
Abstract
Obesity with excessive levels of circulating free fatty acids (FFAs) is tightly linked to the incidence of type 2 diabetes. Insulin resistance of peripheral tissues and pancreatic β-cell dysfunction are two major pathological changes in diabetes and both are facilitated by excessive levels of FFAs and/or glucose. To gain insight into the mitochondrial-mediated mechanisms by which long-term exposure of INS-1 cells to excess FFAs causes β-cell dysfunction, the effects of the unsaturated FFA linoleic acid (C 18:2, n-6) on rat insulinoma INS-1 β cells was investigated. INS-1 cells were incubated with 0, 50, 250 or 500 μM linoleic acid/0.5% (w/v) BSA for 48 h under culture conditions of normal (11.1 mM) or high (25 mM) glucose in serum-free RPMI-1640 medium. Cell viability, apoptosis, glucose-stimulated insulin secretion, Bcl-2, and Bax gene expression levels, mitochondrial membrane potential and cytochrome c release were examined. Linoleic acid 500 μM significantly suppressed cell viability and induced apoptosis when administered in 11.1 and 25 mM glucose culture medium. Compared with control, linoleic acid 500 μM significantly increased Bax expression in 25 mM glucose culture medium but not in 11.1 mM glucose culture medium. Linoleic acid also dose-dependently reduced mitochondrial membrane potential (ΔΨm) and significantly promoted cytochrome c release from mitochondria in both 11.1 mM glucose and 25 mM glucose culture medium, further reducing glucose-stimulated insulin secretion, which is dependent on normal mitochondrial function. With the increase in glucose levels in culture medium, INS-1 β-cell insulin secretion function was deteriorated further. The results of this study indicate that chronic exposure to linoleic acid-induced β-cell dysfunction and apoptosis, which involved a mitochondrial-mediated signal pathway, and increased glucose levels enhanced linoleic acid-induced β-cell dysfunction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes (T2D) is rapidly becoming a global pandemic and is projected to afflict more than 300 million individuals worldwide by the year 2025 [1]. Insulin resistance of peripheral tissues and pancreatic β-cell dysfunction are generally accepted to be the main pathological characteristics of T2D, and the latter is widely believed to be secondary to prolonged exposure to increased glucose and/or excess free fatty acid (FFA) levels, conditions that are often associated with obesity. Importantly, recent studies have indicated that a dysregulation in lipid homeostasis, but not glucose homeostasis, is the primary metabolic defect in T2D, leading to a major paradigm shift in the understanding of T2D from a “glucocentric” to “lipocentric” theory [2–4]. Although this remains controversial [5], excess FFAs are now recognized as significant contributors to insulin resistance pathophysiology, and lipotoxicity is recognized as the primary characteristic of T2D [6, 7]. Our understanding of the mechanism underlying FFA-induced β-cell dysfunction is limited and warrants further investigation.
Increased levels of circulating FFAs, particularly saturated FFAs, are the main factors that induce β-cell apoptosis and dysfunction, although the effects vary with FFA chain length and saturation level through multiple mechanisms [8–15]. However, the effects of unsaturated FFAs on pancreatic islet remain controversial. Some studies have shown that unsaturated FFAs protect pancreatic β cells from the effects of saturated FFAs and/or cytokine-induced apoptosis [14, 16, 17], whereas other studies have indicated that unsaturated FFAs induce β-cell apoptosis [15, 18, 19]. Recently, a growing body of evidence has linked the cytotoxic action of unsaturated FFAs to mitochondrial dysfunction and activation of mitochondrial apoptotic pathways [10, 20]. The mechanisms underlying different effects of unsaturated fatty acids, especially polyunsaturated FFAs, on β-cell viability and function are not clear and reflect the lack of systematic investigations.
On the basis of these observations, we hypothesized that chronic exposure of β cells to unsaturated FFAs induced the mitochondrial dysfunction and attenuated insulin secretion, which subsequently impaired glucose regulation and β-cell dysfunction. The aim of this study was to determine whether exposure of INS-1 β cells to linoleic acid-induced cell apoptosis through a mitochondrial-regulated signal pathway and whether high glucose levels potentiated the lipotoxicity of linoleic acid. To address these questions, the rat insulinoma β-cell line INS-1 was used because it has previously been demonstrated that FFAs and cytokines can cause apoptosis in this cell line [21–23] and it is considered an appropriate model for β-cell apoptosis research. In addition, compared with isolated islets, the large amount of available cellular material from pure β cells allows more biochemical determinations to be performed than with isolated islets of mixed cell types. In our study, INS-1 cells were incubated with various concentrations of linoleic acid for up to 48 h with normal (11.1 mM) and high (25 mM) glucose concentrations, and β-cell viability and function were studied.
Materials and methods
Reagents and chemicals
Rhodamine 123 (Rh 123) and the ApoTarget™ Annexin-V FITC Apoptosis Kit were purchased from Invitrogen (Madison, WI, USA). CellTiter 96® AQueous One Solution Cell Proliferation Assay, Caspase-Glo® 3/7 and Caspase-Glo® 9 Activity Assay were purchased from Promega Corp. (Madison, WI, USA). Linoleic Acid (C 18:2, water soluble), FFA-free BSA, β-mercaptoethanol, and carbonyl cyanide m-chlorophenylhydrazone (CCCP) were obtained from Sigma (Sigma-Aldrich, USA). Sensitive Insulin assay kit (SKI-13) was purchased from Linco Research (Millipore, USA).
Cell culture and incubation
INS-1 β cells (a gift from Dr T. Biden, Garvan Institute, Sydney, Australia) were maintained in RPMI-1640 (11.1 mM glucose, 1 mM sodium pyruvate, and 10 mM HEPES) supplemented with 10% FBS, 2 mM l-glutamine, and 50 μM β-mercaptoethanol containing antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin) at 37°C and 5% CO2, as described previously [24]. A 50 mM linoleic acid stock solution and 10% (w/v) FFA-free BSA solution was prepared in a water bath at 55°C. Altering the proportion of these two solutions when mixed together varied the concentration of FFA compounded to BSA, and the molar ratio of FFA/BSA varied from 1:3.3 to 1:6.6 to ensure that the binding of linoleic acid to BSA was not saturated and the solubility of FFA in water was not exceeded [25–28]. INS-1 β cells were subcultured on six-well plates to 60% confluence. The cells were then incubated in 11.1 and 25 mM glucose RPMI-1640 medium with 50, 250, and 500 μM linoleic acid/0.5% (w/v) BSA compound for 48 h. The incubation time and concentrations of linoleic acid used in the study to mimic high FFA exposure conditions were selected based on previous studies on INS-1 cells [8, 17, 26].
Determination of cell viability
To determine cell viability, INS-1 β cells were seeded at 2 × 104 cells per well in a 96-well plate and grown for 2 days. The absorbance at 490 nm was measured after 48-h treatment with different concentrations of linoleic acid/BSA compound by CellTiter 96® AQueous One Solution Cell Proliferation Assay. Cell viability was assessed as OD value of treatment/OD value of control × 100%.
Annexin-V FITC staining apoptosis assay
INS-1 β cells were seeded at 2 × 105 cells per well in six-well plates, treated with different concentrations of linoleic acid/BSA compound and grown to 70% confluency in 11.1 and 25 mM glucose RMPI-1640 medium for 48 h [8]. After washing twice with cold PBS, cells were stained with Annexin-V fluorescein isothiocyanate (FITC) and propidium iodide (PI) according to the recommended protocol, and apoptosis was assessed by flow cytometry. In the early stages of apoptosis, phosphatidyl serine (PS), which moved from the inner side of the plasma membrane to the outer layer, was stained by Annexin-V FITC, whereas PI staining was used to distinguish apoptotic cells from necrotic cells.
Caspase-Glo® 3/7 and Caspase-Glo® 9 activity assay
INS-1 β cells were seeded at 5 × 103 cells per well in 96-well plates (Corning Inc., Corning, NY) and cultured for 24 h. INS-1 β cells were treated with 500 μM linoleic acid/BSA compound in 11.1 or 25 mM glucose RMPI-1640 medium for 48 h. Caspase activity was analyzed using the commercially available Caspase-Glo® 3/7 and Caspase-Glo® 9 Activity Assays (Promega Corp., WI, USA) according to the manufacturer’s instructions. Luminescence was measured using a luminometer (POLAR Star Omega, BMG LABTECH).
Insulin secretion assay
When the cells reached approximately 70% confluence, INS-1 β cells were washed twice with PBS (pH 7.40, 37°C) and incubated in fresh RPMI-1640 medium containing 11.1 and 25 mM glucose with different concentrations of linoleic acid/BSA compound for 48 h in 24-well plates. Before stimulation, the β cells were washed twice with PBS and pre-incubated in 500 μl of Krebs–Ringer bicarbonate (KRB) buffer containing 115 mM NaCl, 4.7 mM KCl, 2.6 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 20 mM NaHCO3, 16 mM HEPES, and 0.5% BSA (glucose-free and FFA-free) (pH 7.40) at 37°C in a humidified 5% CO2 atmosphere for 60 min [17]. Cells were then washed once with glucose-free KRB buffer and subsequently incubated for 60 min with KRB buffer containing 3 or 25 mM glucose. The incubation buffer was collected and centrifuged at 1.0 × 104 rpm for 10 min at 4°C. Insulin concentrations were measured using Sensitive Insulin assay kits.
Quantitative analysis of mRNA expression by real-time PCR
Total RNA was isolated from INS-1 β cells by TRIzol (Invitrogen, CA, USA) according to the manufacturer’s instructions. First-strand synthesis of complementary DNA (cDNA) was carried outperformed using iScriptase reverse transcriptase following according to the manufacturer’s instructions (Bio-RAD, CA, USA). The real-time PCR amplification reaction was done in 20 μl containing 0.5 μM forward and reverse primers, 10 μl 2 × Fast SYBR® Green Master Mix (Applied Bioscience, USA) and 2 μl cDNA (transcript from 1 μg RNA, totally 20 μl) plus RNase-free water. To select proper housekeeping gene, the stability of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression under different treatment condition was tested [29]. The expression level of the gene of interest was normalized with the housekeeping gene GAPDH by comparison of Ct values (delta − delta Ct) [30]. The specific primers used and their respective PCR fragment lengths were as follows: GAPDH, forward, 5′-AGTTCAACGGCACAGTCAAG-3′, reverse, 5′-TACTCAGCACCAGCATCACC-3′ (118 bp); Bcl-2 forward, 5′-AGGATTGTGGCCTTCTTTGAGT-3′, reverse, 5′-GCCGGTTCAGGTACTCAGTCAT-3′ (115 bp); Bax forward, 5′-GGTTGCCCTCTTCTACTTTGCT-3′, reverse, 5′-TGAGCCCATCTTCTTCCAGA-3′ (247 bp) [31, 32]. Relative expression levels of Bcl-2 and Bax to GAPDH was calculated.
Detecting change of mitochondrial membrane potential
Linoleic acid-induced reduction in mitochondrial membrane potential (MMP, ΔΨm) under different concentrations of glucose was quantified in INS-1 β cells. Incubation medium (11.1 or 25 mM glucose) with or without linoleic acid/BSA was added successively. After 48-h treatment, cells were loaded with 1 μg/ml Rh 123 for 15 min. The mitochondrial uncoupler CCCP (final concentration: 10 μM) was used as a positive control. Fluorescence was excited at 488 nm and measured at 529 nm. PI was used as a viability marker and added to the cell suspension prior to flow cytometric analysis to discard non-viable cells; data analysis was performed only on non-PI-labeled cells. The mean fluorescence intensity (MFI) in the Rh 123 positive cells represented the loss of mitochondrial membrane potential [33].
Cytochrome c release analysis by western blot analysis
Total protein was collected using radio-immunoprecipitation assay (RIPA) lysis buffer containing 50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, pH 8.0 (chelating agent), 1% Triton X-100 (non-ionic detergent), 1% sodium deoxycholate (anionic detergent), 0.1% SDS (anionic detergent), 10 mM sodium fluoride (serine–threonine phosphatase inhibitor), and Complete Mini® protease inhibitor (EDTA-free, Roche, USA). Mitochondria and cytosol were separated using Pierce mitochondrial isolation kit according to the manufacturer’s instructions (Thermo Scientific, Rockford, USA). Then mitochondrial protein and cytosolic protein were then extracted using RIPA lysis buffer. Protein content was determined using a BCA™ Protein Assay kit (Thermo Scientific, Rockford, USA). Total proteins, mitochondrial protein, and cytosolic proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes (Pall Corporation, NY, USA). The blots were incubated with the desired primary antibody overnight at the following dilutions: cytochrome c (1:500; MAB1800, Millipore, MA, USA) and GAPDH (1:100,000; sc-166545; Santa Cruz, CA, USA). Subsequently, the membrane was incubated with appropriate secondary antibodies (anti-mouse, 1:2000; Santa Cruz, CA, USA) for 1 h at room temperature. The bands were detected using an enhanced chemiluminescence system (SuperSignal® West Pico Chemiluminescent Substrate, Thermo Scientific, Rockford, USA) and quantified by densitometry methods.
Statistical analysis
All data were expressed as mean ± S.E. Statistical significance was evaluated using Student’s t test or one-way/two-way ANOVA with a Tukey’s post hoc test. Percentage changes were analyzed by the Kruskal and Wallis test. P < 0.05 was considered statistically significant.
Results
Effects of linoleic acid on cell viability and apoptosis
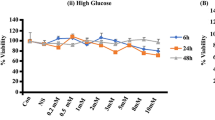
After exposure to linoleic acid for 48 h, cell viability was tested in INS-1 β cells. The concentration of linoleic acid ranged from 0 to 500 μM in 11.1 or 25 mM glucose culture medium. As shown in Fig. 1, there was a significant decrease in cell viability in the 500 μM treatment group at both 11.1 and 25 mM glucose levels. When INS-1 cells were cultured in 11.1 mM glucose RPMI-1640 medium (Fig. 1a), representing the normal glucose conditions for β cells, cell viability showed fluctuations with increasing linoleic acid concentration, but was only markedly decreased with 500 μM linoleic acid treatment (P < 0.05). When INS-1 cells were incubated in 25 mM glucose to mimic the increased glucose conditions in T2D, fluctuation of cell viability was also observed with increasing linoleic acid dose and markedly reduced with 500 μM treatment (P < 0.05; Fig. 1b). Thus, linoleic acid only had significantly effects on INS-1 β-cell viability at a dose of 500 μM.

The effect of chronic exposure to different levels of linoleic acid in 11.1 mM (a) and 25 mM (b) glucose culture media on the viability of cultured INS-1 cells. Each value represents the mean ± S.E. (N = 4). *P < 0.05; **P < 0.01
To confirm possible suppression of cell viability by linoleic acid, cell apoptosis assays were performed with 0, 50, 250, and 500 μM linoleic acid treatments in 11.1 and 25 mM glucose incubation. Cells were double-stained with FITC and PI to examine apoptosis by flow cytometry. Compared with the control group (0 μM linoleic acid treatment group), linoleic acid significantly induced apoptosis in the 500 μM treatment groups under 11.1 or 25 mM glucose conditions (Fig. 2a; P < 0.001). Consistent with the cell viability data shown in Fig. 1, linoleic acid only significantly attenuated cell viability when administered at 500 μM under both glucose culture conditions. Compared with the 11.1 mM glucose culture group, there was no significant increase in cell apoptosis in the 25 mM glucose group.

Linoleic acid-induced apoptosis after chronic exposure of INS-1 cells. The quantitative histogram of different linoleic acid-induced apoptosis in 11.1 and 25 mM glucose culture media (a), and there are significantly increases in apoptosis with 500 μM linoleic acid. To further confirm apoptosis induced by linoleic acid, the caspase-3/7 and caspase-9 activity levels in the 500 μM linoleic acid treatment group are presented in b and c, respectively. Values are the mean ± S.E. (N = 4). *P < 0.05 vs control; ***P < 0.001 vs control
Although cell viability and apoptosis assays showed the cytotoxic effects of 500 μM linoleic acid, they provided little information regarding the mechanism by which the cell population decreased. To further investigate the mechanism that linoleic acid-induced INS-1 cell apoptosis, we tested caspase-9 and caspase-3/7 activity induced by 500 μM linoleic acid under both 11.1 and 25 mM glucose culture conditions by cell-based assays. After 48 h incubation, the relative luminescent unit (RLU) measure, which represented caspase-3/7 and caspase-9 activity, was significantly increased with 500 μM linoleic acid compared with controls (Fig. 2b, c; P < 0.05). Caspase-9 generally receives the apoptosis signal from the mitochondria-regulated pathway, and then combined with caspase-3/7 to execute cell apoptosis [34]. The result indicated that linoleic acid-induced INS-1 cell apoptosis through mitochondria-regulated mechanisms.
Mitochondria-regulated mechanisms of the apoptotic process induced by linoleic acid
A variety of physiological cell death signals, as well as pathological cellular insults, trigger the genetically programmed pathway of apoptosis [35, 36]. Previous studies in several cellular models have implied that mitochondria have an important role in apoptosis induced by lipotoxicity and glucotoxicity [35]. To further determine whether linoleic acid induces cell apoptosis as a result of mitochondrial dysfunction, mitochondria-mediated apoptosis pathways were investigated, including Bcl-2 family gene expression, change of mitochondrial membrane potential (ΔΨm) and release of mitochondrial cytochrome c.
Linoleic acid increase Bax expression
Bcl-2 family members reside upstream of irreversible cellular damage and their effects are mostly located at the level of mitochondria [35, 37]. We investigated Bcl-2 and Bax expression to indicate whether different treatments truly induced mitochondria-mediated apoptosis. Compared with control, linoleic acid significantly increased Bax expression when administered at a dose of 500 μM in 25 mM glucose culture medium (Fig. 3b; P < 0.01). There was, however, no remarkable increase in 11.1 mM glucose culture medium (Fig. 3a), which reflected a close to normal glucose level in rats in vivo. In contrast, incubation with linoleic acid in 11.1 or 25 mM glucose medium had no significant alteration in Bcl-2 expression compared with control. These results indicated that linoleic acid may trigger mitochondrial dysfunction and apoptosis by upregulating Bax expression and abnormal glucose levels may further aggravate its cell toxicity.

Chronic exposure of INS-1 cells to linoleic acid increased Bax expression in 11.1 mM (a) and 25 mM (b) glucose culture media. Incubation with various concentration of linoleic acid caused dose-dependent increases in Bax expression compared with control; the increase was not significant with 11.1 mM but was significantly increased in the 500 μM linoleic treatment group in 25 mM culture medium. Values shown are the mean ± S.E. (N = 4). *P < 0.05 vs control; **P < 0.01 vs control
The loss of mitochondrial membrane potential
Change in mitochondrial membrane potential (ΔΨm) was determined using Rh 123-loaded cells. Rh 123 is a lipophilic dye that selectively concentrates (is quenched) in mitochondria due to their negative transmembrane potential relative to cytoplasm environments. When the mitochondrial trans-membrane potential is reduced, Rh 123 is released (or less quenched) into the cytoplasm and fluorescent level is increased. In other words, at a normal mitochondrial trans-membrane potential, most of the Rh 123 is concentrated in the mitochondrial matrix and quenched to have relatively low fluorescent signal. When ΔΨm is reduced due to dysfunction of mitochondria, Rh 123 is released, leading to an increase in Rh 123 fluorescence. Thus in this experiment, the loss of ΔΨm corresponds to a higher fluorescence density value of Rh 123 [38]. In our study, CCCP, a protonophore causing a complete disruption of the ΔΨm, was included as a positive control in every measurement to discriminate cells with already depolarized mitochondrial potential from healthy cells. Typical ΔΨm data from flow cytometry analysis are presented in Fig. 4. The MFI increased with increasing linoleic acid concentration. Under 11.1 mM glucose culture conditions (Fig. 4a), MFI significantly increased with 50, 250, and 500 μM (P < 0.001) linoleic acid treatment compared with control. When the glucose level was increased from 11.1 to 25 mM, the MFI of all three linoleic acid treatment groups significantly increased (P < 0.001) compared with the control group (Fig. 4b). Compared with apoptosis results (Fig. 2), the loss of ΔΨm occurred earlier and was more sensitive to linoleic acid treatment, even with lower levels of linoleic acid.

Chronic exposure of INS-1 cells to linoleic acid-induced a loss in mitochondrial membrane potential (ΔΨm). Cells were harvested after 48 h culture and incubated in suspension at room temperature with 1 μg/ml Rh 123 for 15 min. Mean fluorescent intensity (MFI) was assessed using BDLSII Flow Cytometry. Compared with control, both 11.1 mM (a) and 25 mM (b) glucose groups showed significant increases in MFI after 50, 250, and 500 μM linoleic acid treatment; 10 μM CCCP treatment was used as a positive control to make sure that INS-1 cell mitochondria show a good response to FFA treatment. Values shown are the mean ± S.E. (N = 4). ***P < 0.001 vs control
Cytochrome c release
Loss of mitochondrial membrane potential suggests that linoleic acid may induce a loss of mitochondrial function and increase mitochondrial membrane permeability, which promotes cytochrome c release into the cytoplasm, which has been implicated in the involvement of mitochondria-regulated apoptotic processes [39, 40]. Therefore, we investigated whether cytochrome c was released from the mitochondrial compartment to the cytosol in INS-1 β cells after chronic exposed to linoleic acid. Figure 5 shows cytochrome c levels assessed by western blot analysis when cells were incubated with 11.1 or 25 mM glucose together with 0 (control) or 500 μM linoleic acid. With increasing linoleic acid concentration, there was no significant change in measurable total cytochrome c (including mitochondria and cytosol) expression level in both 11.1 and 25 mM glucose medium. However, expression of cytochrome c in isolated mitochondria was reduced after treatment with 500 μM linoleic acid in the 11.1 mM glucose culture group and disappeared in the 25 mM glucose treatment group. The results indicated that the release of cytochrome c in the presence of linoleic acid was greater with increasing linoleic acid concentration, reaching a maximum after 48 h exposure with 500 μM group. There was a noticeable increase in intensity of the band in the cytosolic fractions, indicating that linoleic acid was able to accelerate cytochrome c release from mitochondria to cytosolic fractions and to trigger apoptosis via mitochondrial signaling.

Increased fatty acid and glucose levels promote cytochrome c expression and release in INS-1 cells. Loading control GAPDH (G), total protein (T) including mitochondrial and cytosolic protein, mitochondrial protein (M), and cytosolic protein (C) in the left panel were obtained from treated cells after 48 h incubation, as described in “Materials and methods” section. 40-μg protein samples were loaded on a 12% SDS-polyacrylamide gel and detected using a monoclonal cytochrome c antibody. The bands from 11.1 mM glucose treatment (a, total protein; b, cytosol protein) and 25 mM glucose treatment (c, total protein; d, cytosol protein) were quantified by densitometry (right panel). Experimental conditions were 11.1 or 25 mM glucose culture medium containing 500 μM linoleic acid with 0.5% BSA. The immunoblots shown are representative of three independent experiments (N = 4). *P < 0.05 vs control
Linoleic acid suppressed glucose-stimulated insulin secretion
Basal insulin secretion (3 mM glucose KRB buffer) and glucose-stimulated insulin secretion (25 mM glucose KRB buffer) from the INS-1 β cells were tested by radioimmunoassay. Insulin basal secretion level dose-dependently decreased after chronic exposure to linoleic acid and was significantly suppressed by 500 μM linoleic acid (P < 0.05) in both 11.1 and 25 mM glucose culture medium (Fig. 6a). Compared with 11.1 mM culture medium, the level of basal insulin secretion was reduced by long-term culture under high glucose conditions (25 mM glucose). Glucose-stimulated insulin secretion was markedly suppressed with 250 and 500 μM linoleic acid treatment in 11.1 glucose culture medium, which represented normal glucose culture conditions (Fig. 6b; P < 0.05). In the 25 mM glucose culture group, which mimics the high glucose conditions of T2D, the glucose-stimulated insulin secretion level was generally less than seen in the 11.1 mM glucose group and was markedly attenuated with 500 μM linoleic acid (Fig. 6b; P < 0.05). When the two glucose culture medium levels were compared, the 11.1 mM group, which mimicked the normal glucose level, had the highest insulin secretion levels. These results indicate that high glucose is harmful to INS-1 β-cell function and increased glucose suppresses linoleic acid-induced basal and glucose-stimulated insulin secretion.

Effects of chronic exposure of INS-1 cells to linoleic acid on basal and glucose-stimulated insulin secretion. Insulin secretion is shown as the value normalized to cell protein content. Basal insulin secretion (a) was tested in 3 mM glucose KRB buffer and showed dose-dependent decreases after exposure to linoleic acid and was significantly attenuated with 500 μM treatment. Glucose-stimulated insulin secretion (b) was tested in 25 mM glucose KRB buffer, and insulin secretion was markedly suppressed with 250 and 500 μM linoleic acid treatment in 11.1 mM glucose culture medium. In the 25 mM glucose culture group, significant suppression of insulin secretion was also found in 500 μM linoleic acid treatment group. Each value represents the mean ± S.E. (N = 6). *P < 0.05 vs control
Discussion
The purpose of thsi study was to investigate the hypothesis that chronic exposure to unsaturated FFAs in different concentrations of glucose initiates a cascade of changes inside pancreatic β cells that ultimately lead to mitochondrial dysfunction and apoptosis. This study demonstrated that mitochondria-mediated signal pathways contribute to INS-1 β-cell dysfunction and apoptosis induced by linoleic acid. The finding that cell viability is affected by low concentrations and reduced by high concentrations of linoleic acid was probably due to stimulation of cell apoptosis by high level of FFAs. In support of this mechanism, cells apoptosis was significantly increased with 500 μM linoleic acid treatment. High glucose combined with linoleic acid significantly increased cell apoptosis, mimicking obesity-related diabetes. Coincident with linoleic acid-induced apoptosis, Bax expression was significantly increased by linoleic acid when cells were treated with the combination of high glucose and linoleic acid concentrations. Linoleic acid also depolarized the ΔΨm and promoted cytochrome c release from mitochondria to the cytosol in INS-1 cells, clearly indicating mitochondrial dysfunction. In support of this, GSIS, a mitochondrial function-dependent insulin secretion, was significantly attenuated by treatment of cells with high levels of linoleic acid and/or glucose. High glucose conditions did not potentiate cell apoptosis but markedly affected insulin secretion function, which is dependent on mitochondrial function.
Exposure to FFAs and cytokines induces apoptosis of INS-1 β cells, which is similar to that observed in pancreatic islet β cells [21–23]. Therefore, mitochondria-mediated mechanisms involved in apoptosis were studied in INS-1 β cells. In addition, compared with isolated islet cells, the availability of large numbers of pure β-cell populations of this cell line allows RNA, protein, and hormone assays, all of which require large numbers of cells. Recent studies have indicated that FFAs have an important role in regulating β-cell function through mitochondria-mediated pathways [13], and several hypotheses have been proposed to explain lipotoxicity-induced β-cell dysfunction in vivo and in vitro [23, 41]. First, the inner mitochondrial membrane contains a group of pro-apoptotic factors, such as cytochrome c, which could be released from the mitochondria to the cytosol via mitochondrial permeability transition pore (MPTP) in FFA-induced mitochondrial dysfunction and cell apoptosis [42]. Second, mitochondrial DNA damage was reported to occur after chronic exposure of β cells to FFAs [21]. Chronic exposure of β cells to FFAs causes accumulation of intracellular lipid-derived signal molecules, which in turn generate reactive oxygen species (ROS) through the non-oxidation pathway [43]. At the same time, mitochondrial DNA is more vulnerable to ROS damage owing to low expression levels of DNA repair enzymes in the mitochondria [44, 45]. Third, evidence for the involvement of mitochondria in FFA-induced apoptosis has come from the studies of anti-apoptotic genes (such as Bcl-2 and Bcl-XL) and pro-apoptotic genes (such as Bax) of the Bcl-2 family, which could determine the fate of cells to a certain extent [35]. These studies indicate that mitochondrial dysfunction maybe a causative factor of FFA-induced β-cell apoptosis. This study demonstrated that polyunsaturated linoleic acid induces apoptosis and mitochondrial dysfunction by collapsing mitochondrial membrane potential and the loss of capacity to retain Rh 123 after linoleic acid treatment, confirming disturbed mitochondrial function. This, in turn, led to the release of cytochrome c from the mitochondria to the cytosolic space, which then initiated caspase-mediated apoptosis.
The underlying mechanism of mitochondria-mediated β-cell dysfunction, especially by unsaturated FFAs, has not yet been fully determined. Different forms of FFAs have different patterns of apoptosis after chronic exposure [8, 17]. Numerous studies have focused on the effects of saturated FFAs, such as palmitate acid, which markedly induce apoptosis, but little research has focused on the action of unsaturated FFAs [46, 47]. Linoleic acid (18:2, n-6) is an essential fatty acid, and is the main fatty acid in a “Modern Western Diet”. Clinical research has indicated that excessive levels of n-6 fatty acids may increase the risk of obesity and cancer [48]. Although there is little direct and systematic evidence for the role of mitochondria in unsaturated FFA-induced apoptosis, our study provides some clues regarding this process.
Bcl-2 family members reside upstream of irreversible cellular damage and have an important role in regulating cell apoptosis, particularly at the level of mitochondria [35, 49]. It is accepted that the mitochondrial apoptosis pathway begins with permeabilization of the mitochondrial outer membrane, but the mechanisms by which this occurs remain controversial [50]. Previous studies have suggested that mitochondria-dependent apoptosis is hierarchically regulated by Bcl-2 subfamilies, which have major sites of activity on the mitochondrial membrane, alter the permeabilization and disrupt ΔΨm [51]. Comparisons of the expression levels of Bcl-2 and Bax expression after linoleic acid treatment showed an increase in Bax expression with 500 μM linoleic acid treatment in 25 mM glucose medium (Fig. 3b) but not in 11.1 mM glucose; there was also no significant change in Bcl-2 expression, suggesting that linoleic acid may affect downstream mitochondrial function through regulation of Bax activity or translocation. The increase in Bax expression may translocate to the mitochondrial membrane and form the mitochondrial trans-membrane pores; further increases lead to the reduction in ΔΨm and the release of cytochrome c from the mitochondria.
Consistent with this hypothesis, a reduction in ΔΨm was found after linoleic acid treatment in all linoleic acid treatment groups. Previous studies have suggested that loss of ΔΨm may be an early but reversible event of mitochondria-mediated apoptosis [52, 53]. In our study, 50 and 250 μM linoleic acid did not induce apoptosis, but they showed loss of ΔΨm after chronic exposure. These results further indicate that loss of ΔΨm is an early apoptosis event under the conditions of this study. Furthermore, loss of ΔΨm has been demonstrated to be a key regulator of cytochrome c release. After 500 μM linoleic acid treatment, cytochrome c was released from the mitochondria to the cytosolic compartment. Upon receiving an apoptotic stimulus, cytochrome c is released from the intermembrane space of mitochondria into the cytoplasm. Once in the cytosol, cytochrome c binds tightly to Apaf-1 and forms an apoptosome. The only known function of the apoptosome is to recruit and facilitate activation of procaspase-9. Once activated, caspase-9 stays with the apoptosome as a holoenzyme to maintain its catalytic activity and activate caspase-3, which leads to irreversible cell apoptosis [34]. In our study, we found that caspase-9 and caspase-3/7 activity was significantly increased after 500 μM linoleic acid incubation (Fig. 2b, c). These findings indicate that the apoptosis induced by linoleic acid is linked to mitochondrial dysfunction and mediates apoptotic signal pathways.
Increased glucose concentrations potentiated linoleic acid-induced mitochondrial dysfunction and further impaired INS-1 β-cell insulin secretion in this study, but had little effect on β-cell apoptosis. With the increase in glucose concentration in culture medium, linoleic acid-induced apoptosis when administered at a dose of 500 μM at both the normal and high glucose levels. However, compared with normal glucose culture conditions, basal insulin secretion and GSIS level were attenuated. The combination of high glucose culture conditions and high FFA concentrations had significant synergistic effects. Although the underlying mechanism of such synergistic effects of glucose and FFAs is not yet fully understood, some hypotheses have been proposed recently. It is well accepted that glucose and FFAs counteract each other during energy storage and metabolism [54, 55]. Glucose is phosphorylated by glucokinase (GK) and converted to pyruvate by glycolysis. Pyruvate preferentially enters the mitochondria and feeds the tricarboxylic acid (TCA) cycle, resulting in the transfer of reducing equivalents (NADPH and FADH) to the electron transport chain, leading to hyperpolarization of the mitochondrial membrane and generation of ATP. Subsequently, closure of the KATP-channels depolarizes the cytosolic membrane, which opens voltage-gated Ca2+ channels, increasing intracellular calcium levels and triggering insulin exocytosis [54]. FFAs penetrate the plasma membrane and react with ATP to give fatty acyl adenylate, which reacts with free coenzyme A to generate fatty acyl-CoA. Acyl-CoA is then cleaved to acetyl-CoA units plus reducing equivalents (NADPH and FADH) to feed the TCA cycle and the electron transport chain via mitochondrial β-oxidation to stimulate insulin secretion [43]. Under physiological conditions, glucose provision promotes glucose oxidation and storage, and inhibits fatty acid oxidation; increased FFA levels promote fatty acid oxidation and storage, and inhibit glucose oxidation [3]. When this balance is disrupted by an excess of FFAs, excess intracellular glucose (which is not stored as glycogen or oxidized via glycolytic pathways) enters the lipogenic pathways as acetyl-CoA, to undergo carboxylation to malonyl-CoA, which is an intermediate in the synthesis of fatty acids, triacylglycerol, and ceramides [56]. In addition to this, malonyl-CoA can inhibit CPT-1 to obstruct FFA β-oxidation, which further enhances the lipotoxicity of FFAs. Furthermore, β cells are extremely susceptible to hyperglycemia-induced ROS damage because they exhibit low levels of free radical quenching enzymes [45]. Although there is increasing evidence for this molecular mechanism, it remains to be determined how excess FFAs and glucose cause β-cell dysfunction.
The results of this study demonstrate that linoleic acid regulates mitochondrial signal pathways, which lead to an increase in Bax expression, a collapse of ΔΨm, release of the pro-apoptotic factor mitochondrial cytochrome c, activation of caspase-9 and 3/7 activity, and finally reduced levels of both basal insulin secretion and glucose-stimulated insulin secretion. Glucose levels greater than normal potentiate the lipotoxicity of linoleic acid on β cells in vitro.
References
B.B. Lowell, G.I. Shulman, Mitochondrial dysfunction and type 2 diabetes. Science 307, 384–387 (2005)
C.M. Kusminski, S. Shetty, L. Orci, R.H. Unger, P.E. Scherer, Diabetes and apoptosis: lipotoxicity. Apoptosis 14, 1484–1495 (2009)
P.J. Randle, Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab. Rev. 14, 263–283 (1998)
L.O. Schulz, P.H. Bennett, E. Ravussin, J.R. Kidd, K.K. Kidd, J. Esparza, M.E. Valencia, Effects of traditional and western environments on prevalence of type 2 diabetes in Pima Indians in Mexico and the U.S. Diabetes Care 29, 1866–1871 (2006)
V. Poitout, R.P. Robertson, Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr. Rev. 29, 351–366 (2008)
R.H. Unger, L. Orci, Diseases of liporegulation: new perspective on obesity and related disorders. FASEB J. 15, 312–321 (2001)
R.H. Unger, Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol. Metab. 14, 398–403 (2003)
E. Lai, G. Bikopoulos, M.B. Wheeler, M. Rozakis-Adcock, A. Volchuk, Differential activation of ER stress and apoptosis in response to chronically elevated free fatty acids in pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 294, E540–E550 (2008)
J.L. Evans, I.D. Goldfine, B.A. Maddux, G.M. Grodsky, Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 52, 1–8 (2003)
P. Newsholme, E.P. Haber, S.M. Hirabara, E.L. Rebelato, J. Procopio, D. Morgan, H.C. Oliveira-Emilio, A.R. Carpinelli, R. Curi, Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J. Physiol. 583, 9–24 (2007)
A.I. Oprescu, G. Bikopoulos, A. Naassan, E.M. Allister, C. Tang, E. Park, H. Uchino, G.F. Lewis, I.G. Fantus, M. Rozakis-Adcock, M.B. Wheeler, A. Giacca, Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes 56, 2927–2937 (2007)
J.L. Evans, B.A. Maddux, I.D. Goldfine, The molecular basis for oxidative stress-induced insulin resistance. Antioxid. Redox Signal. 7, 1040–1052 (2005)
G. Szabadkai, M.R. Duchen, Mitochondria mediated cell death in diabetes. Apoptosis 14, 1405–1423 (2009)
E. Karaskov, C. Scott, L. Zhang, T. Teodoro, M. Ravazzola, A. Volchuk, Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 147, 3398–3407 (2006)
I. Kharroubi, L. Ladriere, A.K. Cardozo, Z. Dogusan, M. Cnop, D.L. Eizirik, Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 145, 5087–5096 (2004)
K. Eitel, H. Staiger, M.D. Brendel, H. Brandhorst, R.G. Bretzel, H.U. Haring, M. Kellerer, Apoptosis induced by free fatty acids. Med. Klin. 98, 248–252 (2003)
T. Kawai, H. Hirose, Y. Seto, H. Fujita, T. Saruta, Chronic effects of different fatty acids and leptin in INS-1 cells. Diabetes Res. Clin. Pract. 51, 1–8 (2001)
I. Maestre, J. Jordan, S. Calvo, J.A. Reig, V. Cena, B. Soria, M. Prentki, E. Roche, Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawal and fatty acids in the beta-cell line INS-1. Endocrinology 144, 335–345 (2003)
M. Cnop, J.C. Hannaert, D.G. Pipeleers, Troglitazone does not protect rat pancreatic beta cells against free fatty acid-induced cytotoxicity. Biochem. Pharmacol. 63, 1281–1285 (2002)
F.T. Schulthess, S. Katz, A. Ardestani, H. Kawahira, S. Georgia, D. Bosco, A. Bhushan, K. Maedler, Deletion of the mitochondrial flavoprotein apoptosis inducing factor (AIF) induces beta-cell apoptosis and impairs beta-cell mass. PLoS One 4, e4394 (2009)
V. Grishko, L. Rachek, S. Musiyenko, S.P. Ledoux, G.L. Wilson, Involvement of mtDNA damage in free fatty acid-induced apoptosis. Free Radic. Biol. Med. 38, 755–762 (2005)
L.G. Grunnet, R. Aikin, M.F. Tonnesen, S. Paraskevas, L. Blaabjerg, J. Storling, L. Rosenberg, N. Billestrup, D. Maysinger, T. Mandrup-Poulsen, Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta-cells. Diabetes 58, 1807–1815 (2009)
L.I. Rachek, N.P. Thornley, V.I. Grishko, S.P. LeDoux, G.L. Wilson, Protection of INS-1 cells from free fatty acid-induced apoptosis by targeting hOGG1 to mitochondria. Diabetes 55, 1022–1028 (2006)
L.K. Olson, J. Qian, V. Poitout, Glucose rapidly and reversibly decreases INS-1 cell insulin gene transcription via decrements in STF-1 and C1 activator transcription factor activity. Mol. Endocrinol. 12, 207–219 (1998)
S.P. Cousin, S.R. Hugl, C.E. Wrede, H. Kajio, M.G. Myers Jr., C.J. Rhodes, Free fatty acid-induced inhibition of glucose and insulin-like growth factor I-induced deoxyribonucleic acid synthesis in the pancreatic beta-cell line INS-1. Endocrinology 142, 229–240 (2001)
M. Cnop, J.C. Hannaert, A. Hoorens, D.L. Eizirik, D.G. Pipeleers, Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes 50, 1771–1777 (2001)
F. Frigerio, T. Brun, C. Bartley, A. Usardi, D. Bosco, K. Ravnskjaer, S. Mandrup, P. Maechler, Peroxisome proliferator-activated receptor alpha (PPARalpha) protects against oleate-induced INS-1E beta cell dysfunction by preserving carbohydrate metabolism. Diabetologia 53, 331–340 (2010)
C. Warnotte, M. Nenquin, J.C. Henquin, Unbound rather than total concentration and saturation rather than unsaturation determine the potency of fatty acids on insulin secretion. Mol. Cell. Endocrinol. 153, 147–153 (1999)
X. Du, D. Edelstein, S. Obici, N. Higham, M.H. Zou, M. Brownlee, Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J. Clin. Invest. 116, 1071–1080 (2006)
A.K. Cardozo, P. Proost, C. Gysemans, M.C. Chen, C. Mathieu, D.L. Eizirik, IL-1beta and IFN-gamma induce the expression of diverse chemokines and IL-15 in human and rat pancreatic islet cells, and in islets from pre-diabetic NOD mice. Diabetologia 46, 255–266 (2003)
A.M. Brambrink, A. Schneider, H. Noga, A. Astheimer, B. Gotz, I. Korner, A. Heimann, M. Welschof, O. Kempski, Tolerance-Inducing dose of 3-nitropropionic acid modulates bcl-2 and bax balance in the rat brain: a potential mechanism of chemical preconditioning. J. Cereb. Blood Flow Metab. 20, 1425–1436 (2000)
J.E. Brown, S.J. Dunmore, Leptin decreases apoptosis and alters BCL-2: Bax ratio in clonal rodent pancreatic beta-cells. Diabetes Metab. Res. Rev. 23, 497–502 (2007)
G. Juan, M. Cavazzoni, G.T. Saez, J.E. O’Connor, A fast kinetic method for assessing mitochondrial membrane potential in isolated hepatocytes with rhodamine 123 and flow cytometry. Cytometry 15, 335–342 (1994)
A. Masud, S.A. Lakhani, K. Kuida, G.A. Porter Jr, C.J. Booth, W.Z. Mehal, I. Inayat, R.A. Flavell, Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311, 847–851 (2006)
A. Gross, J.M. McDonnell, S.J. Korsmeyer, BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13, 1899–1911 (1999)
D.L. Vaux, S.J. Korsmeyer, Cell death in development. Cell 96, 245–254 (1999)
H. Kim, Y.N. Kim, C.W. Kim, Oxidative stress attenuates Fas-mediated apoptosis in Jurkat T cell line through Bfl-1 induction. Oncogene 24, 1252–1261 (2005)
H.J.M. Brady, Apoptosis methods and protocols (Humana Press, Totowa, NJ, 2004)
D. Zhang, C. Lu, M. Whiteman, B. Chance, J.S. Armstrong, The mitochondrial permeability transition regulates cytochrome c release for apoptosis during endoplasmic reticulum stress by remodeling the cristae junction. J. Biol. Chem. 283, 3476–3486 (2008)
T. Eissing, S. Waldherr, F. Allgower, P. Scheurich, E. Bullinger, Response to bistability in apoptosis: roles of bax, bcl-2, and mitochondrial permeability transition pores. Biophys. J. 92, 3332–3334 (2007)
T. Kudo, J. Wu, Y. Ogawa, S. Suga, N. Hasegawa, T. Suda, H. Mizukami, S. Yagihashi, M. Wakui, Novel mechanism of chronic exposure of oleic acid-induced insulin release impairment in rat pancreatic beta-cells. J. Pharmacol. Exp. Ther. 318, 1203–1210 (2006)
A. Sultan, P.M. Sokolove, Free fatty acid effects on mitochondrial permeability: an overview. Arch. Biochem. Biophys. 386, 52–61 (2001)
C.J. Nolan, M.S. Madiraju, V. Delghingaro-Augusto, M.L. Peyot, M. Prentki, Fatty acid signaling in the beta-cell and insulin secretion. Diabetes 55(Suppl 2), S16–S23 (2006)
V.A. Bohr, T. Stevnsner, N.C. de Souza-Pinto, Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene 286, 127–134 (2002)
S. Lenzen, Oxidative stress: the vulnerable beta-cell. Biochem. Soc. Trans. 36, 343–347 (2008)
K. Maedler, J. Oberholzer, P. Bucher, G.A. Spinas, M.Y. Donath, Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 52, 726–733 (2003)
L.L. Listenberger, X. Han, S.E. Lewis, S. Cases, R.V. Farese Jr., D.S. Ory, J.E. Schaffer, Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl Acad. Sci. USA 100, 3077–3082 (2003)
M. Rodriguez-Cruz, A.R. Tovar, M. del Prado, N. Torres, Molecular mechanisms of action and health benefits of polyunsaturated fatty acids. Rev. Invest. Clin. 57, 457–472 (2005)
J.M. Jurgensmeier, Z. Xie, Q. Deveraux, L. Ellerby, D. Bredesen, J.C. Reed, Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl Acad. Sci. USA 95, 4997–5002 (1998)
D.R. Green, Apoptotic pathways: ten minutes to dead. Cell 121, 671–674 (2005)
H. Kim, M. Rafiuddin-Shah, H.C. Tu, J.R. Jeffers, G.P. Zambetti, J.J. Hsieh, E.H. Cheng, Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 8, 1348–1358 (2006)
P. Bernardi, The permeability transition pore. Control points of a cyclosporin A-sensitive mitochondrial channel involved in cell death. Biochim. Biophys. Acta 1275, 5–9 (1996)
D.R. Green, G. Kroemer, The pathophysiology of mitochondrial cell death. Science 305, 626–629 (2004)
P. Maechler, C.B. Wollheim, Mitochondrial signals in glucose-stimulated insulin secretion in the beta cell. J. Physiol. 529(Pt 1), 49–56 (2000)
C.B. Wollheim, P. Maechler, Beta-cell mitochondria and insulin secretion: messenger role of nucleotides and metabolites. Diabetes 51(Suppl 1), S37–S42 (2002)
C.M. Kusminski, S. Shetty, L. Orci, R.H. Unger, P.E. Scherer, Diabetes and apoptosis: lipotoxicity. Apoptosis 14, 1484–1495 (2009)
Acknowledgments
The authors would like to acknowledge scientific contribution of Drs SH Lee and F Steyn, and editorial help of Mr M Sinclair. Ya Tuo is a recipient of overseas post-graduate research scholarship from the China Scholarship Council (CSC, China) and subsidy scholarship by the University of Queensland; and research was supported by Australian NHMRC and the University of Queensland
Conflict of interest
The authors have nothing to disclose.
Author information
Authors and Affiliations
Corresponding author
Additional information
Shengbin Li and Chen Chen contributed equally to this work.
Rights and permissions
About this article
Cite this article
Tuo, Y., Wang, D., Li, S. et al. Long-term exposure of INS-1 rat insulinoma cells to linoleic acid and glucose in vitro affects cell viability and function through mitochondrial-mediated pathways. Endocr 39, 128–138 (2011). https://doi.org/10.1007/s12020-010-9432-3
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-010-9432-3