART.
Studies that exclude the effects of HAART
As the pathophysiology of IR in HIV involves many factors operating simultaneously, some laboratories have performed in vitro studies to isolate the effects of the HIV virus in T-cells from the other variables. In a recent study of the proteomic composition of HIV-1 infected CD4 cells, Chan et al. found an increase in the concentration of fatty acid synthase (FASN) after HIV-1 infection [29]. Rasheed et al. also performed proteomic analysis of a human T-cell line and reported upregulation of 18 HIV-modulated proteins and their interacting pathways, which would be expected to promote fatty acid synthesis and dysregulate multiple pathways of lipid metabolism. They concluded that HIV replication in human T-cells per se, independently of the effects of antiviral drugs or other factors, can affect lipid metabolism [30]. In support of this hypothesis, recent human studies have shown that untreated HIV-infected individuals have higher levels of circulating FASN than those on ART, and that HCV co-infection can also elevate FASN levels. Serum levels of insulin and inflammatory cytokines were correlated positively with FASN levels [31], indicating that the disruption of lipid metabolism within HIV-infected immune cells could contribute to both systemic lipid metabolic defects and an inflammatory form of whole body IR, and thereby to dysglycemia.
Furthermore, evidence of HIV-mediated inflammatory IR was noted in a number of insulin-regulated pathways prior to the HAART era. The Grunfeld laboratory found that interferon-α levels were higher in patients with AIDS compared to those with uncomplicated HIV infection and matched healthy controls [32]. Concomitantly, AIDS patients had increased triglyceride levels, but not increased TNF levels when compared to HIV patients without AIDS [33]. A more recent cross-sectional study found that TNF-α levels were higher in HAART-naïve HIV patients than their HAART-treated counterparts. Levels of the insulin-sensitizing hormone adiponectin were decreased in both groups compared to healthy controls, and were correlated inversely with IR as measured by the homeostatic model assessment of insulin resistance (HOMA-IR) [34].
Studies in the presence of HAART while elucidating non-antiretroviral mechanisms
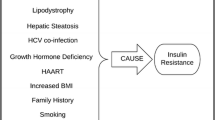
Insulin sensitivity is affected through multiple other mechanisms including lipodystrophy [35], viral effects [36–38], HCV co-infection [19], growth hormone (GH) deficiency [39], low CD4 count [40], and hepatic steatosis [41]. Most studies investigating these mechanisms have been performed on antiretroviral-treated patients. Multiple studies suggest that traditional risk factors such as increased age, positive family history, increased BMI, and smoking are more important than HAART in the development of IR [42, 43].
Lipodystrophy, reflecting profound defects in lipid turnover kinetics [35, 44] clearly induces severe IR and the tendency to hyperglycemia. Hyperinsulinemic-euglycemic clamp studies have demonstrated that compared to non-lipodystrophic patients, HIV patients with lipodystrophy have decreased insulin-stimulated glucose disposal [45, 46], increased intramyocellular lipid [45], and impaired skeletal muscle glucose uptake [46]. Lipodystrophic patients also have increased fasting plasma levels of free fatty acids and insulin [16], and a higher percentage of hepatic fat (associated with increased serum insulin levels) [47]. Clamp studies also suggest that insulin-resistant (but nondiabetic) HIV lipodystrophic individuals may have an increased insulin secretion rate due to both endogenous nonglucose secretatogues (including free fatty acids) and absent negative feedback of insulin on β-cells [48].
During any infectious process, the release of cytokines may affect glucose metabolism. In HIV infection, there is an increased release of TNF-α, IL-6, and IL-8 by both infected T-cells and adipose tissues—these cytokines can induce inflammatory IR and are associated with increased levels of HOMA-IR [36]. Activities of the HIV-1 accessory proteins Vpr and Tat have potential roles in initiating or aggravating IR. Vpr has been shown to inhibit PPAR-γ-mediated transcriptional co-activation in vitro (which might induce an inflammatory or lipotoxic form of IR in adipose tissues and liver) [37] and to decrease specific transcriptional effects of insulin such as inhibition of phosphoenolpyruvate carboxykinase (PEPCK) expression [38], which would promote gluconeogenesis and fasting hyperglycemia. Tat is known to activate NFκB, which would induce TNF-α production, block FFA uptake by adipocytes and suppress insulin receptor signaling—all of which could potentially decrease glucose disposal by blunting Glut4 translocation, and promote IR by increasing serine phosphorylation of insulin receptor substrate-1 (IRS-1) [38, 49]. In HIV patients initiating HAART, levels of TNF-α after 48 weeks of treatment were associated with incident DM [49].
HIV co-infection with HCV is associated with a higher prevalence of hyperglycemia. This correlates with the fact that HIV/HCV co-infected patients on HAART demonstrate greater IR, higher levels of activated platelets, and more endothelial dysfunction than HIV patients without co-infection [50]. When patients with HIV/HCV co-infection are placed on PI-based HAART, they have significantly increased risk of new-onset hyperglycemia [19, 51].
Lipodystrophic HIV patients also have accelerated fatty acid flux to the liver with blunted fat oxidation, factors that predispose them to accelerated re-esterification in the liver and the development of hepatic steatosis [28, 35, 44, 52]. Sutinen et al. suggest that IR in HIV patients is related more closely to fat accumulation in the liver than to intra-abdominal fat [47]. Consistent with this sequence of events leading to IR, Hadigan et al. have shown that blocking lipolysis acutely with acipimox improved insulin sensitivity in this group of patients [53].
The prevalence of hepatic steatosis in HIV-infected patients is high, especially in patients with chronic HCV or on nucleoside reverse transcriptase inhibitors (NRTIs) [41]. Rates of steatosis in HIV/HCV co-infected persons range from 40 to 69% [54, 55], while in HIV-infected patients without viral hepatitis co-infection, the rate is about 30% (although most were diagnosed with ultrasound rather than the gold standard of liver biopsy) [56]. The latter is at the upper end of the prevalence range for the general US population [57]. NRTIs may also induce hepatic steatosis via inhibition of mitochondrial DNA replication; hence use of these agents may exacerbate the tendency toward triglyceride accumulation in the liver [58]. Recent data suggest that fatty liver may cause IR via multiple mechanisms, many involving hepatic adipokines which are involved in the pathogenesis of type 2 diabetes [59].
Partial or complete GH deficiency is common among HIV patients, and is associated with both HIV lipodystrophy and IR [39]. Schwarz et al. treated HIV lipodystrophic men with low-dose (3 mg/day) GH for 6 months, and found that insulin sensitivity, measured by hyperinsulinemic-euglycemic clamp, was initially reduced, but returned to baseline after 6 months of therapy; this was associated with a significant decrease in hepatic lipogenesis and serum triglyceride levels [60]. D’Amico et al. demonstrated that the mechanism underlying this effect is related to the ability of physiologic GH replacement to decrease whole body lipolytic rates [61]. It is critical to understand that the dose of GH must be physiologic, in the setting of some degree of GH deficiency, to obtain a therapeutically beneficial effect. Most studies, generally using supraphysiologic doses of GH, have shown that GH treatment improves visceral adiposity but worsens glycemic control [62, 63]. To illustrate this principle, tesamorelin (GH releasing hormone, or GHRH) administration—which normalizes but does not elevate circulating GH and IGF-I levels—is associated with reduction in truncal and visceral adiposity without change in lipids or glucose tolerance [64]. Recently, another study using hyperinsulinemic-euglycemic clamps reported that 3-month treatment with IGF-1/IGFBP3 improved whole body glucose uptake and glucose tolerance (while increasing hepatic gluconeogenesis) and improved fasting triglycerides, though visceral adiposity remained the same [65].
Another risk factor for developing lipodystrophy and dysglycemia may be a low CD4 count (<200 cells/μl) [40]. Data from the longitudinal HIV Outpatient Study suggest that increased duration of HIV infection, high viral load and low CD4 nadir prior to initiation of HAART, as well as prolonged survival and duration, may be the most significant risks to developing lipodystrophy [66]. Studies exploring the associations of CD4 cell count with abnormal glucose levels in HIV patients show inconsistent results. Low CD4 cell count was associated with IGT and DM in patients co-infected with HIV, hepatitis C and hepatitis B virus [67]. El-Sadr et al. reported an inverse relationship between CD4 counts and IR in HIV patients, but no relationship between CD4 counts and fasting glucose concentrations [2]. Our group recently showed that among multiethnic, hypertriglyceridemic HIV patients on HAART without a history of diabetes, an interaction was observed between CD4 counts and ethnicity that affected glycemic levels (Misra, Balasubramanyam, et al., unpublished data).
Studies in the presence of HAART elucidating mechanisms that are directly related to HAART
Clinically measurable alterations in body fat depots are seen in association with the use of both PIs and NRTIs [68]. In vitro cellular studies suggest that various PIs may induce lipolysis or block adipogenesis via decreased expression of the transcription factor SREB1c [69], ultimately leading to consequences such as decreased expression of critical regulators of lipid metabolism such as lipoprotein lipase and FASN [70]. NRTIs may also promote fat cell apoptosis [71]. However, the clinical relevance of these in vitro data is limited because they employed high concentrations of these drugs, generally exceeding those used in clinical practice [68].
Significant hormonal and cytokine alterations that could affect glucose metabolism occur in HIV patients on HAART. Most studies have focused on the adipokines leptin and adiponectin, though recent studies are uncovering potential roles for many other endocrine and paracrine factors [72].
Hypoleptinemia, along with hypertriglyceridemia and hyperinsulinemia, is observed in HIV patients with lipoatrophy and six or more months of HAART treatment [73]. Leptin’s physiologic effects primarily involve coordinating the body’s physiologic response to starvation. During prolonged fasting its levels decline disproportionally to the fat loss and result in apparently adaptive changes in a variety of diverse hormonal axes, including the thyroid, reproductive, and endocrine-immune axes. Administration of leptin in this state partially or wholly reverses these hormonal changes associated with starvation [74]. Leptin also participates in the regulation of insulin sensitivity. A small randomized, controlled trial by Lee et al. showed that 2 months of physiologic leptin replacement (with r-met-HuLeptin) in hypoleptinemic HIV patients on HAART improved fasting insulin levels and HOMA-IR, along with reductions in body weight and fat mass, with no adverse effects on treatment [75]. A small, non-randomized study by Mulligan et al. in a similar group of patients showed that 6 months of escalating doses of physiologic leptin replacement improved the ability of insulin to suppress endogenous glucose production while decreasing both glycogenolysis and gluconeogenesis [76].
Adiponectin, in addition to its significant anti-inflammatory and anti-atherosclerotic effects, also enhances insulin-mediated suppression of gluconeogenesis and glucose release, and increases liver FFA oxidation and muscle glucose uptake [77, 78]. Its transcription is mediated by the SREBP1/PPARγ pathway [79]. Adiponectin levels are low in HIV-infected individuals with lipodystrophy and hypertriglyceridemia [80, 81]. Hypoadiponectinemia is seen in HIV-infected patients even prior to the initiation of HAART, though it worsens after initiation [34, 82]. Addy et al. demonstrated that serum adiponectin levels correlated negatively with IR as measured by HOMA-IR among HIV patients on HAART, though this relationship became non-significant after adjusting for NRTI use [83]. A study of HIV-infected children with fat redistribution showed that decreased levels of adiponectin were associated with IR (as assessed by the insulin/glucose ratio), even after controlling for HIV treatment [84].
The pathogenesis of IR in HIV can also be classified with regard to the class of antiretroviral treatment (ART). Most effects are related to the use of NRTIs and PIs [1, 85].
NRTIs include thymidine analogs (e.g., stavudine, zidovudine) which, in addition to inhibiting HIV reverse transcriptase, also inhibit DNA polymerase-γ, active in mitochondrial replication [86]. This leads to mitochondrial dysfunction; in the muscle and liver, this is linked to lipotoxic IR from the inability of these tissues to oxidize fat [87]. NRTIs also promote lipoatrophy in adipose tissue [88].
In an animal model, Murata et al. showed that the PI indinavir-induced IR by selectively inhibiting the function of the Glut4 transporter, which is responsible for insulin-stimulated glucose uptake into muscle and adipose tissue [89, 90]. The effect was then demonstrated in human subjects, via use of hyperinsulinemic-euglycemic clamp [91, 92]. It has also been suggested, based on a 60% homology of the HIV-1 protease with the cytoplasmic retinoic-binding protein type-1 (CRABP-1) and the low-density lipoprotein receptor-like protein (LRP), that PIs could interfere with normal triglyceride metabolism, thus inducing lipotoxic IR in tissues [93]. There is currently no experimental evidence to support this mechanism in vivo. There is better evidence for specific effects of PIs on glucose transporters. By binding and inhibiting Glut4, the first-generation PIs indivavir and ritonavir acutely impair insulin-regulated glucose uptake in muscle [94] and also inhibit glucose-stimulated insulin secretion in cultured pancreatic beta cells and in mice [95]. However, newer PIs such as atazanavir and tipranavir show little to no acute effect on insulin sensitivity [96, 97]. Chronically, longer exposure to PIs may disturb insulin signaling and indirectly lead to impaired glucose uptake via mechanisms dependent on IRS-1, Akt, and/or SOCS-1 [85].
There are no consistent data showing the development of IR following treatment with NNRTIs, fusion inhibitors, CCR5 antagonists, or integrase inhibitors [1].
Screening and diagnosis
The Infectious Diseases Society of America (IDSA) recommends the assessment of fasting glucose and fasting lipids prior to and within 4–6 weeks after starting HAART [98]. In addition, the International Association of AIDS-USA recommends repeating these measurements at the time of switching therapy, 3–6 months after switching therapy, and at least annually thereafter while on stable therapy [99]. Patients with pre-existing DM should have hemoglobin A1C (HgbA1C) monitored at least every 6 months with a goal of <7%, following American Diabetes Association (ADA) Guidelines [98, 100].
The diagnosis of DM should be made in accordance with ADA Guidelines [100, 101]. HbA1c should not be used for diagnostic purposes in HIV patients as it may underestimate glycemia, especially among patients on NRTIs; this may be a result of NRTI-induced macrocytosis [102]. In addition, the oral glucose tolerance test may be used to detect IGT or DM in selected HIV patients who have a normal fasting glucose [67].
While it is still important to screen for DM as a risk factor for coronary heart disease (CHD), DM might not be a CHD “risk equivalent” among HIV patients as it is in the general population. The D:A:D study showed a 7.6% incidence of MI in HIV patients with preexisting DM and no pre-existing CHD, and a 31.1% incidence of MI in patients with preexisting CHD but no pre-existing DM [103].
Management
DM and IGT should be managed according to current ADA guidelines [100], which also apply to non-HIV populations. To date, there are no large clinical trials that deal specifically with glucose management in a HIV population. However, several studies on metabolic abnormalities in HIV do shed some light in the area of HIV-specific glycemic management.
The substitution of thymidine analogs (e.g., stavudine and zidovudine) with other ARTs has been successful as a therapeutic approach to decreasing the severity and clinically observed frequency of lipoatrophy [104–106]. As this class of drugs as well as PIs has been associated with increased IR, it is suggested that similar substitutions could be instituted to improve insulin sensitivity and dysglycemia. While this is plausible, there are currently no data suggesting that switching ARTs is beneficial to HIV patients with hyperglycemia [98].
The IDSA recommends lifestyle modifications (exercise, diet, and weight loss) as the first step in managing hyperglycemia [98]. Earlier studies on lifestyle modifications showed equivocal results, with only some showing improvements in insulin sensitivity [107–110]; in these studies most patients were receiving thymidine analog NRTI therapy. One of these studies was repeated from 2003 to 2007, with fewer subjects taking thymidine analogs. In that study, diet and exercise-induced weight loss led to decrease in visceral and subcutaneous fat and improved insulin sensitivity [111]. The Heart Positive study recently showed a small but significant decrease in HbA1c in the arm randomized to receive only intensive diet and supervised exercise among hypertriglyceridemic but euglycemic HIV patients on HAART [23].
When oral medications are necessary to treat diabetes, metformin and thiazolidinediones (TZDs) have insulin-sensitizing properties that are beneficial in patients with IR and dysglycemia. The use of these medications must be balanced with the potential for adverse reactions.
Among HIV patients on HAART, metformin treatment reduced serum fasting insulin levels [112] and insulin area under the curve on oral glucose tolerance testing [113]. There is a very small risk of lactic acidosis associated with the use of metformin in the general population; however, since the incidence of lactic acidosis is higher in HIV patients due to the use of NRTIs (especially stavudine and didanosine) [114, 115], plasma lactate levels should be measured if the patient develops symptoms of acidemia while on metformin. If the patient has a serum creatinine or venous lactate concentration more than twice the upper normal limit, metformin should not be prescribed [99].
Rosiglitazone, a TZD, improved insulin sensitivity as measured by hyperinsulinemic-euglycemic clamp [116], decreased serum insulin levels [117, 118], increased adiponectin levels [116, 118], and reduced free fatty acid levels [116] in HIV patients. However, rosiglitazone was also associated with detrimental effects on serum total cholesterol, triglycerides, HDL-C, and LDL-C [119]. Since a meta-analysis by Nissen et al. raised concern about increased risk of MI and cardiovascular death in diabetic patients treated with rosiglitazone [120], the US FDA has restricted its use [121]. Pioglitazone may be a useful alternative, as it generally elevates serum HDL-C levels with a neutral impact on other lipid markers [119], and may decrease the risk of MI in diabetic patients [122]. However, there are only two large published studies of the effects of pioglitazone in HIV patients [123]. In one study, pioglitazone treatment demonstrated no benefits on fasting glucose or insulin levels [124]. In another study, pioglitazone reduced IR and increased serum adiponectin levels [125]. It appears that pioglitazone should be used with some caution in hyperglycemic HIV patients, given the well-known TZD adverse effect of fluid retention. TZDs should be avoided in HIV patients with NYHA Class III and IV heart failure [126].
There are currently no large trials exploring the use of sulfonylureas, meglitinides, GLP-1 agonists, DPP-IV inhibitors, or α-glucosidase inhibitors on HIV patients taking HAART [1]. A single case report shows that treatment with a combination of exenatide, metformin, and repaglinide improved insulin sensitivity and led to significant weight loss in a diabetic HIV patient on HAART [127]. GLP-1 agonists (like exenatide) control diabetes through several mechanisms, including glucose-dependent stimulation of insulin secretion, suppression of inappropriate glucagon secretion, slowing of gastric emptying, appetite suppression, and resulting weight loss [128]. A large trial would be useful in the assessment of diabetes management via GLP-1 agonists in this population. Nevertheless, if glycemic management cannot be optimized with oral agents or GLP-1 agonists, insulin therapy should be initiated [129].
Conclusions
HIV infection and HAART presents challenges to the diagnosis and management of dysglycemia. Most epidemiological data suggest that dysglycemia is associated with HAART use. However, the mechanisms of dysglycemia appear to be both HAART-dependent and HAART-independent. Much of the literature focuses on the role of underlying lipid metabolic defects and inflammation, showing that lipotoxicity and inflammatory disorders lead to IR and eventually dysglycemia [28]. However, there is a paucity of information regarding the glucose metabolic defects in this population, and more studies would be helpful. At this point, DM or IGT management is best guided by ADA recommendations [100], with a few modifications as described above. Switching antiretrovirals may help in theory, but we do not as yet have sufficient evidence to support this approach. Physiologic leptin replacement may be useful to treat dysglycemia, but this agent is still investigational. A comparison of current diabetic therapies via randomized controlled trials would be helpful in designing more specific treatment strategies for dysglycemia in the HIV population.
References
I.J. Paik, D.P. Kotler, The prevalence and pathogenesis of diabetes mellitus in treated HIV-infection. Best Pract. Res. Clin. Endocrinol. Metab. 25, 469–478 (2011)
W.M. El-Sadr et al., Effects of HIV disease on lipid, glucose and insulin levels: results from a large antiretroviral-naive cohort. HIV Med. 6, 114–121 (2005)
F. Visnegarwala, L. Chen, S. Raghavan, E. Tedaldi, Prevalence of diabetes mellitus and dyslipidemia among antiretroviral naive patients co-infected with hepatitis C virus (HCV) and HIV-1 compared to patients without co-infection. J. Infect. 50, 331–337 (2005)
P.C. Tien et al., Antiretroviral therapy exposure and incidence of diabetes mellitus in the Women’s Interagency HIV Study. AIDS 21, 1739–1745 (2007)
C. Hadigan et al., Metabolic abnormalities and cardiovascular disease risk factors in adults with human immunodeficiency virus infection and lipodystrophy. Clin. Infect. Dis. 32, 130–139 (2001)
T.T. Brown et al., Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch. Intern. Med. 165, 1179–1184 (2005)
M. Beregszaszi et al., Longitudinal evaluation and risk factors of lipodystrophy and associated metabolic changes in HIV-infected children. J. Acquir. Immune Defic. Syndr. 40, 161–168 (2005)
R. Palacios et al., Incidence of and risk factors for insulin resistance in treatment-naive HIV-infected patients 48 weeks after starting highly active antiretroviral therapy. Antivir. Ther. 11, 529–535 (2006)
S. De Wit et al., Incidence and risk factors for new-onset diabetes in HIV-infected patients: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) study. Diabetes Care 31, 1224–1229 (2008)
V. Estrada et al., Lipodystrophy and metabolic syndrome in HIV-infected patients treated with antiretroviral therapy. Metabolism 55, 940–945 (2006)
C. Jerico et al., Metabolic syndrome among HIV-infected patients: prevalence, characteristics, and related factors. Diabetes Care 28, 132–137 (2005)
M.E. Sobieszczyk et al., Prevalence and predictors of metabolic syndrome among HIV-infected and HIV-uninfected women in the Women’s Interagency HIV Study. J. Acquir. Immune Defic. Syndr. 48, 272–280 (2008)
K. Samaras et al., Prevalence of metabolic syndrome in HIV-infected patients receiving highly active antiretroviral therapy using International Diabetes Foundation and Adult Treatment Panel III criteria: associations with insulin resistance, disturbed body fat compartmentalization, elevated C-reactive protein, and [corrected] hypoadiponectinemia. Diabetes Care 30, 113–119 (2007)
K. Mondy et al., Metabolic syndrome in HIV-infected patients from an urban, midwestern US outpatient population. Clin. Infect. Dis. 44, 726–734 (2007)
C. Gazzaruso et al., Hypertension among HIV patients: prevalence and relationships to insulin resistance and metabolic syndrome. J. Hypertens. 21, 1377–1382 (2003)
G. Meininger et al., Elevated concentrations of free fatty acids are associated with increased insulin response to standard glucose challenge in human immunodeficiency virus-infected subjects with fat redistribution. Metabolism 51, 260–266 (2002)
C. Hadigan et al., Fasting hyperinsulinemia and changes in regional body composition in human immunodeficiency virus-infected women. J. Clin. Endocrinol. Metab. 84, 1932–1937 (1999)
A.A. Howard et al., Abnormal glucose metabolism among older men with or at risk of HIV infection. HIV Med. 7, 389–396 (2006)
S.H. Mehta, R.D. Moore, D.L. Thomas, R.E. Chaisson, M.S. Sulkowski, The effect of HAART and HCV infection on the development of hyperglycemia among HIV-infected persons. J. Acquir. Immune Defic. Syndr. 33, 577–584 (2003)
M.J. Glesby et al., Glycated haemoglobin in diabetic women with and without HIV infection: data from the Women’s Interagency HIV Study. Antivir. Ther. 15, 571–577 (2010)
F.A. Ramirez-Marrero et al., Prevalence of cardiometabolic risk factors in Hispanics living with HIV. Ethn. Dis. 20, 423–428 (2010)
S.L. Samson et al., Heart positive: design of a randomized controlled clinical trial of intensive lifestyle intervention, niacin and fenofibrate for HIV lipodystrophy/dyslipidemia. Contemp. Clin. Trials 27, 518–530 (2006)
A. Balasubramanyam et al., Combination of niacin and fenofibrate with lifestyle changes improves dyslipidemia and hypoadiponectinemia in HIV patients on antiretroviral therapy: results of “heart positive,” a randomized, controlled trial. J. Clin. Endocrinol. Metab. 96, 2236–2247 (2011)
M. Mary-Krause et al., Increased risk of myocardial infarction with duration of protease inhibitor therapy in HIV-infected men. AIDS 17, 2479–2486 (2003)
N. Friis-Moller et al., Combination antiretroviral therapy and the risk of myocardial infarction. N. Engl. J. Med. 349, 1993–2003 (2003)
V.A. Triant, H. Lee, C. Hadigan, S.K. Grinspoon, Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J. Clin. Endocrinol. Metab. 92, 2506–2512 (2007)
S.W. Worm et al., Risk of myocardial infarction in patients with HIV infection exposed to specific individual antiretroviral drugs from the 3 major drug classes: the data collection on adverse events of anti-HIV drugs (D:A:D) study. J. Infect. Dis. 201, 318–330 (2010)
F. Magkos, C.S. Mantzoros, Body fat redistribution and metabolic abnormalities in HIV-infected patients on highly active antiretroviral therapy: novel insights into pathophysiology and emerging opportunities for treatment. Metabolism 60, 749–753 (2011)
E.Y. Chan et al., Quantitative analysis of human immunodeficiency virus type 1-infected CD4+ cell proteome: dysregulated cell cycle progression and nuclear transport coincide with robust virus production. J. Virol. 81, 7571–7583 (2007)
S. Rasheed, J.S. Yan, A. Lau, A.S. Chan, HIV replication enhances production of free fatty acids, low density lipoproteins and many key proteins involved in lipid metabolism: a proteomics study. PLoS One 3, e3003 (2008)
G. Aragones et al., Infection with HIV and HCV enhances the release of fatty acid synthase into circulation: evidence for a novel indicator of viral infection. BMC Gastroenterol. 10, 92 (2010)
C. Grunfeld et al., Circulating interferon-alpha levels and hypertriglyceridemia in the acquired immunodeficiency syndrome. Am. J. Med. 90, 154–162 (1991)
C. Grunfeld et al., Lipids, lipoproteins, triglyceride clearance, and cytokines in human immunodeficiency virus infection and the acquired immunodeficiency syndrome. J. Clin. Endocrinol. Metab. 74, 1045–1052 (1992)
S. Das et al., In treatment-naive and antiretroviral-treated subjects with HIV, reduced plasma adiponectin is associated with a reduced fractional clearance rate of VLDL, IDL and LDL apolipoprotein B-100. Diabetologia 49, 538–542 (2006)
R.V. Sekhar et al., Metabolic basis of HIV-lipodystrophy syndrome. Am. J. Physiol. Endocrinol. Metab. 283, E332–E337 (2002)
P. Limone et al., Insulin resistance in HIV-infected patients: relationship with pro-inflammatory cytokines released by peripheral leukocytes. J. Infect. 47, 52–58 (2003)
S. Shrivastav et al., Human immunodeficiency virus (HIV)-1 viral protein R suppresses transcriptional activity of peroxisome proliferator-activated receptor gamma and inhibits adipocyte differentiation: implications for HIV-associated lipodystrophy. Mol. Endocrinol. 22, 234–247 (2008)
T. Kino, M. Mirani, S. Alesci, G.P. Chrousos, AIDS-related lipodystrophy/insulin resistance syndrome. Horm. Metab. Res. 35, 129–136 (2003)
P. Koutkia, K. Eaton, S.M. You, J. Breu, S. Grinspoon, Growth hormone secretion among HIV infected patients: effects of gender, race and fat distribution. AIDS 20, 855–862 (2006)
C. Grady, M. Ropka, R. Anderson, H.C. Lane, Body composition in clinically stable men with HIV infection. J. Assoc. Nurses AIDS Care 7, 29–38 (1996)
V. Soriano et al., Antiretroviral drugs and liver injury. AIDS 22, 1–13 (2008)
D.A. Wohl et al., Current concepts in the diagnosis and management of metabolic complications of HIV infection and its therapy. Clin. Infect. Dis. 43, 645–653 (2006)
J.S. Currier, D.V. Havlir, Complications of HIV disease and antiretroviral therapy. Highlights of the 11th Conference on Retroviruses and Opportunistic Infections, February 8–11, 2004, San Francisco, California, USA. Top. HIV Med. 12, 31–45 (2004)
R.V. Sekhar et al., Severely dysregulated disposal of postprandial triacylglycerols exacerbates hypertriacylglycerolemia in HIV lipodystrophy syndrome. Am. J. Clin. Nutr. 81, 1405–1410 (2005)
S.K. Gan et al., Altered myocellular and abdominal fat partitioning predict disturbance in insulin action in HIV protease inhibitor-related lipodystrophy. Diabetes 51, 3163–3169 (2002)
G.M. Behrens et al., Impaired glucose phosphorylation and transport in skeletal muscle cause insulin resistance in HIV-1-infected patients with lipodystrophy. J. Clin. Invest. 110, 1319–1327 (2002)
J. Sutinen et al., Increased fat accumulation in the liver in HIV-infected patients with antiretroviral therapy-associated lipodystrophy. AIDS 16, 2183–2193 (2002)
S.B. Haugaard et al., Insulin secretion in lipodystrophic HIV-infected patients is associated with high levels of nonglucose secretagogues and insulin resistance of beta-cells. Am. J. Physiol. Endocrinol. Metab. 287, E677–E685 (2004)
T.T. Brown, K. Tassiopoulos, R.J. Bosch, C. Shikuma, G.A. McComsey, Association between systemic inflammation and incident diabetes in HIV-infected patients after initiation of antiretroviral therapy. Diabetes Care 33, 2244–2249 (2010)
G.F. de Larranaga, S.D. Wingeyer, L.M. Puga, B.S. Alonso, J.A. Benetucci, Relationship between hepatitis C virus (HCV) and insulin resistance, endothelial perturbation, and platelet activation in HIV-HCV-coinfected patients under highly active antiretroviral treatment. Eur. J. Clin. Microbiol. Infect. Dis. 25, 98–103 (2006)
M. Duong et al., Association between insulin resistance and hepatitis C virus chronic infection in HIV-hepatitis C virus-coinfected patients undergoing antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 27, 245–250 (2001)
A. Balasubramanyam, R.V. Sekhar, HIV-associated lipodystrophy syndrome: an accelerated form of the metabolic syndrome of insulin resistance due to altered fat distribution. Res. Initiat. Treat. Action 12, 5–11 (2006)
C. Hadigan, J. Liebau, M. Torriani, R. Andersen, S. Grinspoon, Improved triglycerides and insulin sensitivity with 3 months of acipimox in human immunodeficiency virus-infected patients with hypertriglyceridemia. J. Clin. Endocrinol. Metab. 91, 4438–4444 (2006)
B.H. McGovern et al., Hepatic steatosis is associated with fibrosis, nucleoside analogue use, and hepatitis C virus genotype 3 infection in HIV-seropositive patients. Clin. Infect. Dis. 43, 365–372 (2006)
M.S. Sulkowski et al., Hepatic steatosis and antiretroviral drug use among adults coinfected with HIV and hepatitis C virus. AIDS 19, 585–592 (2005)
N. Crum-Cianflone et al., Nonalcoholic fatty liver disease among HIV-infected persons. J. Acquir. Immune Defic. Syndr. 50, 464–473 (2009)
G.C. Farrell, C.Z. Larter, Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 43, S99–S112 (2006)
A.E. Ogedegbe, D.L. Thomas, A.M. Diehl, Hyperlactataemia syndromes associated with HIV therapy. Lancet Infect. Dis. 3, 329–337 (2003)
N. Stefan, H.U. Haring, The metabolically benign and malignant fatty liver. Diabetes 60, 2011–2017 (2011)
J.M. Schwarz et al., Effects of recombinant human growth hormone on hepatic lipid and carbohydrate metabolism in HIV-infected patients with fat accumulation. J. Clin. Endocrinol. Metab. 87, 942 (2002)
S. D’Amico et al., Physiologic growth hormone replacement improves fasting lipid kinetics in patients with HIV lipodystrophy syndrome. Am. J. Clin. Nutr. 84, 204–211 (2006)
J.C. Lo et al., The effects of recombinant human growth hormone on body composition and glucose metabolism in HIV-infected patients with fat accumulation. J. Clin. Endocrinol. Metab. 86, 3480–3487 (2001)
D.P. Kotler et al., Effects of growth hormone on abnormal visceral adipose tissue accumulation and dyslipidemia in HIV-infected patients. J. Acquir. Immune Defic. Syndr. 35, 239–252 (2004)
P. Koutkia, B. Canavan, J. Breu, S. Grinspoon, Effects of growth hormone-releasing hormone on bone turnover in human immunodeficiency virus-infected men with fat accumulation. J. Clin. Endocrinol. Metab. 90, 2154–2160 (2005)
M.N. Rao et al., Effects of insulin-like growth factor (IGF)-I/IGF-binding protein-3 treatment on glucose metabolism and fat distribution in human immunodeficiency virus-infected patients with abdominal obesity and insulin resistance. J. Clin. Endocrinol. Metab. 95, 4361–4366 (2010)
K.A. Lichtenstein et al., Clinical assessment of HIV-associated lipodystrophy in an ambulatory population. AIDS 15, 1389–1398 (2001)
N. Gianotti et al., Detecting impaired glucose tolerance or type 2 diabetes mellitus by means of an oral glucose tolerance test in HIV-infected patients. HIV Med. 12, 109–117 (2011)
A. Balasubramanyam, R.V. Sekhar, F. Jahoor, P.H. Jones, H.J. Pownall, Pathophysiology of dyslipidemia and increased cardiovascular risk in HIV lipodystrophy: a model of ‘systemic steatosis’. Curr. Opin. Lipidol. 15, 59–67 (2004)
J.M. Lenhard et al., HIV protease inhibitors block adipogenesis and increase lipolysis in vitro. Antivir. Res. 47, 121–129 (2000)
A.R. Miserez, P.Y. Muller, V. Spaniol, Indinavir inhibits sterol-regulatory element-binding protein-1c-dependent lipoprotein lipase and fatty acid synthase gene activations. AIDS 16, 1587–1594 (2002)
D. Nolan, M. John, S. Mallal, Antiretoviral therapy and the lipodystrophy syndrome, part 2: concepts in aetiopathogenesis. Antivir. Ther. 6, 145–160 (2001)
S. Tsiodras, A. Perelas, C. Wanke, C.S. Mantzoros, The HIV-1/HAART associated metabolic syndrome—novel adipokines, molecular associations and therapeutic implications. J. Infect. 61, 101–113 (2010)
G.S. Nagy et al., Human immunodeficiency virus type 1-related lipoatrophy and lipohypertrophy are associated with serum concentrations of leptin. Clin. Infect. Dis. 36, 795–802 (2003)
J.L. Chan, K. Heist, A.M. DePaoli, J.D. Veldhuis, C.S. Mantzoros, The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J. Clin. Invest. 111, 1409–1421 (2003)
J.H. Lee, J.L. Chan, E. Sourlas, V. Raptopoulos, C.S. Mantzoros, Recombinant methionyl human leptin therapy in replacement doses improves insulin resistance and metabolic profile in patients with lipoatrophy and metabolic syndrome induced by the highly active antiretroviral therapy. J. Clin. Endocrinol. Metab. 91, 2605–2611 (2006)
K. Mulligan et al., The effects of recombinant human leptin on visceral fat, dyslipidemia, and insulin resistance in patients with human immunodeficiency virus-associated lipoatrophy and hypoleptinemia. J. Clin. Endocrinol. Metab. 94, 1137–1144 (2009)
T. Yamauchi et al., Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 8, 1288–1295 (2002)
T. Yamauchi et al., Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423, 762–769 (2003)
M. Iwaki et al., Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes 52, 1655–1663 (2003)
L. Kosmiski, D. Kuritzkes, K. Lichtenstein, R. Eckel, Adipocyte-derived hormone levels in HIV lipodystrophy. Antivir. Ther. 8, 9–15 (2003)
K. Falasca et al., Associations between hypertriglyceridemia and serum ghrelin, adiponectin, and IL-18 levels in HIV-infected patients. Ann. Clin. Lab. Sci. 36, 59–66 (2006)
C. Vernochet et al., Human immunodeficiency virus protease inhibitors accumulate into cultured human adipocytes and alter expression of adipocytokines. J. Biol. Chem. 280, 2238–2243 (2005)
C.L. Addy et al., Hypoadiponectinemia is associated with insulin resistance, hypertriglyceridemia, and fat redistribution in human immunodeficiency virus-infected patients treated with highly active antiretroviral therapy. J. Clin. Endocrinol. Metab. 88, 627–636 (2003)
R. Verkauskiene et al., Serum adiponectin and leptin concentrations in HIV-infected children with fat redistribution syndrome. Pediatr. Res. 60, 225–230 (2006)
P.W. Hruz, Molecular mechanisms for insulin resistance in treated HIV-infection. Best Pract. Res. Clin. Endocrinol. Metab. 25, 459–468 (2011)
W. Lewis, B.J. Day, W.C. Copeland, Mitochondrial toxicity of NRTI antiviral drugs: an integrated cellular perspective. Nat. Rev. Drug Discov. 2, 812–822 (2003)
C.M. Shikuma, L.J. Day, M. Gerschenson, Insulin resistance in the HIV-infected population: the potential role of mitochondrial dysfunction. Curr. Drug Targets Infect. Disord. 5, 255–262 (2005)
M. Gerschenson, K. Brinkman, Mitochondrial dysfunction in AIDS and its treatment. Mitochondrion 4, 763–777 (2004)
H. Murata, P.W. Hruz, M. Mueckler, The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J. Biol. Chem. 275, 20251–20254 (2000)
H. Murata, P.W. Hruz, M. Mueckler, Indinavir inhibits the glucose transporter isoform Glut4 at physiologic concentrations. AIDS 16, 859–863 (2002)
M.A. Noor et al., Metabolic effects of indinavir in healthy HIV-seronegative men. AIDS 15, F11–F18 (2001)
M.A. Noor et al., Indinavir acutely inhibits insulin-stimulated glucose disposal in humans: a randomized, placebo-controlled study. AIDS 16, F1–F8 (2002)
A. Carr, K. Samaras, D.J. Chisholm, D.A. Cooper, Abnormal fat distribution and use of protease inhibitors. Lancet 351, 1736 (1998)
A.K. Vyas, J.C. Koster, A. Tzekov, P.W. Hruz, Effects of the HIV protease inhibitor ritonavir on GLUT4 knock-out mice. J. Biol. Chem. 285, 36395–36400 (2010)
J.C. Koster, M.S. Remedi, H. Qiu, C.G. Nichols, P.W. Hruz, HIV protease inhibitors acutely impair glucose-stimulated insulin release. Diabetes 52, 1695–1700 (2003)
M.A. Noor, O.P. Flint, J.F. Maa, R.A. Parker, Effects of atazanavir/ritonavir and lopinavir/ritonavir on glucose uptake and insulin sensitivity: demonstrable differences in vitro and clinically. AIDS 20, 1813–1821 (2006)
P.W. Hruz, Q. Yan, Tipranavir without ritonavir does not acutely induce peripheral insulin resistance in a rodent model. J. Acquir. Immune Defic. Syndr. 43, 624–625 (2006)
J.A. Aberg et al., Primary care guidelines for the management of persons infected with human immunodeficiency virus: 2009 update by the HIV medicine Association of the Infectious Diseases Society of America. Clin. Infect. Dis. 49, 651–681 (2009)
M. Schambelan et al., Management of metabolic complications associated with antiretroviral therapy for HIV-1 infection: recommendations of an International AIDS Society-USA panel. J. Acquir. Immune Defic. Syndr. 31, 257–275 (2002)
D.M. Nathan et al., Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 32, 193–203 (2009)
International Expert Committee, International Expert Committee report on the role of the A1C assay in the diagnosis of diabetes. Diabetes Care 32, 1327–1334 (2009)
P.S. Kim et al., A1C underestimates glycemia in HIV infection. Diabetes Care 32, 1591–1593 (2009)
S.W. Worm et al., Diabetes mellitus, preexisting coronary heart disease, and the risk of subsequent coronary heart disease events in patients infected with human immunodeficiency virus: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D Study). Circulation 119, 805–811 (2009)
A. Carr et al., Abacavir substitution for nucleoside analogs in patients with HIV lipoatrophy: a randomized trial. JAMA 288, 207–215 (2002)
A. Martin et al., Reversibility of lipoatrophy in HIV-infected patients 2 years after switching from a thymidine analogue to abacavir: the MITOX Extension Study. AIDS 18, 1029–1036 (2004)
G.A. McComsey et al., Improvement in lipoatrophy associated with highly active antiretroviral therapy in human immunodeficiency virus-infected patients switched from stavudine to abacavir or zidovudine: the results of the TARHEEL study. Clin. Infect. Dis. 38, 263–270 (2004)
A. Gavrila et al., Exercise and vitamin E intake are independently associated with metabolic abnormalities in human immunodeficiency virus-positive subjects: a cross-sectional study. Clin. Infect. Dis. 36, 1593–1601 (2003)
K.E. Yarasheski et al., Resistance exercise training reduces hypertriglyceridemia in HIV-infected men treated with antiviral therapy. J. Appl. Physiol. 90, 133–138 (2001)
K.V. Fitch et al., Effects of a lifestyle modification program in HIV-infected patients with the metabolic syndrome. AIDS 20, 1843–1850 (2006)
E.S. Engelson et al., Body composition and metabolic effects of a diet and exercise weight loss regimen on obese, HIV-infected women. Metabolism 55, 1327–1336 (2006)
J.B. Albu, C.M. Kim, E.S. Engelson et al., Effects of diet and exercise and/or rosiglitazone on body composition and glucose metabolism in HIV+ and HIV− subjects. Antivir. Ther. 13(Suppl. 4), A31 (2008)
T. Saint-Marc, J.L. Touraine, Effects of metformin on insulin resistance and central adiposity in patients receiving effective protease inhibitor therapy. AIDS 13, 1000–1002 (1999)
C. Hadigan et al., Increased PAI-1 and tPA antigen levels are reduced with metformin therapy in HIV-infected patients with fat redistribution and insulin resistance. J. Clin. Endocrinol. Metab. 86, 939–943 (2001)
Lactic Acidosis International Study Group, Risk factors for lactic acidosis and severe hyperlactataemia in HIV-1-infected adults exposed to antiretroviral therapy. AIDS 21, 2455–2464 (2007)
L.T. Matthews et al., A risk-factor guided approach to reducing lactic acidosis and hyperlactatemia in patients on antiretroviral therapy. PLoS One 6, e18736 (2011)
C. Hadigan et al., Metabolic effects of rosiglitazone in HIV lipodystrophy: a randomized, controlled trial. Ann. Intern. Med. 140, 786–794 (2004)
J. Sutinen et al., Rosiglitazone in the treatment of HAART-associated lipodystrophy—a randomized double-blind placebo-controlled study. Antivir. Ther. 8, 199–207 (2003)
A. Carr et al., No effect of rosiglitazone for treatment of HIV-1 lipoatrophy: randomised, double-blind, placebo-controlled trial. Lancet 363, 429–438 (2004)
J. Sutinen, The effects of thiazolidinediones on metabolic complications and lipodystrophy in HIV-infected patients. PPAR Res. 2009, 373524 (2009)
S.E. Nissen, K. Wolski, Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 356, 2457–2471 (2007)
FDA Drug Safety Communication: Updated Risk Evaluation and Mitigation Strategy (REMS) to Restrict Access to Rosiglitazone-containing Medicines including Avandia, Avandamet, and Avandaryl. http://www.fda.gov/Drugs/DrugSafety/ucm255005.htm (2011). Accessed 4 November 2011
A.M. Lincoff, K. Wolski, S.J. Nicholls, S.E. Nissen, Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA 298, 1180–1188 (2007)
S.H. Sheth, R.J. Larson, The efficacy and safety of insulin-sensitizing drugs in HIV-associated lipodystrophy syndrome: a meta-analysis of randomized trials. BMC Infect. Dis. 10, 183 (2010)
L. Slama et al., Effect of pioglitazone on HIV-1-related lipodystrophy: a randomized double-blind placebo-controlled trial (ANRS 113). Antivir. Ther. 13, 67–76 (2008)
A. Gavrila et al., Improvement in highly active antiretroviral therapy-induced metabolic syndrome by treatment with pioglitazone but not with fenofibrate: a 2 × 2 factorial, randomized, double-blinded, placebo-controlled trial. Clin. Infect. Dis. 40, 745–749 (2005)
R.W. Nesto et al., Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. October 7, 2003. Circulation 108, 2941–2948 (2003)
P. Oriot, M.P. Hermans, P. Selvais, M. Buysschaert, X. de la Tribonniere, Exenatide improves weight loss insulin sensitivity and beta-cell function following administration to a type 2 diabetic HIV patient on antiretroviral therapy. Ann. Endocrinol. (Paris) 72, 244–246 (2011)
M. Briones, M. Bajaj, Exenatide: a GLP-1 receptor agonist as novel therapy for type 2 diabetes mellitus. Expert Opin. Pharmacother. 7, 1055–1064 (2006)
J.D. Lundgren et al., European AIDS Clinical Society (EACS) guidelines on the prevention and management of metabolic diseases in HIV. HIV Med. 9, 72–81 (2008)
Acknowledgments
AG is supported by T32 HL66991, and AB by RO1 DK081553 and R21 082827, all from the National Institutes of Health.
Conflict of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gutierrez, A.D., Balasubramanyam, A. Dysregulation of glucose metabolism in HIV patients: epidemiology, mechanisms, and management. Endocrine 41, 1–10 (2012). https://doi.org/10.1007/s12020-011-9565-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-011-9565-z