
Abstract
Obesity and obesity-related illnesses are global epidemics impacting the health of adults and children. The purpose of the present work is to evaluate a genetically intact obese mouse model that more accurately reflects the impact of aging on diet-induced obesity and type 2 diabetes in humans. Male C57Bl/6J mice consumed either a control diet or one in which 60% kcal were due to lard beginning at 5–6 weeks of age. Body weight and fat measurements were obtained and necropsy performed at 15, 20, 30, and 40 weeks of age. Serum chemistry, histopathology, gene expression of the liver, and renal and hepatic function were also evaluated. In concert with significant increases in percent body fat and weight, mice fed the high-fat versus control diet had significantly increased levels of serum cholesterol. At ages 20 and 30 weeks, serum glucose was significantly higher in obese versus controls, while serum insulin levels were ≥4-fold higher in obese mice at ages 30 and 40 weeks. The effect of age exacerbated the effects of consuming a high-fat diet. In addition to being hyperinsulinemic and leptin resistant, older obese mice exhibited elevated hepatic PAI-1 and downregulation of GLUT4, G6PC, IGFBP-1, and leptin receptor mRNA in the liver, steatosis with subsequent inflammation, glomerular mesangial proliferation, elevated serum ALT, AST, and BUN, and increased numbers of pancreatic islets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The global prevalence of obesity and obesity-related illnesses is increasing at staggering rates. Between the years 1995 and 2000, the number of diagnosed cases of obesity (BMI > 30 kg/m2) increased approximately 50%, exceeding 300 million persons aged 15 and older worldwide [1].
Obese individuals are susceptible to the development of type 2 diabetes, a metabolic disorder that is primarily characterized by insulin resistance and hyperglycemia. In addition to increased susceptibility to type 2 diabetes, obese individuals are more prone to cardiovascular diseases, dyslipidemia, sleep apnea, hepatic and renal diseases, hypertension, and stroke [2–4]. It has also been suggested that obesity increases susceptibility to various cancers [5–7].
Studies have identified insulin resistance as a prerequisite biomarker to the onset of metabolic maladies associated with obesity [8, 9]. Inflammation is another factor contributing to the pathogenesis of certain diseases in obese patients. Of particular interest are increased blood levels of inflammatory markers: C-reactive protein (CRP), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α) [10–14]. In addition to inflammation, obese and type 2 diabetic patients have also exhibited increased activity of many hepatic metabolizing enzymes including cytochrome P450s (CYPs). Evidence of CYP2E1 induction has been substantiated in association with obesity and type 2 diabetes [15, 16]. It was reported that CYP2E1 plays an important role in the positive feedback relationship between inflammation and reactive oxygen species.
Numerous in vivo models have been introduced in order to replicate obesity and type 2 diabetes. Many of these animal models have achieved this phenotype through genetic engineering [17, 18]. A primary target of these mutations has been the leptin receptor gene. Evidence has shown that adipose tissue is not only fundamental in metabolism, but is also an organ within the endocrine system responsible for the secretion of adipokines, such as leptin [19, 20]. Leptin is an essential hormone involved in the regulation of body weight, metabolism, and reproductive function. In both animal models and humans, data have indicated that leptin deficiency does not automatically lead to the onset of diabetes or adiposity in humans. The overall objective of the present work was to evaluate a genetically intact obese mouse model that resembled the progressive deterioration associated with diet-induced obesity in humans as a result of sustained consumption of a high-fat diet. The presented research was conducted using genetically intact C57Bl/6J male mice fed (ad libitum consumption) either a control (10% fat) or high-fat (60% fat) diet. The temporal progression of obesity and type 2 diabetes through the consumption of a high-fat/high calorie diet should more aptly represent the onset of these diseases in humans. Prior studies have utilized diet-induced obese C57Bl/6J mouse models; however, these studies have primarily focused on the development of this phenotype in mice ≤20 weeks of age and/or were females [21–26]. Thus, the present work will extend the current insights on this model especially in regards to the influence of aging on the pathogenesis of diet-induced obesity.
Materials and methods
Animals
Ten-week-old male C57Bl/6J mice, fed a diet containing 10% (control) or 60% (high-fat) of the caloric contents from fat, were purchased from Jackson Laboratories (Bar Harbor, ME). Both high-fat and control diets, D12492i and D12450Bi respectively, were obtained from Research Diets, Inc. (New Brunswick, NJ) and provided to mice at Jackson Labs beginning at 5–6 weeks of age. C57Bl/6J mice were maintained on either the control or high-fat diets ad libitum through 40 weeks of age at the National Institute of Environmental Health Sciences. Animals were housed individually in facilities with a 12-h light-dark cycle with unfettered access to food and water. All animal care and experimentation were conducted according to NIH guidelines [27].
Body fat assessment
Mice were weighed once a week and body fat percentages were determined at ages 15, 20, 30, and 40 weeks. Body fat composition was acquired by dual-energy X-ray absorptiometry (DEXA) using the Piximus® densitometer (GE Medical Systems, Waukesha, WI). Mice were anesthetized using a mixture of 2.5–3% isoflurane. This mixture enabled the quick recovery of each animal once removed from the densitometer.
Pathology
Complete necropsy was performed on subsets of C57Bl/6J mice at ages 15, 20, 30, and 40 weeks. Mice were sedated using CO2 and blood was drawn for clinical chemistry from the retro-orbital venous sinus using heparinized capillary tubes. After blood was obtained, mice were euthanized via CO2 asphyxiation, tissues were collected immediately, fixed in 10% formalin for 18 h, and transferred to 70% ethanol for histopathological analysis. Histopathologic evaluation was performed on all mice at ages 15, 20, 30, and 40 weeks. Following routine H&E evaluation, 3 μm sections of kidney were prepared and evaluated with H&E and Periodic-Acid Schiff (PAS). The following tissues were examined via routine 5 μm H&E staining: brain, spinal cord, eye (with optic nerve, if possible), harderian glands, Zymbal’s glands, lacrimal glands, nasal cavity, pituitary gland, adrenal glands, thyroid gland, parathyroid glands, trachea, esophagus, salivary glands (submandibular, sublingual, and parotid), thymus, lungs, heart, aorta, spleen, kidney, liver, gallbladder, stomach, pancreas, duodenum, jejunum, ileum, cecum, colon, lymph nodes (mandibular and mesenteric), urinary bladder, skin, bone marrow, muscle, testes, epididymides, prostate gland, seminal vesicles, and preputial glands.
Evaluation of hepatic lipidosis, glomerular mesangial proliferation, and chronic inflammation of the pancreas was conducted using a 4-point grading scheme. Increases of cytoplasmic fat observed in the hepatocytes or mesangial cells (hyperplasia) and/or matrix deposition in glomeruli were scored as 0 (no change), 1+ (minimal) 1–25% of the tissue was affected, 2+ (mild) 25–50%, 3+ (moderate) 50–75%, and 4+ (marked) 75–100%. Kidney severity was graded based on the number and percentage of glomeruli affected. Mesangial cell counts from PAS-stained kidney sections were performed on 10 arbitrarily selected glomeruli/mouse/group at ages 30 and 40 weeks. Glomeruli with a diameter of 100–150 µm were evaluated. For each section of the pancreas, islets of Langerhans were counted ranging from smallest to largest. Islet dimensions were estimated based upon the size of a single red blood cell (RBC), 1 RBC averages to approximately 6 µm in diameter. Size estimates were verified by photographing select islets and then measuring them with the scale bar.
Blood chemistry
Whole blood collected from the retro-orbital venous sinus was captured in BD Microtainer® Serum Separator Tubes (Suwanee, GA) between 9:00 and 10:30 AM and centrifuged for serum collection. Biochemical variables were assessed in all serum samples on an Olympus AU400e Clinical Chemistry Analyzer, Olympus America Inc (Irvin, TX). Using reagents obtained from the instrument manufacturer, serum glucose, triglycerides, cholesterol, blood urea nitrogen (BUN) concentrations, and alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were determined. Insulin concentrations were determined using two immunoassay methods, radioimmunoassay (RIA) using reagents obtained from Linco Research (St Charles, MO) on an Apex Gamma Counter, ICN Micromedic Systems Inc. (Huntsville, Al) and electrochemiluminescence using a Sector Imager 2400 and reagents from Meso Scale Discovery (MSD) (Gaithersburg, MD). Serum leptin concentrations were generated simultaneously with the serum insulin concentrations using the MSD technique. Mouse-specific reagents and procedures for insulin and leptin were obtained from the instrument manufacturer. Urine was also collected at this time and tested for glucose and ketones using Roche Chemstrip uGK Urine Test Strips for Glucose and Ketones distributed by DH Medical (Brooklyn, NY).
Glucose tolerance test
Animals fed control or high-fat diet for 30 weeks were fasted 16 h with full access to water. Fasted animals were administered D-glucose solution at 2 g/10 ml saline/kg body weight. D-glucose dosing solution was prepared in 0.9% saline and administered (ip). Control animals received vehicle (ip) at 10 ml/kg body weight. At 30, 60, or 120 min after administration, mice were anesthetized with CO2/O2 and blood drawn from the retro-orbital venous sinus. Serum was used to measure glucose, insulin, and leptin.
Hepatic gene expression
RNA preparation and quantitative RT-PCR
Total RNA was isolated from liver using the TRIzol® Reagent (Invitrogen, Carlsbad, CA) and purified with the RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The total RNA yield and quality was evaluated by measuring its absorbance at A260/A280. The first strand cDNA was synthesized by using RT2 First Strand Kit (SuperArray Bioscience Corporation, Frederick, MD) with 1 μg of total RNA in each reaction. The cDNA was amplified by PCR using RT2 Real-Time SYBR Green PCR master mix (SuperArray, Frederick, MD) according to the manufacturer’s instructions. The expression profile for each gene was determined using the comparative CT method. Normalization was established by subtracting the endogenous control or reference (an average of 4 genes), from the CT values each gene in the array to get ΔCT. Calculation of ΔΔCT was the difference in the ΔCT of genes expressed in high-fat versus control diet fed mice. The final gene expression levels were reported as fold change = 2−ΔΔCT. Gene expressions were presented if fold changes were ±2.0 with accompanying P-values ≤0.05. All measurements were done in triplicate.
Statistical analysis
All data presented are expressed as means ± standard error. Group mean comparisons were achieved using Microsoft Excel Student’s two-tailed t-test. Trend tests on serum chemistry were performed using ANOVA and also included a linear contrast to test for age-related linear trends. In order to determine the area under the curve (AUC) for serum concentrations of glucose in mice used for the glucose tolerance test as well as determine standard deviation for each AUC, we used the formula published by Yuan [28] and then employed a z-test for pairwise comparison of the AUCs to determine significance [29]. Repeated measures analysis of variance was used to assess mesangial cell counts and Kruskal–Wallis analysis of variance, a non-parametric ANOVA method was used to compare liver and kidney lesion severity. Pancreatic islet counts were analyzed using t-tests (one-sided P-values) to compare diets at each week and pairs of weeks within each diet. Trend tests were also performed on the islets counts for each diet using linear regression. Values were considered statistically significant at P ≤ 0.05.
Results
Body weight and body fat
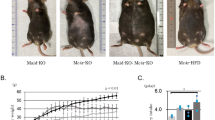
Body weights were significantly higher in male mice consuming high-fat diet in comparison to age-matched littermates eating the control diet as early as 12 weeks of age and throughout the study (P < 0.01). Male C57Bl/6J mice on the high-fat diet from ages 12–23 weeks experienced weight gain at a rate faster than mice of the same age consuming the control diet (Fig. 1a). At age 40 weeks, mice consuming the high-fat diet were approximately 41% heavier than their age-matched littermates on the control diet (Fig. 1a). In addition to weight, percent body fat between the two groups was notably different (Fig. 1b). At all time points, body fat composition of mice on the high-fat diet was significantly higher than mice on the control diet (Fig. 1b). In agreement with the pattern of weight gain observed in mice fed the high-fat diet, percent body fat between ages 15–20 and 30–40 weeks increased rapidly, while only a mere increase was determined in these mice between ages 20 and 30 weeks (Fig. 1b). In contrast, mice fed the control diet exhibited a deliberate increase in percent body fat from ages 15–30 weeks. The variable gains seen over time in mice on either diet after age 30 weeks indicated the contribution of an age-related response.

a Body weight of male C57Bl/6J mice with ad libitum consumption of either control or high-fat diets through age 40 weeks. A significant difference in weight gain was observed in mice fed the high-fat diet versus age-matched littermates on the control diet and throughout 40 weeks. Data points represent the mean ± SE of 24 mice per group. b Body fat percentage of male C57Bl/6J mice ad libitum consumption of either control or high-fat diets through age 40 weeks. A significant difference in body fat composition was observed in mice fed the high-fat diet at 15, 20, 30, and 40 weeks. Data points represent the mean ± SE of 24 mice per group
Serum biochemistry: glucose, insulin, triglycerides, and cholesterol
High-fat versus control diet mice
Compared to age-matched controls, obese mice demonstrated statistically significant differences in various biochemical parameters of carbohydrate and lipid metabolism (Figs. 2 and 3). Overall glucose levels in obese mice were higher at ages 15–30 weeks in comparison to mice of the same age fed the control diet (Fig. 2a). Statistically relevant differences in serum glucose, however, were seen at 20 and 30 weeks of age (P < 0.01) and (P < 0.05), respectively (Fig. 2a). As mice approached 40 weeks of age, serum glucose was statistically similar, 190.3 ± 14.1 versus 205.2 ± 11.0 mg/dl, in obese and control mice, respectively (Fig. 2a). Statistical increases in serum cholesterol were determined in obese versus control mice at all ages (P < 0.01) (Fig. 3a). Similarly, while serum insulin was increased in obese versus control mice at all ages, it was significantly higher at ages 30 and 40 weeks (approximately 5- and 4-fold, respectively) (P < 0.01) (Fig. 2a). Triglycerides in obese mice were also significantly higher at age 40 weeks, approximately 1.6-fold versus age-matched controls (P < 0.01).

a Relationship between serum glucose and insulin concentrations in male C57Bl/6J mice fed the control (CD) or high-fat (HF) diets at 15, 20, 30 and 40 weeks of age. ω Significant differences in glucose levels were determined at 20 and 30 weeks of age in high-fat versus age-matched control mice. Mice on the high-fat diet exhibited a significant age-related negative trend in serum glucose (P < 0.05), while no significant trend in regards to age was determined in mice on the control diet (P = 0.115). † Serum insulin concentrations were statistically significant in mice fed the high-fat diet at ages 30 and 40 weeks versus age-matched control mice. a At age 40 weeks insulin levels in both control and high-fat mice were statistically different in comparison to mice on the same diet at 15, 20, and 30 weeks of age. n = 12, 12, 18, 12 mice/diet at 15, 20, 30, and 40 weeks, respectively. A positive age-related trend was determined in control and high-fat diet mice, P < 0.01 and P < 0.01, respectively. b Serum insulin and leptin concentrations from fasted and fed mice at age 30 weeks. b Denotes significant increases in both serum insulin and leptin concentrations in fasted mice on the high-fat versus control diet (P < 0.01). c Denotes significant increases in serum insulin in fed mice on the high-fat diet versus insulin levels in both fasted mice on the same diet and fed mice on the control diet. # Denotes significant increases in serum leptin in fed mice on the high-fat diet versus fed mice on the control diet. Data points represent the mean ± SE of 4 mice per group

a Serum cholesterol in male C57Bl/6J mice fed the control or high-fat diet at ages 15, 20, 30, and 40 weeks. * Denotes significant increases in serum cholesterol of mice on the high-fat versus age-matched mice on the control diet. † Denotes serum cholesterol concentrations that were statistically significant in mice fed the high-fat diet at ages 15 versus 20, 30, and 40 weeks and at ages 20 versus 30 and 40 weeks. # Denotes serum cholesterol concentrations that were statistically significant in mice fed the control diet at ages 15 versus 20 and 40 weeks. α Denotes serum cholesterol concentrations that were statistically significant in mice fed the control diet at ages 20 versus 40 weeks. Both groups of mice, control and high-fat diet exhibited significantly positive trends in serum cholesterol from ages 15–40 weeks, control and high-fat diet, P < 0.05 and P < 0.01, respectively. b Serum triglycerides in male C57Bl/6J mice fed the control or high-fat diet at ages 15, 20, 30, and 40 weeks. * At 40 weeks of age significant increases in serum triglycerides were determined in mice on the high-fat versus age-matched controls. † Denotes a significant difference in serum triglycerides of mice on the high-fat at ages 15 versus 20, 30, and 40 weeks. An age-related trend could not be established for serum triglycerides in high-fat diet fed mice. # Denotes a significant decline in triglycerides in mice on the control diet at ages 15 versus 30 and 40 weeks. Serum triglycerides exhibited a negative age-related trend in mice on the control diet (P < 0.05)
Mice on high-fat diet
A measurable effect resulting from a combination of age and consumption of a high-fat diet was determined in mice at age 40 weeks (Figs. 2 and 3). An approximate 20% decline in serum glucose was determined at age 40 weeks versus measured concentrations at age 15 weeks (Fig. 2a), indicating a negative age-related trend (P < 0.05). In contrast, serum insulin levels escalated throughout the study demonstrating a positive trend (P < 0.01) (Fig. 2a). By age 40 weeks, serum insulin concentration increased approximately 6-fold versus insulin levels determined in mice at age 15 weeks, indicating a positive age-related trend (P < 0.01). Statistical increases in serum cholesterol were also assessed at ages 20, 30, and 40 weeks in comparison to initial concentrations at age 15 weeks (Fig. 3a). Obese mice at age 40 weeks presented with serum cholesterol concentrations that were approximately 68% higher than at age 15 weeks (Fig. 3a). These data, like serum insulin, yielded a significantly positive age-related trend (P < 0.01). In contrast, a significant age-related trend could not be established for serum triglycerides in mice consuming the high-fat diet. From age 15–40 weeks triglyceride levels declined approximately 20% (Fig. 3b). However, these mice exhibited an increase in serum triglycerides between the ages of 20–40 weeks (Fig. 3b).
Mice on control diet
As control diet fed mice progressed in age, changes in several biochemical parameters were evident. Through age 30 weeks, insulin levels remained relatively unchanged. However, at age 40 weeks an approximate 2-fold increase in serum insulin (Fig. 2a) was determined, yielding a significantly positive age-related trend (P < 0.01). Slight fluctuations in the concentration of serum glucose were also observed throughout the study; however, absent was any significant age-related variability (Fig. 2a). A positive trend in serum cholesterol was also determined in control mice (P < 0.05) (Fig. 3a). By age 40 weeks, serum cholesterol had increased approximately 20% from initial values at age 15 weeks (P < 0.05), indicating a positive age-related trend. Conversely, serum triglycerides declined in mice consuming the control diet by age 40 weeks. An estimated 25% decline in serum triglycerides from ages 15 to 40 weeks (Fig. 3b) was established yielding a negative age-related trend (P < 0.05).
Blood biochemistry: fasted versus fed
Distinctions in blood chemistry between fasted and fed animals on either diet were determined using mice at 30 weeks old in an attempt to alleviate the impact of age-related responses. Meso scale discovery (MSD) electrochemiluminescent technique was used to confirm and complement insulin results initially determined using radioimmunoassay (RIA). Fasting significantly influenced the serum concentrations of both leptin and insulin in mice fed the control diet with differences approximating 2- and 2.3-fold (P < 0.01 and P < 0.01), respectively. Similarly, obese mice exhibited a 3.6-fold increase in the concentration of serum insulin when food was available (P < 0.05) (Fig. 2b). However, in contrast to control mice, fasting did not affect serum leptin concentrations of obese mice. At age 30 weeks, serum leptin in fasted and fed obese mice was nearly identical (Fig. 2b). However, in comparison to fasted controls, serum leptin and insulin were significantly higher in fasted obese mice, approximately 5-fold (P < 0.05) (Fig. 2b). Interestingly, the serum concentration of insulin in fasted obese mice was higher than serum insulin in control mice that had continuous access to food. Serum glucose, cholesterol, and triglycerides were also determined in fasted mice at 30 weeks of age. However, there were no significant differences in serum cholesterol or triglycerides between fasted and fed mice within each diet group. Analysis of urine collected from obese and control mice revealed no evidence of glycosuria or elevated ketones in either group.
Glucose tolerance test
At 30 weeks of age, obese mice exhibited significantly elevated fasted glucose levels in comparison to age-matched mice consuming the control diet, 187.4 ± 8.0 versus 214.2 ± 7.5 mg/dl (P < 0.05) respectively (Fig. 4). As expected 30 min following intraperitoneal administration of D-glucose, animals on both diets exhibited serum glucose levels high above pre-administration values. Elevated concentrations of glucose, however, persisted in obese versus control mice and statistically important differences between the two groups were identified at 0 min (P < 0.05) and 120 min (P < 0.05) after dosing using t-test analysis (Fig. 4). The shape of the curve indicates that an earlier time point, such as 15 min post D-glucose administration, may have provided additional information. However, comparison of the standard deviations determined for each AUC, obese versus control mice revealed a z observation of 18.6 that indicated a P value <0.01. Therefore, from this calculation serum glucose levels in obese mice remained significantly elevated from 0 to 120 min after the administration of D-glucose in comparison to control mice (Fig. 4).

Glucose tolerance test in C57Bl/6J mice fed the control or high-fat diet at age 30 weeks. * Statistical differences were determined between control and high-fat mice at 0 (fasted) and 120 min post (ip) administration of 2-D glucose (P < 0.05). Data points represent the mean ± SE of 4–8 mice per group. Estimation and comparison of variance for AUCs created by obese and control mice indicated a significant difference between the two curves (P < 0.01)
Liver enzyme activity and renal function
At 30 weeks of age BUN was comparatively similar in obese and control mice, 21.7 ± 0.9 mg/dl and 23.8 ± 0.6 mg/dl, respectively (Table 1). At age 40 weeks, a slight but significant increase in BUN (P < 0.01) was detected in obese mice, 26.3 ± 0.4 mg/dl, while control mice maintained BUN levels at 20.3 ± 1.1 mg/dl (Table 1). Despite an increase in BUN in obese mice from age 30 to 40 weeks, statistically there were no significant changes in urea clearance within the diet group (Table 1). Serum levels of alanine aminotransferase (ALT) were significantly higher, greater than 3- and 2-fold, respectively, in obese mice at ages 30 and 40 weeks versus age-matched controls (P < 0.01 and P < 0.01) (Table 1). Serum levels of aspartate aminotransferase (AST) reflected a similar pattern with approximately 2-fold higher activity in obese versus control mice at both 30 and 40 weeks of age (P < 0.01 and P < 0.01).
Histopathology
Liver
As early as age 20 weeks, 50% (4/8) of mice consuming the high-fat diet developed hepatic lipidosis (steatosis) (Table 2). By age 30 weeks, the numbers of mice presenting with this lesion increased to 100% (8/8) and by age 40 weeks, a progression in the severity of the lesion was characterized by minimal inflammation associated with steatosis (6/6) (Table 2). The lipidosis or steatosis was in the form of microvesicular vacuolation of hepatocyte cytoplasm and primarily identified in the centrilobular region but often expanded to include hepatocytes of midzonal regions (Fig. 5b). Microvesicular vacuolation was characterized by multiple, small, round, clear vacuoles with distinct margins within the cytoplasm of hepatocytes, which are features consistent with accumulation of lipids (Fig. 5b). The severity of hepatic steatosis was graded according to the percent of liver involved and generally increased in severity with age (Table 2). Lesion severity in obese mice ranged from mild to marked at age 30 weeks, only marked at age 40 weeks, and only moderate at age 20 weeks. By 40 weeks of age, the lipidosis was accompanied by minimal pericellular collagen deposition and scattered infiltrates of mixed inflammatory cells (mononuclear and occasionally granulocytic) with rare degenerate hepatocytes, possibly an indication of early steatohepatitis (Fig. 5c–e). Also within the liver some hepatocytes contained large, round, clear vacuoles (macrovesicular vacuolation, primarily periportal and presumably lipid), sporadic occurrences of inflammatory infiltrate unrelated to steatosis, as well as lacy vacuolation (consistent with glycogen), but these findings did not appear to be diet- or age-related (data not shown).

Photomicrographs of a, b normal liver from a control diet mouse at age 30 weeks (40×, left; 200×, right). c, d Microvesicular vacuolation (steatosis) of hepatocyte cytoplasm in high-fat, 30-week-old mouse characterized by clear, round, microvesicular vacuoles (arrow) and centrilobular distribution (40×, left; 200×, right). e Biliary epithelial cell hyperplasia (arrow) (400×); f mononuclear cell inflammation (arrow) (400×); g, h pericellular collagen deposition between hepatocytes (Masson’s trichrome special stain 400×, left, and 600×, right) (arrow) in 40-week-old mice fed high-fat diet
Kidney
At 30 weeks of age, only obese mice (8/8) developed minimal to moderate mesangial cell and matrix proliferation (Table 2, Fig. 6b, c). At age 40 weeks this lesion was identified as minimal to mild in 5/6 control mice and marked in all (6/6) obese mice, indicating a progression in the pathology with age and treatment (Table 2). PAS staining of 3 μm kidney sections provided for a more sensitive evaluation of the glomeruli in comparison to H&E analysis. The lesion was characterized by variably increased numbers (10–30%) of mesangial cells with segmental multifocal areas of expansion of the mesangium by pale, homogeneous, eosinophilic material (Fig. 6b, c). Glomeruli in obese mice had approximately 30% increased numbers of mesangial cells with expanded mesangium compared to age-matched controls at age 30 weeks (P < 0.01) (Table 2). Consistent with glomerular age-associated growth, at 40-week-old mice on the control diet demonstrated an approximate 40% increase in mean mesangial cell counts versus control mice at age 30 weeks (P < 0.01) (Fig. 6c). Hyperplasia of mesangial cells identified in 85% (5/6) 40-week control mice was characterized as minimal to mild relative to mice age 30 weeks (Table 2). The hyperplasia of mesangial cells identified in (6/6) obese mice at age 40 weeks was of increased severity compared to age-matched controls 2.7 versus 1.2, respectively (P < 0.01) (Table 2).

Photomicrographs of a normal glomerulus from a control diet mouse age at 30 weeks (600×) (3 μm H&E, left; 3 μm PAS, right). Defined capillary loops and thin glomerular basement membrane (arrows). b Mesangial cell hyperplasia with mesangial expansion in high-fat diet age 30-week-old mice (600×) (H&E, left; PAS, right). Indicative of the hyperplasia and expansion were increased numbers of mesangial cells and expanded matrix (arrows). c Mean mesangial cell counts of glomeruli from mice fed either control or high-fat diets. # Denotes statistical differences in mice at age 30 versus 40 weeks consuming either diet (P < 0.01) and (P < 0.01), respectively. † Denotes statistical differences in mice on the control versus high-fat diet (P < 0.01) and (P < 0.01), respectively
Pancreas
Minimal, scattered inflammation was observed in approximately 42% of mice independent of diet and age. Pancreatic lesions consisted of focal interstitial accumulations of lymphocytes and plasma cells and occasional necrosis. The inflammatory infiltrate was associated with interlobular and intralobular ducts and adjacent structures, including blood vessels and rarely islets of Langerhans. This lesion was ultimately considered a background finding. As mice progressed in age from 15 to 40 weeks, a statistically significant trend consisting of an increase in the number of islets of Langerhans was determined in both high-fat and control diet fed mice, P < 0.01 and P < 0.01, respectively (Fig. 7). Although overall mean counts of islets were higher in high-fat versus control diet fed mice, significant differences were only determined within each diet group as a function of age and not as a result of ad libitum consumption of diet.

Mean counts for islets of Langerhans in C57Bl/6J mice fed the high-fat versus control diet from age 15–40 weeks. † Denotes a significant increase in pancreatic islets in control mice at 15 versus 20, 30, and 40 weeks of age (P < 0.05, <0.01, and <0.05) respectively. # Denotes a significant increase in pancreatic islets in mice on the high-fat diet at 15 versus 30 and 40 weeks of age (P < 0.05 and <0.05), respectively. α Denotes a significant increase in pancreatic islets in mice on the high-fat diet 40 versus 20 and 30 weeks of age (P < 0.05 and <0.05), respectively
Significant increases in the pancreatic islets were measured in mice on the control diet at ages 15 versus 40 weeks (P < 0.05) and 40 versus 20 and 30 weeks, P < 0.01 and P < 0.05, respectively (Fig. 7). Mice on the high-fat diet demonstrated significant differences at ages 15 versus 30 and 40 weeks, P < 0.05 and P < 0.05, respectively, and 40 versus 20 and 30 weeks, P < 0.05 and P < 0.05, respectively (Fig. 7).
Hepatic gene expression determined by RT-PCR arrays
In order to gain further insight into the mechanisms contributing to the pathogenesis of diet-induced obesity, hepatic mRNA of a number of genes involved in glucose, insulin, and lipid homeostasis was evaluated. Gene expressions were presented if fold changes were ±2.0 with accompanying P-values ≤0.05. Using these criteria, obese mice at 30 weeks of age exhibited notable changes in several genes in comparison to age-matched mice fed the control diet (Table 3). Plasminogen activated receptor-1 (PAI-1) was upregulated 6.2-fold in obese versus control mice (Table 3). In contrast, downregulation of leptin receptor (LEPR), facilitated glucose transporter, member 4 (GLUT4), insulin-like growth factor binding protein 1 (IGFBP1), and glucose 6-phosphatase catalytic unit (GP6C) was determined in obese versus control mice, −5.3, −6.7, −2.3, and −6.7, respectively (Table 3).
Discussion
According to the World Health Organization (WHO), more than 1 billion adults worldwide are considered overweight (BMI > 25 kg/m2) and approximately one-third of this population has been diagnosed as clinically obese (BMI > 30 kg/m2) [1]. Still, the global burden of obesity is projected to further escalate with 2.3 billion adults anticipated to be overweight by 2015 and 700 million of these individuals classified as obese [1]. Numerous studies have suggested that obese individuals are at greater risk in developing an array of chronic illnesses including metabolic disorders such as type 2 diabetes and cancer [2, 6]. In vivo animal models, both diet-induced and genetically altered, have been employed in order to study the development of obesity and type 2 diabetes. Many of the studies that investigated the impact of diet-induced obesity, however, have assessed this disorder in female and male mice at younger ages (approximately 20 weeks) and/or consumed a diet high in fat for a shorter duration of time than what has been presented in the current work [21–26, 30]. Therefore, primary aim of this research is to evaluate the temporal relationships between age and sustained consumption of a high-fat diet on the pathogenesis of diet-induced obesity.
Dietary composition played a significant role in the pathophysiology of obesity within the current study. Daily consumption of the high-fat diet in which 60 kcal% were attributed to fat imparted notable changes in body weight, body fat composition, serum biochemistry, gene expression in the liver, histological aberrations of the liver kidney, and pancreas, and altered hepatic and renal function in mice. The added variable of age appeared to exacerbate these maladies in obese mice in comparison to age-matched controls. Using a genetically intact mouse model, the presented work effectively replicated the onset of diet-induced obesity in humans as a chronic disorder characterized as a time-dependent and/or age-influenced progressive disease. As early as 12 weeks of age, C57Bl/6J mice consuming the high-fat diet exhibited significant increases in body weight in comparison to age-matched mice fed the control diet and were considered obese. Along with body weight, percent body fat was consistently higher in mice consuming the high-fat diet. Exacerbated by excess weight and rising body fat percentages, circulating leptin levels at age 30 weeks were markedly elevated in obese animals (>5-fold). Studies conducted in both humans and rodents have demonstrated a direct relationship between fasted serum leptin levels and body fat [31, 32]. With diet as the dependent variable, current work supports earlier findings showing that as weight and body fat increased, sensitivity to leptin decreased thus fostering leptin resistance. Further evidence of leptin resistance was demonstrated as leptin receptors in the liver were downregulated 5.3-fold in obese versus control mice at age 30 weeks. Earlier observations have reported an inverse relationship between hyperleptinemia and the expression of leptin receptors in both the liver and hypothalamus [33]. It has been suggested that leptin downregulates its receptors and through this action promotes weight gain [33]. Studies have also proposed that obesity inhibits the body’s response to the regulatory actions of leptin through impaired leptin transport across the blood–brain barrier [34–37]. Banks et al. [34] identified elevated triglycerides as the principal cause for leptin’s failure to penetrate into the CNS and bind to specific receptors located in the hypothalamus. Within the current work, in concert with hyperleptinemia and hyperlipidemia, triglyceride levels in obese mice were elevated in comparison to mice fed the control diet throughout the study and significantly higher as obese mice aged to 40 weeks old. An important extra-neuronal function of leptin is its direct involvement in the reduction of serum triglycerides [38]. This action of leptin is apparently achieved by inhibiting free fatty acid synthesis, while promoting triglyceride hydrolysis and fatty acid oxidation [38]. Results of the present work showed a persistent decline in serum triglycerides in control mice over time, demonstrating a significantly negative trend as well as an indicator that control mice remained sensitive to the actions of leptin even as they increased in age.
Differing hypotheses have been proposed regarding the influence of leptin on insulin secretion and vice versa [20, 39–42]. Collectively, studies have shown a continuous interplay between the two hormones. Leptin acts as both an indirect and direct antagonist to insulin secretion and biosynthesis within the sympathetic nervous system and pancreatic β-cells, respectively [43]. The lipogenic actions of insulin have been shown to induce leptin biosynthesis and secretion from white adipose tissue [43]. In the present work, a positive correlation between obesity and the ballooning levels of both leptin and insulin was clearly evident. The reduced efficacy of peripheral leptin correlates with the hypersecretion of insulin and presumed insulin resistance observed in mice fed the high-fat diet. Further indication of insulin resistance in obese mice was the downregulation of hepatic insulin-like growth factor binding protein 1 (IGFBP-1), 6.7-fold. Normally, insulin regulates the production of IGFBP-1 through insulin responsive elements [44]. However, studies have inversely related adiposity and elevated serum insulin with IGFBP-1 expression [44, 45].
It has been proposed that the inability of leptin to curtail glucose-stimulated insulin secretion from pancreatic β-cells and foster insulin resistance in obese patients promotes chronic hyperinsulinemia and is a factor in the pathophysiology of type 2 diabetes [46]. In the current model, consumption of a high-fat diet induced mild hyperglycemia as early as 20 weeks of age in concert with elevated serum insulin. Although serum glucose remained significantly higher in obese mice through age 30 weeks in comparison to age-matched controls, glucose levels in obese mice fell below their peak attained at 20 weeks of age. Undoubtedly, declining glucose can be attributed to the aggressive escalation of insulin in obese mice, while surging levels of serum leptin encouraged hyperinsulinemia and leptin resistance. In contrast, control mice at age 30 weeks exhibited significantly reduced levels of circulating leptin in parallel with consistently lower and stable serum insulin and glucose concentrations. Only as control mice aged to 40 weeks old was there a notable increase in serum insulin.
The downregulation of hepatic glucose-6-phosphate catalytic subunit (G6PC), a critical enzyme involved in hepatic glycogenolysis and gluconeogensis, was likely a response to the hyperinsulinemia exhibited in obese mice at age 30 weeks. G6PC mediates the hydrolysis of glucose-6-phosphate to glucose which then binds to facilitated glucose transporter 4 (GLUT4), enabling the uptake of glucose from the plasma and deposition into fat and muscle tissues [47]. Within the current work, in addition to G6PC, GLUT4 was also significantly downregulated (6.7-fold) in obese mice. GLUT4 is an insulin-sensitive protein and its expression has shown to be inhibited as both insulin insensitivity and adiposity increases [47, 48]. The downregulation of these genes involved in the mobilization of glucose bolster the results of the glucose tolerance test showing reduced clearance of glucose in 30-week-old obese versus control mice 2 h after D-glucose had been administered. Interestingly, the number of pancreatic islets in both control and obese mice progressively increased as a function of age. Although there were no statistical differences in islet number as a result of diet, older obese mice exhibited higher mean counts than controls. The increase in islets of Langerhans reflected the increase in serum insulin in both control and obese mice. Islets of Langerhans are composed of cells responsible for the production and secretion of pancreatic hormones including glucagon and insulin. Further analysis is needed in order to determine if larger islet size correlates with increased size or number of glucagon-secreting α- and/or insulin-secreting β-cells. Although mild, obese mice were hyperglycemic and therefore stores of glucagon maybe increased in obese versus control mice. Studies have ascribed both an increase in α-cells and an ablation of β-cell function as an affirmation of type 2 diabetes pathology in both humans and mice [30, 49, 50].
Sustained exposure to hyperlipidemia in the blood and the accumulation of fatty acids in non-adipose tissues as a consequence of obesity and leptin resistance significantly amplifies the risk for the development of metabolic abnormalities and organ damage [51–54]. Studies have shown that an important function of leptin is to shield non-adipose tissues from retaining excessive fat stores through increased oxidation of fatty acids and decreased lipogenesis [55]. The intimate relationship amongst adipose tissues, leptin, and fat stores suggests leptin resistance is involved in the pathogenesis of nonalcoholic fatty liver disease (NAFLD) and other progressive disorders including renal failure. In turn, the potentiation of insulin resistance through hyperinsulinemia and lipolysis encourages the accumulation of fat within liver cells and the emergence of NAFLD [56]. Within the current studies, in association with increased incidence and severity of hepatocellular cytoplasmic microvesicular vacuolation consistent with NAFLD in obese versus control mice, plasminogen activator inhibitor (PAI-1) was also upregulated. Studies have positively correlated increased PAI-1 to adiposity, elevated serum insulin and glucose levels, and type 2 diabetes [57–59]. Furthermore, elevated PAI-1 has been associated with hepatic injury including NAFLD [60]. It has been postulated that lipoproteins can directly induce the expression of PAI-1 in the liver by binding to response elements within the PAI-1 promoter [61, 62]. Therefore, continued consumption of a diet high in fat in the present model, elevated lipoproteins (triglycerides and cholesterol) contributed to the upregulation of PAI-1 and development of NAFLD in obese mice.
A more severe liver damage was observed in obese mice as they aged to 40 weeks in comparison to mice on the same diet at 30 weeks of age or younger. Mononuclear inflammation was juxtaposed with rare individual necrotic hepatocytes, biliary epithelial cell hyperplasia, and collagen deposition between hepatocytes consistent with a progression towards early steatohepatitis and in combination with pronounced hyperinsulinemia and hyperleptinemia suggests a pre-type 2 diabetic state in 40-week-old obese mice. Inflammatory infiltrates and collagen associated with microvesicular vacuolation of hepatocytes indicates a response to hepatocellular injury, perhaps, peroxidation of lipid accumulation. Additional evidence of hepatocellular damage was demonstrated through significantly elevated levels of alanine (ALT) and aspartate (AST) aminotransferases in obese versus control mice at 30 and 40 weeks of age. It is likely that age played a role in the increased expression of these liver enzymes in 40-week-old control mice. However, despite this heightened enzymatic activity, control mice demonstrated no phenotypic changes in hepatocellular integrity at age 40 weeks, consistent with minor toxicity. In contrast, the combined effect of age and diet in obese mice produced a synergistic reaction characterized by increased ALT and AST levels in association with progressive liver injury.
It is possible that dysregulation of hepatocellular function leads to increased susceptibility to environmental insult as a result of an inflammatory response and/or altered expression of metabolizing enzymes, particularly cytochrome P450s (CYPs). Localization of the initial lesion, microvesicular vacuolation within the centrilobular region of the liver coincides with an abundance of CYPs, including CYP2E1. In the current study, CYP2E1 expression was approximately 40% higher in obese versus control mice at ages 30 and 40 weeks (data not shown). These data confirm previous findings demonstrating that hepatic CYP2E1 is upregulated in animals and patients with nonalcoholic steatohepatitis (NASH), the subsequent disorder to NAFLD [63, 64]. CYP2E1 has also been shown to have a high inclination to generate reactive oxygen radicals and promote lipid and protein peroxidation in the presence or absence of substrates [65–67]. Studies investigating the role of CYP2E1 in the generation of oxidative stress within an animal model representative of NASH have shown a significant increase in the activity of microsomal NADPH-dependent lipid peroxidation in comparison to control animals. In addition to the production of oxygen radicals via CYP2E1, another local source of oxidative stress within the current model can be directly attributed to increased levels of fatty acids and triglycerides sequestered within the hepatocytes of obese mice. Moreover, the generation of reactive oxygen species and oxidative damage has been strongly associated with biological aging [68]. Therefore, the collective effects of CYP2E1 generated oxidative stress coupled with the innate oxidative actions of hepatocellular fatty acids and normal cellular aging may have contributed to the progression in severity of the hepatic lesions in our study.
Obesity-related glomerulosclerosis has also emerged as a significant problem affecting the health of both adults and children [53]. Studies have shown a positive correlation amongst elevated serum leptin levels, hyperinsulinemia and the development of mesangial glomerulosclerosis [69]. In the current study, glomerular mesangial cell hyperplasia was characterized in only 30- and 40-week-old obese mice as a significant increase in the number of mesangial cells and an expansion of the mesangial matrix. These findings differ from earlier studies that have identified similar renal lesions in younger C57Bl/6J mice fed diets high in fat [21]. In the current work, male C57Bl/6J mice began ad libitum consumption of the high-fat diet during puberty (5–6 weeks of age) and continued through age 40 weeks, while postpubertal consumption of the same high-fat diet was administered in the prior work and only given for 12 weeks. It remains to be determined if differences in the time of the initial exposure to the high-fat diet contributed to either delayed or early manifestation of obesity-related renal pathology. Although an assessment of BUN did not indicate significant changes in renal function in obese versus control mice at age 30 weeks, a significant increase in the concentration of BUN was determined at age 40 weeks. Elevated BUN may have been in response to the increased severity of glomerular lesions identified in older obese mice. However, in control mice at age 40 weeks, the variable of age appears to have limited influence on renal function. Despite hyperplastic growth of mesangial cells, the severity of the phenotype and serum BUN was significantly less in comparison to the shared effect of age and diet exemplified in 40-week-old obese mice. Though the analysis of urine indicated an absence of renal damage in obese mice at both 30 and 40 weeks of age, the assessment of urine microalbuminemia could have provided additional knowledge regarding the ability of the glomeruli to retard the leakage of albumin into the urine especially as the severity of the lesion increased and serum BUN elevated in older 40 week old mice. Ultimately, the fate of these glomerular lesions is uncertain as they may be reversible or a precursor to nephropathy characterized by thickening of the glomerular basement membrane (GBM) and capillary walls leading to chronic glomerulosclerosis. Patients diagnosed with diabetic nephropathy have exhibited glomerular lesions characterized by mesangial cell proliferation, expansion of the mesangial matrix, endothelial damage discernible as thickened GBM, and podocyte damage [70, 71]. The development of these glomerular lesions in mice exhibiting hyperleptinemia and hyperinsulinemia lend further support to the suggestion that manipulations in the dynamic between leptin and insulin prompt a cascade of detrimental biological processes within in the kidney as well in other tissues.
In conclusion, the current model is efficacious in presenting diet-induced obesity as an age-influenced chronic progressive disease in a manner that reflects the onset of obesity and obesity-related disorders in humans. Diet-induced obesity was associated with dominating hyperinsulinemia and hyperleptinemia, altered expression of hepatic genes, as well as elevated circulating triglycerides and cholesterol levels in comparison to mice fed a control diet. Aging was also an important dynamic when associated with the consumption of high-fat diet. As mice grew older, serum biochemistry, hepatic and renal structures as well as function were further compromised, and a progressive alteration in pancreatic islet morphology was determined. Although mild hyperglycemia was evident in obese mice, serum glucose levels remained controlled as a reaction to surging levels of serum insulin and leptin. This may indicate that the manifestation and progressive nature of lesions identified in obese mice were most likely due to the dysregulation of the relationship between leptin and insulin rather than insulin and glucose. It is interesting to note that at age 20 weeks in mice consuming the high-fat diet, hepatic mRNA of genes GLUT4, G6PC, and LEPR were downregulated and PAI-1 upregulated as they were at age 30 weeks, however, to a notably lesser extent (data not shown). In contrast to 30-week-old obese mice, hepatic IGFBP-1 was upregulated in mice that consumed the high-fat diet at age 20 weeks (data not shown). A possible explanation for this difference is that at age 20 weeks mice on the high-fat diet were not hyperinsulinemic and the dynamic between serum leptin and insulin was still functional. We anticipate that this progressive dysregulation of genes as well as others involved in lipid, insulin, and glucose metabolism will continue to occur as obese mice advance in age. Proinflammatory actions of leptin and aging in concert with insulin and leptin resistance are in need of further study, but are undoubtedly essential in the pathogenesis and progression of metabolic disorders, obesity and type 2 diabetes, and may contribute to the development of other obesity-related illnesses. In addition, this animal model may prove valuable to assess whether metabolic disorders, in particular, diet-induced obesity exacerbates susceptibility to environmental chemical pollutants.
References
World Health Organization, Joint WHO/FAO Expert Consultation on Diet, Nutrition, and the Prevention of Chronic Diseases (World Health Organization, Geneva, 2003)
D.M. Muoio, C.B. Newgard, Annu. Rev. Biochem. 75, 367–401 (2006)
P. Poirier, T.D. Giles, G.A. Bray, Y. Hong, J.S. Stern, F.X. Pi-Sunyer, R.H. Eckel, Arterioscler. Thromb. Vasc. Biol. 26(5), 968–976 (2006)
R. Wolk, V.K. Somers, Diabetes Obes. Metab. 8(3), 250–260 (2006)
J.A. Meyerhardt, D. Niedzwiecki, D. Hollis, L.B. Saltz, R.J. Mayer, H. Nelson, R. Whittom, A. Hantel, J. Thomas, C.S. Fuchs, J. Clin. Oncol. 26(25), 4109–4115 (2008)
E.E. Calle, M.J. Thun, Oncogene 23(38), 6365–6378 (2004)
F. Bianchini, R. Kaaks, H. Vainio, Lancet Oncol. 3(9), 565–574 (2002)
G. Reaven, Diabetes Care 26(2), 539–540 (2003)
G. Wilcox, Clin. Biochem. Rev. 26(2), 19–39 (2005)
M. Bullo, P. Garcia-Lorda, I. Megias, J. Salas-Salvado, Obes. Res. 11(4), 525–531 (2003)
T. McLaughlin, F. Abbasi, C. Lamendola, L. Liang, G. Reaven, P. Schaaf, P. Reaven, Circulation 106(23), 2908–2912 (2002)
L. Roytblat, M. Rachinsky, A. Fisher, L. Greemberg, Y. Shapira, A. Douvdevani, S. Gelman, Obes. Res. 8(9), 673–675 (2000)
J.S. Yudkin, C.D. Stehouwer, J.J. Emeis, S.W. Coppack, Arterioscler. Thromb. Vasc. Biol. 19(4), 972–978 (1999)
J.S. Yudkin, S.W. Coppack, K. Bulmer, A. Rawesh, V. Mohamed-Ali, Thromb. Res. 96(1), 1–9 (1999)
A. Dey, A.I. Cederbaum, Hepatology 45(6), 1355–1365 (2007)
Z. Wang, S.D. Hall, J.F. Maya, L. Li, A. Asghar, J.C. Gorski, Br. J. Clin. Pharmacol. 55(1), 77–85 (2003)
K. Yoshinari, S. Takagi, J. Sugatani, M. Miwa, Biol. Pharm. Bull. 29(8), 1634–1638 (2006)
M. Rossmeisl, J.S. Rim, R.A. Koza, L.P. Kozak, Diabetes 52(8), 1958–1966 (2003)
S. Cinti, Prostaglandins Leukot. Essent. Fatty Acids 73(1), 9–15 (2005)
M.W. Schwartz, R.L. Prigeon, S.E. Kahn, M. Nicolson, J. Moore, A. Morawiecki, E.J. Boyko, D. Porte Jr., Diabetes Care 20(9), 1476–1481 (1997)
T. Jiang, Z. Wang, G. Proctor, S. Moskowitz, S.E. Liebman, T. Rogers, M.S. Lucia, J. Li, M. Levi, J. Biol. Chem. 280(37), 32317–32325 (2005)
P.I. Parekh, A.E. Petro, J.M. Tiller, M.N. Feinglos, R.S. Surwit, Metabolism 47(9), 1089–1096 (1998)
S. Lin, T.C. Thomas, L.H. Storlien, X.F. Huang, Int. J. Obes. Relat. Metab. Disord. 24(5), 639–646 (2000)
R.S. Surwit, C.M. Kuhn, C. Cochrane, J.A. McCubbin, M.N. Feinglos, Diabetes 37(9), 1163–1167 (1988)
A.S. Muhlfeld, M.W. Spencer, K.L. Hudkins, E. Kirk, R.C. LeBoeuf, C.E. Alpers, Kidney Int. 66(4), 1393–1402 (2004)
K.L. Townsend, M.M. Lorenzi, E.P. Widmaier, Endocrine 33(2), 176–188 (2008)
National Research Council, Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, D.C., 1996)
J. Yuan, J. Pharm. Sci. 82(7), 761–763 (1993)
A.J. Bailer, J. Pharmacokinet. Biopharm. 16(3), 303–309 (1988)
H. Karasawa, S. Nagata-Goto, K. Takaishi, Y. Kumagae, Metabolism 58(3), 296–303 (2009)
C.T. Montague, I.S. Farooqi, J.P. Whitehead, M.A. Soos, H. Rau, N.J. Wareham, C.P. Sewter, J.E. Digby, S.N. Mohammed, J.A. Hurst, C.H. Cheetham, A.R. Earley, A.H. Barnett, J.B. Prins, S. O’Rahilly, Nature 387(6636), 903–908 (1997)
S. Grinspoon, T. Gulick, H. Askari, M. Landt, K. Lee, E. Anderson, Z. Ma, L. Vignati, R. Bowsher, D. Herzog, A. Klibanski, J. Clin. Endocrinol. Metab. 81(11), 3861–3863 (1996)
Z.J. Liu, J. Bian, J. Liu, A. Endoh, Horm. Metab. Res. 39(7), 489–494 (2007)
W.A. Banks, A.B. Coon, S.M. Robinson, A. Moinuddin, J.M. Shultz, R. Nakaoke, J.E. Morley, Diabetes 53(5), 1253–1260 (2004)
W.A. Banks, C.R. DiPalma, C.L. Farrell, Peptides 20(11), 1341–1345 (1999)
S.M. Hileman, D.D. Pierroz, H. Masuzaki, C. Bjorbaek, K. El-Haschimi, W.A. Banks, J.S. Flier, Endocrinology 143(3), 775–783 (2002)
J. Wilsey, S. Zolotukhin, V. Prima, P.J. Scarpace, Am. J. Physiol. Regul. Integr. Comp. Physiol. 285(5), R1011–R1020 (2003)
G.R. Steinberg, A. Bonen, D.J. Dyck, Am. J. Physiol. Endocrinol. Metab. 282(3), E593–E600 (2002)
R.B. Ceddia, H.A. Koistinen, J.R. Zierath, G. Sweeney, FASEB J. 16(10), 1163–1176 (2002)
T.H. Schindler, J. Cardenas, J.O. Prior, A.D. Facta, M.C. Kreissl, X.L. Zhang, J. Sayre, M. Dahlbom, J. Licinio, H.R. Schelbert, J. Am. Coll. Cardiol. 47(6), 1188–1195 (2006)
H.C. Fehmann, C. Peiser, H.P. Bode, M. Stamm, P. Staats, C. Hedetoft, R.E. Lang, B. Goke, Peptides 18(8), 1267–1273 (1997)
Y. Tanizawa, S. Okuya, H. Ishihara, T. Asano, T. Yada, Y. Oka, Endocrinology 138(10), 4513–4516 (1997)
J. Seufert, Diabetes 53(Suppl 1), S152–S158 (2004)
S. Soderberg, B. Ahren, M. Eliasson, B. Dinesen, K. Brismar, T. Olsson, Eur. J. Endocrinol. 144(3), 283–290 (2001)
J.M. Holly, A.M. Cotterill, R.C. Jemmott, D. Shears, S. al-Othman, T. Chard, J.A. Wass, Clin. Endocrinol. (Oxf) 34(4), 275–280 (1991)
J. Seufert, T.J. Kieffer, C.A. Leech, G.G. Holz, W. Moritz, C. Ricordi, J.F. Habener, J. Clin. Endocrinol. Metab. 84(2), 670–676 (1999)
D. Chandrasagar, K. Amira, EMBRO Rep. 6(12), 1137–1142 (2005)
L. Qi, M. Saberi, E. Zmuda, Y. Wang, J. Altarejos, X. Zhang, R. Dentin, S. Hedrick, G. Bandyopadhyay, T. Hai, J. Olefsky, M. Montminy, Cell Metab. 9(3), 277–286 (2009)
S. Deng, M. Vatamaniuk, X. Huang, N. Doliba, M.M. Lian, A. Frank, E. Velidedeoglu, N.M. Desai, B. Koeberlein, B. Wolf, C.F. Barker, A. Naji, F.M. Matschinsky, J.F. Markmann, Diabetes 53(3), 624–632 (2004)
K.H. Yoon, S.H. Ko, J.H. Cho, J.M. Lee, Y.B. Ahn, K.H. Song, S.J. Yoo, M.I. Kang, B.Y. Cha, K.W. Lee, H.Y. Son, S.K. Kang, H.S. Kim, I.K. Lee, S. Bonner-Weir, J. Clin. Endocrinol. Metab. 88(5), 2300–2308 (2003)
M. Murray, Curr. Drug Metab. 7(1), 67–81 (2006)
M. Nangaku, Y. Izuhara, N. Usuda, R. Inagi, T. Shibata, S. Sugiyama, K. Kurokawa, C. van Ypersele de Strihou, T. Miyata, Nephrol. Dial. Transplant. 20(12), 2661–2669 (2005)
N. Kambham, G.S. Markowitz, A.M. Valeri, J. Lin, V.D. D’Agati, Kidney Int. 59(4), 1498–1509 (2001)
C. Garcia-Monzon, E. Martin-Perez, O.L. Iacono, M. Fernandez-Bermejo, P.L. Majano, A. Apolinario, E. Larranaga, R. Moreno-Otero, J. Hepatol. 33(5), 716–724 (2000)
R.H. Unger, Y.T. Zhou, Diabetes 50(Suppl 1), S118–S121 (2001)
P. Angulo, Ann. Hepatol. 1(1), 12–19 (2002)
E.Y. Dimova, T. Kietzmann, Thromb. Haemost. 100(6), 992–1006 (2008)
Y. Aso, S. Matsumoto, Y. Fujiwara, K. Tayama, T. Inukai, Y. Takemura, Metabolism 51(4), 471–476 (2002)
K. Landin, L. Stigendal, E. Eriksson, M. Krotkiewski, B. Risberg, L. Tengborn, U. Smith, Metabolism 39(10), 1044–1048 (1990)
M.C. Alessi, D. Bastelica, A. Mavri, P. Morange, B. Berthet, M. Grino, I. Juhan-Vague, Arterioscler. Thromb. Vasc. Biol. 23(7), 1262–1268 (2003)
R. Zhao, X. Ma, G.X. Shen, Mol. Cell. Biochem. 317(1–2), 197–204 (2008)
G.D. Norata, C. Banfi, A. Pirillo, E. Tremoli, A. Hamsten, A.L. Catapano, P. Eriksson, Br. J. Haematol. 127(1), 97–104 (2004)
I.A. Leclercq, J. Field, A. Enriquez, G.C. Farrell, G.R. Robertson, Biochem. Biophys. Res. Commun. 268(2), 337–344 (2000)
M.D. Weltman, G.C. Farrell, P. Hall, M. Ingelman-Sundberg, C. Liddle, Hepatology 27(1), 128–133 (1998)
I.M. Danko, N.A. Chaschin, Exp. Oncol. 27(4), 248–256 (2005)
A.A. Caro, A.I. Cederbaum, Annu. Rev. Pharmacol. Toxicol. 44, 27–42 (2004)
C.S. Lieber, Physiol. Rev. 77(2), 517–544 (1997)
K.C. Kregel, H.J. Zhang, Am. J. Physiol. Regul. Integr. Comp. Physiol. 292(1), R18–R36 (2007)
D.C. Han, M. Isono, S. Chen, A. Casaretto, S.W. Hong, G. Wolf, F.N. Ziyadeh, Kidney Int. 59(4), 1315–1323 (2001)
G. Remuzzi, A. Schieppati, P. Ruggenenti, N. Engl. J. Med. 346(15), 1145–1151 (2002)
S. Zheng, E.C. Carlson, L. Yang, P.M. Kralik, Y. Huang, P.N. Epstein, J. Am. Soc. Nephrol. 19(11), 2077–2085 (2008)
Acknowledgements
We extend our thanks to Drs. Retha Newbold and Susan Elmore for their commentary and careful review of this manuscript. We also sincerely appreciate Dr. Grace Kissling for her assistance with the statistical analyses. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hoffler, U., Hobbie, K., Wilson, R. et al. Diet-induced obesity is associated with hyperleptinemia, hyperinsulinemia, hepatic steatosis, and glomerulopathy in C57Bl/6J mice. Endocr 36, 311–325 (2009). https://doi.org/10.1007/s12020-009-9224-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-009-9224-9