
Abstract
MicroRNAs (miRNAs) regulate gene expression post-transcriptionally by interacting with the 3′ untranslated regions of their target mRNAs. Previously, miRNAs have been shown to regulate genes involved in cell growth, apoptosis, and differentiation, but their role in ovarian granulosa cell follicle-stimulating hormone (FSH)-stimulated steroidogenesis is unclear. Here we show that expression of 31 miRNAs is altered during FSH-mediated progesterone secretion of cultured granulosa cells. Specifically, 12 h after FSH treatment, miRNAs mir-29a and mir-30d were significantly down-regulated. However, their expression increased after 48 h. Bioinformatic analysis used to predict potential targets of mir-29a and mir-30d revealed a wide array of potential mRNA target genes, including those encoding genes involved in multiple signaling pathways. Taken together, our results pointed to a novel mechanism for the pleiotropic effects of FSH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The primary trigger of immature preantral follicle development into a preovulatory follicle is follicle-stimulating hormone (FSH) stimulation of granulosa cell differentiation [1]. FSH controls the development of granulosa cells during folliculogenesis by stimulating their proliferation and differentiation, and by promoting the formation of the follicular antrum [2]. In particular, FSH enhances the biosynthesis of female sex hormones estradiol and progesterone, which control the estrous cycle and reproduction [3–5]. FSH transcriptionally regulates multiple granulosa cell genes during FSH-induced steroidogenesis, including decay accelerating factor GPI-form precursor (DAF), insulin-like growth factor binding protein 3 (IGFBP3), steroidogenic acute regulatory protein (StAR), bone morphogenetic proteins (BMPs) [6–9]. Previously, we demonstrated that the expression of some microRNAs (miRNAs) (let-7a, mir-125b and mir-143) were inhibited in FSH-treated KK-1 cells [10]. However, the regulation of primary granulosa cell miRNA expression by FSH has not been thoroughly investigated.
miRNAs are highly conserved, ~21 nt long RNA molecules that post-transcriptionally regulate the expression of mRNAs via interaction with their 3′ untranslated regions (3′ UTR) [11]. miRNAs have been implicated in many processes in vertebrates, including cell proliferation, apoptosis, fat metabolism, and neuronal patterning [12]. Moreover, some miRNAs have been shown to differentially regulate translation, inhibiting under conditions of active cell proliferation and promoting under conditions of cell cycle arrest [13]. Though miRNAs have been implicated in hormonal regulation [14–16], the effect of FSH on regulation of miRNAs in ovarian granulosa cells is still unclear. Therefore, we have identified several FSH-regulated miRNAs in primary rat granulosa cells treated with FSH for 12 h. Additionally, we have examined the expression of two miRNAs significantly down-regulated by FSH, and have analyzed their potential targets.
Results
Identification of miRNA expression in FSH-treated granulosa cells
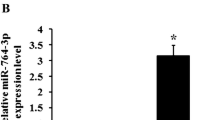
Microarray analysis of granulosa cell miRNA expression revealed that 260 and 236 detectable miRNA transcripts were present after 0 and 12 h of FSH treatment, respectively, in at least 2 of 3 samples (n = 3). Each miRNA transcript was categorized based on its fluorescence levels, which were indicative of its relative expression level within the cells. We therefore compared the expression data of the 0 and 12 h conditions to determine the effect of FSH treatment on miRNA expression. Differentially expressed miRNAs in each chip were listed separately with P value < 0.01 by t test. miRNAs differentially expressed significantly in at least 2 of 3 chips were listed in Table 1 with mean values of log2ratio. A positive log2 value indicates the up-regulation of miRNA expression and a negative log2 value indicates down-regulation. After 12 h of FSH exposure, 17 miRNAs were up-regulated and 14 miRNAs were down-regulated (Table 1). Progesterone levels in the medium were up-regulated by FSH at 12 h (Fig. 1a). Three miRNAs (mir-23b, mir-29a and mir-30d), all of which showed stably significant change under FSH stimulation, were detected by qRT–PCR to validate the microarray results. Mir-23b was up-regulated between 0 and 12 h of FSH treatment (fold change of 1.32), whereas mir-29a and mir-30d were down-regulated during the same period (fold change of 0.48 and 0.60, respectively) in the microarray results. These results were validated by qRT–PCR, and the fold change expression seen for the mature forms of mir-23b (1.94 ± 0.42), mir-29a (0.51 ± 0.09) and mir-30d (0.54 ± 0.03) (Fig. 1b–d) were similar to the expression changes predicted by miRNA microarray analysis.

miRNA expression profiling of granulosa cells after 12 h of FSH treatment (50 ng/ml). a RIA analysis of progesterone production after 12 h FSH treatment. Data were shown as mean ± S.E. (n = 3). Granulosa cell samples treated without FSH were used as controls. * P < 0.05 compared with controls by t test. b, c, d qRT-PCR analyses of mir-23b (b), mir-29a (c), or mir-30d (d). Expression was normalized to U6, and granulosa cell samples treated without FSH were used as controls. Data were shown as mean ± S.E. (n = 3). ** P < 0.01, *** P < 0.001 compared with controls by t test
FSH regulation of mir-29a and mir-30d
Examination of mir-29a and mir-30d levels demonstrated that their expression was down-regulated 12 h post-FSH treatment. To better understand the potential role of miRNAs on FSH-induced progesterone production of cultured granulosa cell, we next investigated the expression of mir-29a and mir-30d after 0, 6, 12, 24, and 48 h of FSH treatment. The time course of mir-29a (Fig. 2a) and mir-30d (Fig. 2b) expression levels during FSH-induced progesterone production of cultured granulosa cells revealed a significant decrease in their expression during the first 12 h. Subsequently, FSH induced a 2 and 3-fold increase in expression of mir-29a and mir-30d at 48 h, respectively.

qRT-PCR analysis of mir-29a expression (a) and mir-30d expression (b) in rat granulosa cells treated with FSH (50 ng/ml) for 0, 6, 12, 24, or 48 h. Data were shown as mean ± S.E. (n = 3). Different letters denote statistical significance (P < 0.05) by one-way ANOVA
Bioinformatic analysis of potential mir-29a and mir-30d targets
We utilized three different software suites to identify potential targets of mir-29a and mir-30d. The complete results of these analyses are shown in Table S1 and S2. A total of 222 potential mir-29a targets and 294 potential mir-30d targets were predicted by at least two of the analysis programs. These predicted target genes were then mapped to the Gene Ontology (GO) function and KEGG pathway databases. Of the predicted mir-29a targets, “Collagen” was the most enriched GO function (P = 3.84E-18) (Fig. 3a). In addition, five KEGG pathways (extracellular matrix (ECM)-receptor interaction, P = 0.0002; Focal adhesion, P = 0.0009; Cell Communication, P = 0.0334; Melanoma, P = 0.0355; and Small cell lung cancer, P = 0.0429) (Fig. 4a) were significantly enriched. Furthermore, for the potential targets of mir-30d, five GO functions (protein binding, P = 1.78E-08; retinoic acid receptor activity, P = 3.08E-07; dystrophin-associated glycoprotein complex, P = 4.18E-07; positive regulation of transcription from RNA polymerase II promoter, P = 3.15E-06; neurofilament, P = 4.16E-06), and two KEGG pathways (Circadian rhythm, P = 0.0108; O-Glycan biosynthesis, P = 0.0202) were significantly enriched in (Figs. 3b, 4b).

Targets of mir29a and mir-30d mapping in a function enrichment graph. Each bar represents the targets of mir-29a (a) or mir-30d (b), which were annotated according to the GO database. Bars with values above the threshold line are significant (P < 0.05). CC cellular component, MF molecular function, BP biological process

Targets of mir29a (a) and mir-30d (b) mapped in a pathway enrichment graph. Each bar represents the targets of mir-29a (a) or mir-30d (b), which were annotated according to the KEGG database. Bars with values above the threshold line are significant (P < 0.05)
Protein expression levels of potential mir-29a and mir-30d targets in FSH-treated granulosa cells
There are several putative targets regulated by mir-29a (Table S1) and mir-30d (Table S2). To gain insight into the possible targets of mir-29a and mir-30d, two respective potential targets of mir-29a (Col4A1 and BMF) and mir-30d (RNF2 and EED) were examined to determine if they were differently expressed in granulosa cells after FSH treatment (Fig. 5). Levels of these possible targets decreased in response to FSH treatment, almost consistent with the expression of mir-29a and mir-30d. These results suggested that mir-29a and mir-30d may contribute to translational repression of these putative targets in granulosa cells after FSH treatment.

The expression profiling of potential mir-29a (a) and mir-30d (b) targets after treatment of 50 ng/ml FSH for 0, 6, 12, 24, 48 h. Similar results were obtained in three different replicates
Discussion
Constitutive miRNA expression is employed by differentiating cells to tightly control the expression levels of specific genes [17]. However, while the role of miRNAs themselves is well established, the mechanisms controlling the regulation of miRNA expression are more obscure. Granulosa cell is likely a prime target for miRNAs mediated changes in gene expression due to the rapid conversion this cell must undergo following the FSH exposure. This phenotypic change contains the FSH-induced estradiol and progesterone production and its attendant gene expression. It has been reported that the function of ovarian corpus luteum may be regulated by miRNAs, suggesting that the secretion of progesterone is associated with miRNAs [18]. In this study, we investigated the differential expression pattern of miRNAs in the process of FSH-induced progesterone production of cultured rat granulosa cell.
We identified 260 mature miRNA transcripts in rat granulosa cells via microarray analysis, which is quite different from the previous result containing 122 miRNAs cloned from whole ovarian tissues [19]. The three differentially expressed miRNAs detected in our experiments (mir-23b, mir-29a, and mir-30d) were also detected as clones in the Ro et al. study. While this validates our findings, this was expected, given that granulosa cells account for a high proportion of ovarian cells. Furthermore, our results are similar to a previous microarray study which identified 212 miRNAs expressed in mouse granulosa cells [15]. Taken together, we believe that miRNA microarray approach may provide more extensive result of miRNA expression in primary cultured granulosa cells compared with cloning.
MiRNAs typically function as negative regulators of a set of target genes. The spatiotemporal expression of miRNAs is in part regulated by transcription factors that bind miRNA promoters [20–22]. Recent studies have demonstrated that miRNAs and transcription factors can form feedback or feedforward loops, which play critical roles in various biological processes [20, 23–26]. These miRNAs is regulated in several cell types, where they control multiple processes. For example, their mis-expression was previously shown to effect cancer [27, 28], cardiovascular disease [29], diabetes [30, 31], and lung development [32]. Recent reports also indicate that both mir-29a and mir-30d are induced by high glucose levels and may regulate insulin production [30, 31], suggesting that their expression may be associated with endocrine signaling. The wide-ranging role of these miRNAs, combined with their temporally specific induction by FSH, suggested that they are important in the process of FSH-induced progesterone production in cultured granulosa cells. The physiological responses to FSH are accomplished by the activation of many different target genes in granulosa cells, including intracellular signaling proteins, transcription factors, cell cycle proteins, and autocrine factors [33]. Here, a time course study of mir-29a and mir-30d indicated that the expression of these miRNAs changed significantly in response to FSH, which suggested that these miRNAs, as a part of gene regulatory networks, is fine-tuned in FSH-medicated progesterone biosynthesis of granulosa cells.
We have also mapped the targets of mir-29a and mir30d to gene function and signaling pathway databases. This approach clearly illustrated that the potential targets of these miRNAs perform integrated regulatory functions. The mRNA targets of mir-29a were primarily associated with ECM-receptor interaction, focal adhesion and cell communication. For example, the most enriched GO function of potential mir-29a targets was “collagen.” Previous study demonstrated that signaling through adhesion molecules such as collagen increased FSH receptor expression and progesterone production in granulosa cells from immature porcine ovarian follicles [34]. Nakano et al. reported that the granulosa cell layer of the mouse ovarian follicle has a basement membrane like matrix containing Col4A1, Col4A2, Col4A5, Col4A6, and the expression of type IV collagen shows the decreasing trend in mouse follicular development [35]. Here we found that the expression pattern of Col4A1 was consistent with mir-29a expression with FSH treatment broadly. These results imply that cell-to-cell communication, a characteristic feature of the cumulus-oocyte complex [36], may be affected by mir-29a. Polycomb group (PcG) complexes maintain epigenetically repressed states that need to be reprogrammed when cells become committed to differentiation [37]. Our findings showed that the kinetics of both RNF2 and EED, as members of PcG family [38], down-regulated in granulosa cells during FSH-treated parallels the upregulation of mir-30d. This implies that mir-30d may play a role in the progesterone production by repressing the PcG members. We can reasonably conclude, then, that FSH-induced miRNA expression is likely to contribute to the pleiotropic effects of FSH on a variety of cellular and physiological processes. However, although we have provided novel information regarding the link between FSH and miRNA expression, future study will be needed to determine the direct link between these miRNAs, their downstream targets, and the resulting consequences for granulosa cell steroidogenesis.
Taken together, we identified differential miRNAs during the process of FSH-induced progesterone production of cultured rat granulosa cells. These results provided a clue to understanding the molecular mechanism of FSH-stimulated progesterone biosynthesis in primary granulosa cells.
Materials and methods
Animals and granulosa cell isolation
Female Sprague–Dawley rats (23 days old) were purchased from the Department of Laboratory Animal Science of Peking University (Beijing, CN). All experiments in this study were conducted in accordance with the requirements established under the Guide for the Care and Use of Laboratory Animals. The animals were injected intraperitoneally with 20 IU pregnant mare serum gonadotropin (PMSG) to stimulate follicular growth. After 48 h, the animals were killed and ovaries were collected. Granulosa cells were harvested by puncturing the individual follicle with 25 gauge needles and cultured in a humidified incubator at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium: Nutrient Mixture F-12 (DMEM/F12) (Hyclone, Logan, UT) supplemented with 15% fetal bovine serum (Hyclone) for 1 day before treatment. Cells were serum-starved for 16 h and subsequently incubated with 50 ng/ml FSH (Sigma, Saint Louis, USA) for selected time intervals.
MiRNA microarray and temporal expression analysis
All procedures were carried out according to the manufacturer’s protocol. Briefly, total RNA was extracted from the cells by using a combination of Trizol (Invitrogen, Carlsbad, CA) and RNeasy mini kit (Qiagen, Valencia, CA). Total RNA was quantified using a NanoDrop-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) and quality was assessed by analysis of 18S and 28S peaks on an Agilent 2001 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Three independent replicates of rat granulosa cells stimulated with or without FSH were collected and analyzed by microarray analysis (LC Sciences, Houston, TX). 2 to 5 μg total RNA was size fractionated using a YM-100 Microcon centrifugal filter (Millipore, Billerica, MA) and the small RNAs (<300 nt) were 3′-extended with a poly(A) tail using poly(A) polymerase. Granulosa cells without FSH were labeled with Cy3, and FSH-stimulated granulosa cells (50 ng/ml for 12 h) were labeled with Cy5. Each granulosa cells replicate with or without FSH was then hybridized as a pair onto an individual Atactic μParaFlo microfluidics chip (LC Sciences, Houston, TX) according to the manufacturer’s instructions. Hybridization was performed in 100 μl 6× SSPE buffer (0.90 M NaCl, 60 mM Na2HPO4, 6 mM EDTA, pH 6.8) containing 25% formamide at 34°C. Hybridization detection was performed using tag-specific Cy3 and Cy5 dyes. Data were analyzed by subtracting the background and normalizing the signals using a LOWESS filter (Locally weighted Regression). For two color experiments, the ratio of the two sets of detected signals (log2 transformed, balanced) and P values of the t test were calculated. Differentially detected signals were those with P values of less than 0.01.
Progesterone assay
Granulosa cells (5 × 105 cells/vial) were cultured in a 24-well plate with 500 μl medium. Cells were treated with or without FSH (50 ng/ml) for 12 h. Media was then collected and stored at −20°C. Media progesterone levels were measured by standard radioimmunoassay (RIA) procedures as previously described [39].
Quantitative RT-PCR for single miRNAs
Quantitative RT-PCR (qRT-PCR) was performed using mature miRNA assay kits for specific miRNAs (Applied Biosystems, Foster City, CA). Total RNA was diluted to a 100 ng/μl working dilution, and 500 ng was reverse transcribed according to the manufacturer’s protocol, with the following modifications. Briefly, 500 ng of total RNA was mixed with 1 U MultiScribe Reverse Transcriptase, 0.25 U RNase Inhibitor, 3 μl hairpin-loop miRNA-specific RT primer, 1 mM dNTPs, and 1× Reverse Transcription Buffer in a total volume of 15 μl. The RT primer contained DNA sequence complementary to the 3′ end of the miRNA to be assayed, and additional DNA sequence at the 5′ hairpin-loop side of the primer. This sequence provided an additional priming site for PCR amplification of the first strand cDNA from the miRNA template. The mixture was incubated at 16°C for 30 min, 42°C for 30 min, and stopped by heating to 85°C for 5 min. Real-time PCR was performed using the Applied Biosystems 7000 Sequence Detection System. 20 μl PCR reaction mixtures included 1.33 μl of RT product, 1× TaqMan Universal PCR Master Mix and 1 μl of primers according to the TaqMan MicroRNA Assay protocol (PE Applied Biosystems). After activation of the AmpliTaq Gold DNA polymerase at 95°C for 10 min, 40 cycles of two-step PCR were run (95°C for 15 s and 60°C for 60 s). Data were collected and analyzed with SDS v.2.2.2 software (Applied Biosystems). The threshold cycle data were determined using default threshold settings. The expression levels of the miRNAs were normalized against the housekeeping gene U6, and presented as the mean normalized expression level. The Ct values for U6 were analyzed by one-way ANOVA, and there is no significant difference between groups treated with or without FSH (P > 0.05). Error bars denote standard error of the mean of three replicate experiments.
Mir-29a and mir-30d potential target prediction
Three miRNA target prediction software programs, TargetScan (http://www.tagetscan.org), PicTar (http://pictar.mdc-berlin.de) and miRanda (http://www.microrna.org/microrna/home.do), were used to predict mir-29a/mir-30d targets. Genes present in the intersection of at least two of three datasets were considered predicted targets. The GO [40] and KEGG [41] packages in R (http://www.r-project.org/) were used to annotate the functions and pathways of the miRNA targets, respectively. If any function or pathway was mapped by more than 5 targets of mir-29a/mir-30d, then it was selected. A hypergeometric test was used to validate the significance (P < 0.05) of the annotated information.
Western blotting
Granulosa cells were lysed cell lysis buffer containing 10 mM Tris–Cl (pH 7.5), 150 mM NaCl, 1 mM EDTA (pH 8.0), 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10 mM sodium fluoride, 0.2 mM sodium orthovanadate, 1 mM PMSF, and protease inhibitor cocktail (Roche Applied Science). Proteins were separated on a 12% SDS–PAGE gel and transferred to nitrocellulose membrane and probed with Col4A1 (Abgent), BMF (Cell Signaling Technology), RNF2 (MBL), and EED (Santa Cruz) antibodies. Detection of signals was performed with an Enhanced Chemiluminescence Western blotting detection system.
Statistical analysis
All the qRT-PCR experiments were repeated three times with granulosa cells preparations obtained from separate groups. The values were presented as the mean ± S.E. (n = 3). Differences between the groups were analyzed by statistical significance using t test or one-way ANOVA. P value < 0.05 was accepted as statistical significance.
References
T.R. Kumar, Y. Wang, N. Lu, M.M. Matzuk, Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat. Genet. 15, 201–204 (1997)
A. Amsterdam, N. Selvaraj, Control of differentiation, transformation, and apoptosis in granulosa cells by oncogenes, oncoviruses, and tumor suppressor genes. Endocr. Rev. 18, 435–461 (1997)
A. Amsterdam, S. Rotmensch, A. Ben-Ze’ev, Coordinated regulation of morphological and biochemical differentiation in a steroidogenic cell: the granulosa cell model. Trends Biochem. Sci. 14, 377–382 (1989)
A. Amsterdam, R.S. Gold, K. Hosokawa, Y. Yoshida, R. Sasson, Y. Jung, F. Kotsuji, Crosstalk among multiple signaling pathways controlling ovarian cell death. Trends Endocrinol. Metab. 10, 255–262 (1999)
R.L. Robker, D.L. Russell, S. Yoshioka, S.C. Sharma, J.P. Lydon, B.W. O’Malley, L.L. Espey, J.S. Richards, Ovulation: a multi-gene, multi-step process. Steroids 65, 559–570 (2000)
N.A. Grieshaber, C. Ko, S.S. Grieshaber, I. Ji, T.H. Ji, Follicle-stimulating hormone-responsive cytoskeletal genes in rat granulosa cells: class I beta-tubulin, tropomyosin-4, and kinesin heavy chain. Endocrinology 144, 29–39 (2003)
R. Sasson, A. Dantes, K. Tajima, A. Amsterdam, Novel genes modulated by FSH in normal and immortalized FSH-responsive cells: new insights into the mechanism of FSH action. FASEB J. 17, 1256–1266 (2003)
M. Tanaka, J.D. Hennebold, K. Miyakoshi, T. Teranishi, K. Ueno, E.Y. Adashi, The generation and characterization of an ovary-selective cDNA library. Mol. Cell. Endocrinol. 202, 67–69 (2003)
S. Shimasaki, R.J. Zachow, D. Li, H. Kim, S. Iemura, N. Ueno, K. Sampath, R.J. Chang, G.F. Erickson, A functional bone morphogenetic protein system in the ovary. Proc. Natl. Acad. Sci. USA 96, 7282–7287 (1999)
N. Yao, C.L. Lu, J.J. Zhao, H.F. Xia, D.G. Sun, X.Q. Shi, C. Wang, D. Li, Y. Cui, X. Ma, A network of miRNAs expressed in the ovary are regulated by FSH. Front Biosci. 14, 3239–3245 (2009)
M. Lagos-Quintana, R. Rauhut, W. Lendeckel, T. Tuschl, Identification of novel genes coding for small expressed RNAs. Science 294, 853–858 (2001)
A.M. Cheng, M.W. Byrom, J. Shelton, L.P. Ford, Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 33, 1290–1297 (2005)
S. Vasudevan, Y. Tong, J.A. Steitz, Switching from repression to activation: microRNAs can up-regulate translation. Science 318, 1931–1934 (2007)
A. Cohen, M. Shmoish, L. Levi, U. Cheruti, B. Levavi-Sivan, E. Lubzens, Alterations in micro-ribonucleic acid expression profiles reveal a novel pathway for estrogen regulation. Endocrinology 149, 1687–1696 (2008)
S.D. Fiedler, M.Z. Carletti, X. Hong, L.K. Christenson, Hormonal regulation of MicroRNA expression in periovulatory mouse mural granulosa cells. Biol. Reprod. 79, 1030–1037 (2008)
T. Yuen, F. Ruf, T. Chu, S.C. Sealfon, Microtranscriptome regulation by gonadotropin-releasing hormone. Mol. Cell. Endocrinol. 302, 12–17 (2009)
G. Stefani, F.J. Slack, Small non-coding RNAs in animal development. Nat. Rev. Mol. Cell. Biol. 9, 219–230 (2008)
M. Otsuka, M. Zheng, M. Hayashi, J.D. Lee, O. Yoshino, S. Lin, J. Han, Impaired microRNA processing causes corpus luteum insufficiency and infertility in mice. J. Clin. Invest. 118, 1944–1954 (2008)
S. Ro, R. Song, C. Park, H. Zheng, K.M. Sanders, W. Yan, Cloning and expression profiling of small RNAs expressed in the mouse ovary. RNA 13, 2366–2380 (2007)
N.J. Martinez, M.C. Ow, M.I. Barrasa, M. Hammell, R. Sequerra, L. Doucette-Stamm, F.P. Roth, V.R. Ambros, A.J. Walhout, A C. elegans genome-scale microRNA network contains composite feedback motifs with high flux capacity. Genes Dev. 22, 2535–2549 (2008)
S.M. Johnson, S.Y. Lin, F.J. Slack, The time of appearance of the C. elegans let-7 microRNA is transcriptionally controlled utilizing a temporal regulatory element in its promoter. Dev. Biol. 259, 364–379 (2003)
K.A. O’Donnell, E.A. Wentzel, K.I. Zeller, C.V. Dang, J.T. Mendell, c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435, 839–843 (2005)
Y. Sylvestre, V. De Guire, E. Querido, U.K. Mukhopadhyay, V. Bourdeau, F. Major, G. Ferbeyre, P. Chartrand, An E2F/miR-20a autoregulatory feedback loop. J Biol. Chem. 282, 2135–2143 (2007)
M. Yamakuchi, C.J. Lowenstein, MiR-34, SIRT1 and p53: the feedback loop. Cell Cycle 8, 712–715 (2009)
B.P. Lewis, C.B. Burge, D.P. Bartel, Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 (2005)
P.K. Rao, R.M. Kumar, M. Farkhondeh, S. Baskerville, H.F. Lodish, Myogenic factors that regulate expression of muscle-specific microRNAs. Proc. Natl. Acad. Sci. USA 103, 8721–8726 (2006)
S. Marton, M.R. Garcia, C. Robello, H. Persson, F. Trajtenberg, O. Pritsch, C. Rovira, H. Naya, G. Dighiero, A. Cayota, Small RNAs analysis in CLL reveals a deregulation of miRNA expression and novel miRNA candidates of putative relevance in CLL pathogenesis. Leukemia 22, 330–338 (2008)
C.A. Gebeshuber, K. Zatloukal, J. Martinez, miR-29a suppresses tristetraprolin, which is a regulator of epithelial polarity and metastasis. EMBO Rep. 10, 400–405 (2009)
J. Wang, R. Xu, F. Lin, S. Zhang, G. Zhang, S. Hu, Z. Zheng, MicroRNA: novel regulators involved in the remodeling and reverse remodeling of the heart. Cardiology 113, 81–88 (2009)
A. He, L. Zhu, N. Gupta, Y. Chang, F. Fang, Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3T3–L1 adipocytes. Mol. Endocrinol. 21, 2785–2794 (2007)
X. Tang, L. Muniappan, G. Tang, S. Ozcan, Identification of glucose-regulated miRNAs from pancreatic beta cells reveals a role for miR-30d in insulin transcription. RNA 15, 287–293 (2009)
A.E. Williams, S.A. Moschos, M.M. Perry, P.J. Barnes, M.A. Lindsay, Maternally imprinted microRNAs are differentially expressed during mouse and human lung development. Dev. Dyn. 236, 572–580 (2007)
M. Hunzicker-Dunn, E.T. Maizels, FSH signaling pathways in immature granulosa cells that regulate target gene expression: branching out from protein kinase A. Cell. Signal. 18, 1351–1359 (2006)
C.K. Sites, B. Kessel, A.R. LaBarbera, Adhesion proteins increase cellular attachment, follicle-stimulating hormone receptors, and progesterone production in cultured porcine granulosa cells. Proc. Soc. Exp. Biol. Med. 212, 78–83 (1996)
K. Nakano, I. Naito, R. Momota, Y. Sado, H. Hasegawa, Y. Ninomiya, A. Ohtsuka, The distribution of type IV collagen alpha chains in the mouse ovary and its correlation with follicular development. Arch. Histol. Cytol. 70, 243–253 (2007)
N.B. Gilula, M.L. Epstein, W.H. Beers, Cell-to-cell communication and ovulation. A study of the cumulus-oocyte complex. J. Cell Biol. 78, 58–75 (1978)
C. Kohler, C.B. Villar, Programming of gene expression by Polycomb group proteins. Trends Cell Biol. 18, 236–243 (2008)
L.A. Boyer, K. Plath, J. Zeitlinger, T. Brambrink, L.A. Medeiros, T.I. Lee, S.S. Levine, M. Wernig, A. Tajonar, M.K. Ray, G.W. Bell, A.P. Otte, M. Vidal, D.K. Gifford, R.A. Young, R. Jaenisch, Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353 (2006)
P.F. Terranova, F. Garza, Relationship between the preovulatory luteinizing hormone (LH) surge and androstenedione synthesis of preantral follicles in the cyclic hamster: detection by in vitro responses to LH. Biol. Reprod. 29, 630–636 (1983)
M. Ashburner, C.A. Ball, J.A. Blake, D. Botstein, H. Butler, J.M. Cherry, A.P. Davis, K. Dolinski, S.S. Dwight, J.T. Eppig, M.A. Harris, D.P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J.C. Matese, J.E. Richardson, M. Ringwald, G.M. Rubin, G. Sherlock, Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29 (2000)
M. Kanehisa, S. Goto, M. Hattori, K.F. Aoki-Kinoshita, M. Itoh, S. Kawashima, T. Katayama, M. Araki, M. Hirakawa, From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34, D354–D357 (2006)
Acknowledgments
This study was supported by National Basic Research Program of China (973) (No. 2010CB529504), Important National Science & Technology Specific Projects (No. 2009ZX09308-006), National Nonprofit Institute Research Grant of NRIFP, and the Denaturing High-performance Liquid chromatography System Update and its Application in Chinese Genetic Resource (No. 2006JG006100). Core facilities used in this research were provided by the Department of Genetics, National Research Institute for Family Planning. The authors would like to thank Prof. Yixun Liu and Prof. Jian Xu for help in primary granulosa cell culture.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Bai-Qing Yang has contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Yao, N., Yang, BQ., Liu, Y. et al. Follicle-stimulating hormone regulation of microRNA expression on progesterone production in cultured rat granulosa cells. Endocr 38, 158–166 (2010). https://doi.org/10.1007/s12020-010-9345-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-010-9345-1