
Abstract
It was recently proposed that, in rat pancreatic islets, the production of bicarbonate accounts for the major fraction of the carbon dioxide generated by the oxidative catabolism of nutrient insulin secretagogues. In search of the mechanism(s) supporting the membrane transport of bicarbonate, the possible role of the electrogenic Na+–HCO3 −-cotransporters NBCe1-A and NBCe1-B in rat pancreatic islet cells was investigated. Expression of NBCe1-A and NBCe1-B in rat pancreatic islet cells was documented by RT-PCR, western blotting, and immunocytochemistry. The latter procedure suggested a preferential localization of NBCe1-B in insulin-producing cells. Tenidap (3–100 μM), previously proposed as an inhibitor of NBCe1-A-mediated cotransport in proximal tubule kidney cells, caused a concentration-related inhibition of glucose-stimulated insulin secretion. It also inhibited 2-ketoisocaproate-induced insulin release and to a relatively lesser extent, the secretory response to l-leucine. Tenidap (50–100 μM) also inhibited the metabolism of d-glucose in isolated islets, increased 22Na net uptake by dispersed islet cells, lowered intracellular pH and provoked hyperpolarization of plasma membrane in insulin-producing cells. This study thus reveals the expression of the electrogenic Na+–HCO3 −-cotransporters NBCe1-A and NBCe1-B in rat pancreatic islet cells, and is consistent with the participation of such transporters in the process of nutrient-stimulated insulin secretion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Insulin secretion by pancreatic beta cells is coupled to glucose metabolism, which results in carbon dioxide production. It was recently proposed that, in rat pancreatic islets, carbon dioxide is converted to a large extent to bicarbonate (HCO3 −) [1]. However, the mechanism by which HCO3 − leaves the islet cells remains to be elucidated. The bicarbonate ion cannot permeate lipid bilayers, implying some facilitated transport.
In the last 10 years, several new bicarbonate transporters have been cloned including five Na+-HCO3 −-cotransporters (NBC) either electroneutral or electrogenic [2]. Their amino acid sequence is 30–35% identical to that of the anion exchangers (AE1-3) mainly involved in Cl−–HCO3 − exchange and together, they define a new superfamily of HCO3 − transporters, the solute carrier 4 family (SLC4) functioning for exit or entry of HCO3 −. The first evidence for Na+–HCO3 − cotransport was provided by Boron and Boulpaep [3] in the salamander kidney proximal tubule and they later expression cloned this first NBC (NBC1 also called SLC4A4) from the salamander kidney [4]. Rat and human NBC1 were homology cloned 1 year later [5]. In the kidney, NBC1 was localized to the basolateral membrane of the proximal tubule where it mediates Na+ and HCO3 − efflux, with a stoichiometry of 1 Na+ equivalent to 3 HCO3 −. Since the cloning of NBC1 gene in the kidney, several splice variants of this cotransporter were identified. Two major variants NBCe1-A (formerly called kNBC) and NBCe1-B (formerly called pNBC) are differentially expressed in a cell and tissue specific manner [2]. NBCe1-B is the most ubiquitous variant, being expressed, e.g., in exocrine pancreas, brain, heart, prostate, small and large intestine, stomach, and epididymis, while the NBCe1-A variant is more restricted, being most highly expressed in the kidney. Functional studies in exocrine pancreatic ducts established that NBCe1B mediates the influx of 1 Na+ with 2 HCO3 −. Structurally, these two variants differ in their amino (NH2) terminal where the first 41 amino acids of the NBCe1-A are replaced by 85 amino acids in the NBCe1-B. A third NBCe1 variant has also been described in the rat brain and has been named NBCe1-C.
In this study, the presence of NBCe1-A and NBCe1-B variants was identified in rat pancreatic islets by RT-PCR, western blotting and immunocytochemistry. Furthermore, an inhibitor of NBCe1-A, tenidap [6], was found to inhibit nutrient-stimulated insulin release from isolated rat islets, to increase 22Na+ uptake by dispersed islet cells, to inhibit glucose catabolism in intact islets and to cause both acidification and plasma membrane hyperpolarization in islet cells.
Results
RT-PCR
Figure 1 summarizes RT-PCR data. Amplicons corresponding to the expected pair of bases were observed for NBCe1-B variant, the universal NBC1 primers and β-actin in all specimen tissues, i.e., kidney, pancreas, and pancreatic islets. The NBCe1-A variant, highly expressed in the kidney, was observed at a lower level in the pancreas and pancreatic islets. Amplification seemed similar in all tissues in the case of both the universal NBCe1 and β-actin. While pancreatic tissue offered apparently a similar level of amplification for both A and B variants, pancreatic islets yielded a stronger amplification signal for NBCe1-B compared to NBCe1-A. These differences in amplification could be indicative of higher levels of NBCe1-B mRNA as compared to NBCe1-A mRNA in rat pancreatic islets.

RT-PCR of kidney, pancreas, and isolated pancreatic islet using the primer sets corresponding to NBCe1-A, NBCe1-B, universal NBCe1 isoforms, and ß-actin
Western blotting
Protein membranes from isolated pancreatic islets were first probed with an anti-NBCe1 antibody recognizing all three variants of the cotransporter. The band detected in rat pancreatic islets corresponded to the lower extremity of the band detected in rat kidney (unglycosylated band), used as a positive control (data not shown). Immunoblotting with antibodies specific for each variant confirmed the expression of NBCe1-A and NBCe1-B in rat pancreatic islets (Fig. 2). The NBCe1-A band in the islets was less intense than that in the kidney, despite the fact that the amount of proteins used for the pancreatic islets was 20 times higher. Likewise, using islet and pancreatic homogenates, containing equivalent amounts of protein, the staining of a 150 kDa band corresponding to NBCe1-B was more pronounced in islets than in pancreas (data not shown). The presence of NBCe1-B in rat pancreas was confirmed by using a plasma membrane-enriched preparation (Fig. 2). These findings on the expression of NBCe1-A and NBCe1-B are superimposable to those obtained by RT-PCR.

Western blot analysis of kidney and islet crude homogenates and a pancreas plasma-membrane enriched preparation with specific antibodies for NBCe1-A and NBCe1-B. The protein content of each sample is indicated at the bottom of the figure
Immunocytochemical localization of NBCe1
In rat pancreatic section, the antibody recognizing all three variants of the cotransporter stained much more intensely pancreatic islets than the surrounding exocrine tissue (Fig. 3). This commercial anti-NBC1 antibody stained heavily the rat kidney proximal tubules while the staining of the exocrine pancreas was more patchy in both acini and ductules (data not shown).

Immunodetection of NBC by anti-NBC1 rabbit antibody (1:100 dilution) in rat pancreatic secretion. Immunohistochemical labelling was performed using avidin–biotin-peroxidase complex with diaminobenzidine (a) or using fluorescein-labeled secondary antibody (b)
Using the same antibody, NBC was localized in an isolated rat pancreatic islet: both insulin and glucagon producing cells appeared to express NBCe1 (Fig. 4).

Labeling of isolated rat pancreatic islets with anti-NBC1 (a and d), anti-insulin (b), antiglucagon (e) documenting NBC (green), glucagon and insulin (red) positive cells. Panels c and f refer to double labeling of NBC-insulin and NBC-glucagon, respectively
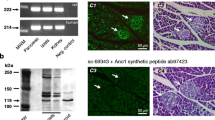
To distinguish whether the labeling observed with this antibody could be attributed to either NBCe1-A or NBCe1-B variant, variant-specific antibodies were used. NBCe1-A immunolabeling (Fig. 5a) of weak intensity was observed in pancreatic islets (I), whereas pancreatic acinar cells (A) were completely devoid of NBCe1-A immunoreactivity, confirming previous observations [6]. Immunoreactivity for NBCe1-B (Fig. 5b) was found in both pancreatic acinar cells (A) and islets (I), yet with a labeling intensity considerably stronger in islets than in exocrine pancreas, confirming immunoblotting data. To underpin the specific localization of each variant in the islet cells, double labeling using anti-insulin and anti-glucagon antibodies was performed. NBCe1-A showed partial co-localization with insulin (Fig. 5c), whereas in glucagon-expressing cells NBCe1-A immunoreactivity was absent (Fig. 5e). The NBCe1-B antibody clearly labeled insulin-producing cells located at the center of the islets (Fig. 5d), but failed to do so in the glucagon-producing cells located at the periphery of the islets (Fig. 5f).

Immunofluorescence for NBCe1-A and NBCe1-B in islets (I) and acinar tissue (A) of rat pancreatic sections (a, b), and double labeling of NBCe1-A (red, c, e) or NBCe1-B (red, d, f) and either insulin (green, c, d) or glucagon (green, e, f)
Insulin secretion
Tenidap (3–100 μM) caused a concentration-related inhibition of insulin secretion evoked over 90 min incubation by d-glucose (16.7 mM) in rat isolated islets, with a half-maximal inhibition close to 50 μM (Fig. 6). At a concentration of 0.1 mM, tenidap failed to affect significantly (P > 0.55) the basal release of insulin recorded in the presence of 5.6 mM d-glucose, abolished (P < 0.01) the secretory response at 8.3 mM d-glucose, and severely decreased (P < 0.001) the insulinotropic action of 16.7 mM d-glucose (Fig. 6). In the presence of tenidap (0.1 mM), the output of insulin recorded at 8.3 mM d-glucose, indeed failed to differ significantly (P > 0.29) from that found at 5.6 mM d-glucose, while remaining lower (P < 0.006) than that measured at 16.7 mM d-glucose.

a Effect of increasing concentrations of tenidap upon insulin output from rat pancreatic islets incubated in the presence of 16.7 mM d-glucose. The oblique line corresponds to the regression line. The horizontal arrow corresponds to a half-maximal inhibition of the secretory response to the hexose and the vertical dotted line to the corresponding tenidap concentration (50 μM). Mean values (±SEM) refer to the number of separate determinations indicated at the bottom of the figure. b Effects of increasing concentrations of d-glucose upon insulin output from rat pancreatic islets incubated in the absence or presence of tenidap (0.1 mM). Mean values (±SEM) refer to the number of separate number of determinations indicated at the top of the figure
The effect of tenidap was also investigated in islets exposed to non-glucidic nutrient secretagogues. Tenidap (0.1 mM) significantly decreased (P < 0.001) insulin secretion provoked by 10.0 mM 2-ketoisocaproate from 85.5 ± 4.4 to 49.7 ± 5.3 μU/90 min per islet (n = 19 in both cases). These results indicate that 10.0 mM 2-ketoisocaproate stimulated insulin secretion as efficiently (P > 0.35) as 8.3 mM d-glucose, and that the relative extent of tenidap inhibitory action was also comparable (P > 0.3) with the 2-keto acid (41.9 ± 8.1%; df = 36) and the hexose (57.0 ± 10.4%; df = 72). l-leucine (20.0 mM) also stimulated insulin secretion (71.2 ± 4.3 μU/90 min per islet) as efficiently (P > 0.65) as 8.3 mM d-glucose. However, the inhibitory action of tenidap upon leucine-stimulated insulin secretion, albeit achieving statistical significance (P < 0.03), corresponded to no more than a 23.6 ± 10.4% (df = 35) relative decrease in insulin output, the latter percentage representing less than half (P < 0.05) of that recorded in islets exposed to 8.3 mM d-glucose.
The effect of tenidap (0.1 mM) on the insulinotropic action of non-nutrient secretagogues was tested over 90 min incubation in the presence of 8.3 mM d-glucose (Table 1). In this set of experiments, tenidap again decreased insulin output recorded in the sole presence of the hexose to 45.6 ± 4.2% (n = 38) of the mean corresponding value found within the same experiment in the absence of tenidap (100.0 ± 4.7%; n = 37). At 8.3 mM d-glucose, theophylline (1.4 mM), forskolin (5.0 μM), glibenclamide (5.0 μM) and cytochalasin B (0.2 mM) all significantly increased (P < 0.005) insulin secretion by islets incubated in the absence of tenidap. In the presence of tenidap, however, the release of insulin failed to differ significantly in the islets incubated in the presence of d-glucose alone or in the concomitant presence of the hexose and a non-nutrient secretagogue.
22Na+ handling
After 10 min of incubation in the absence of ouabain and tenidap, the net uptake of 22Na+ by dispersed rat pancreatic islet cells averaged 194 ± 37 pmol/103 cells (n = 12). Under similar experimental conditions, the extracellular volume, as judged from the apparent distribution space of l-[1-14C]glucose (2.0 mM) averaged 46.4 ± 3.3% (n = 12; P < 0.001) of 3HOH distribution space (100.0 ± 8.9%; n = 12). The intracellular volume taken as the difference between the 3HOH and l-[1-14C]glucose distribution spaces, averaged 1.11 ± 0.19 nl/103 cells (df = 20). Hence, after correction for extracellular contamination, the net uptake of 22Na+ mentioned above corresponded to an estimated intracellular Na+ concentration of 35.9 ± 5.7 mM.
The time course for 22Na+ net uptake was comparable under all experimental conditions. After 20 min incubation, the measurements averaged 132.8 ± 14.2% (n = 41; P < 0.02) of the mean values recorded within the same experiments and under the same experimental conditions after only 5–10 min incubation (100.0 ± 5.8%; n = 90).
Relative to the mean corresponding value recorded within the same experiment after the same length of incubation (5, 10, and 20 min), the cell pellet 22Na+ content of dispersed islet cells incubated in the absence of ouabain averaged, in the presence of tenidap (0.1 mM), 126.5 ± 13.0% (n = 36) as compared to 100.0 ± 8.6% (n = 34; P < 0.10) in its absence. When ouabain was present during both pre-incubation (2.0 mM) and incubation (1.0 mM), the results recorded in the presence of tenidap averaged 145.6 ± 19.6% (n = 30), as compared (P < 0.05) to a mean reference value of 100.0 ± 9.4% (n = 30). In the presence of tenidap, such normalized percentages thus failed to differ significantly (P > 0.4) in the presence or absence of ouabain, yielding an overall mean value of 135.2 ± 11.3% (n = 66), as compared (P < 0.01) to a mean reference value of 100.0 ± 6.3% (n = 64).
d-glucose metabolism
When isolated rat islets were incubated for 90 min in the presence of 16.7 mM d-glucose, tenidap, tested at a 30 μM concentration, inhibited (P < 0.05 or less) both the utilization of d-[5-3H]glucose from 134.8 ± 22.3 (n = 9) to 63.0 ± 25.1 (n = 8) pmol/islet and the oxidation of d-[U-14C]glucose from 24.3 ± 2.8 to 7.9 ± 1.5 pmol/islet (n = 10 in both cases). The relative extent of such an inhibition failed to differ significantly (P > 0.6) for the two metabolic variables under consideration, with an overall mean value of 61.0 ± 13.2% (df = 33; P < 0.004).
Intracellular pH
As illustrated in Fig. 7, tenidap (50–100 μM) provoked a rapid and slowly reversible cellular acidification. As judged from the F440/480 ratio, the relative magnitude of such a decrease in intracellular pH was inversely related (r = −0.989) to the concentration of d-glucose (4, 8, and 16 mM).

a–c Effect of tenidap (50–100 μM), during the period indicated by the horizontal lines, on intracellular pH as judged from the 440 nm to 480 nm fluorescence ratio in rat islet cells loaded with BCECF. d, e Effects of d-glucose (4 and 16 mM) and tenidap (0.1 mM) administered during the period(s) indicated by the horizontal lines on membrane potential in isolated insulin-producing islet cells, using the perforated-patch technique
Membrane potential
Tenidap (0.1 mM) provoked within 1–2 min, a pronounced hyperpolarization of the β-cell membrane potential, whether in the presence of 4 or 16 mM d-glucose (Fig. 7). At the higher concentration of the hexose, this coincided with the suppression of spiking activity. The effect of tenidap on membrane potential was, in most cases, reversible, but this required a prolonged washout period of at least 10 min.
Discussion
The present study demonstrates the expression of both NBCe1-A and NBCe1-B in rat pancreatic islets. The RT-PCR, western blot, and immunocytochemistry data were consistent with a higher level of expression of the NBCe1-B variant, as compared to the NBCe1-A variant, in rat pancreatic islets. Double labeling of islets in pancreatic sections using variant-specific antibodies further indicated that the most abundant NBCe1-B variant is mainly and perhaps exclusively located in insulin-producing cells. The NBCe1-A variant was apparently restricted to some insulin-containing cells. Glucagon-producing cells were indeed devoid of both NBCe1-A and NBCe1-B immunolabeling. The co-localization observed with glucagon and the commercial NBC antibody suggests, therefore, that glucagon-secreting cells may express another NBC isoform recognized by this universal antibody.
The functional significance of the Na+–HCO3 − co-transporters was investigated by exposing isolated pancreatic islets or dispersed islet cells to an alleged selective inhibitor of the renal Na+–HCO3 − cotransporter, i.e., tenidap [6]. It cannot be ignored, however, that the specificity of tenidap action could be questioned. In this respect, a second approach using a knock-down model is now considered.
At concentrations up to 0.1 mM, tenidap caused a concentration-related decrease of glucose-stimulated insulin release with a half maximal inhibitory effect at about 50 μM tenidap. In experiments conducted in Xenopus laevis oocytes expressing the renal Na+–HCO3 − cotransporter, an IC50 of 13 μM was found for the inhibition by tenidap of cotransport current [6]. The inhibition of glucose-stimulated insulin release by tenidap coincided with a decrease of both d-[5−3H]glucose utilization and d-[U-14C]glucose oxidation by isolated rat pancreatic islets. Tenidap also inhibited insulin secretion evoked by 2-ketoisocaproate and l-leucine. As already observed in other cell types [7, 8], tenidap also lowered intracellular pH in dispersed rat islet cells. In the light of the results of a prior study conducted in leucine-stimulated rat pancreatic islets [9], the latter finding might well account for the relatively less marked inhibition by tenidap of the secretory response to this amino acid, as distinct from that to either d-glucose or 2-ketoisocaproate. Furthermore, tenidap provoked in dispersed islet cells, plasma membrane hyperpolarization with the suppression of bioelectrical spiking activity, otherwise recorded in cells exposed to a high concentration of d-glucose. Last, tenidap augmented the net uptake of 22Na+ by dispersed islet cells exposed to 16.7 mM d-glucose. Like in BRIN-BD11 cells [10], such an increase was most obvious in cells also exposed to ouabain. At this point, it could be objected that the prevailing NBCe1-B variant expressed in islet cells is currently considered to work following an influx mode, e.g., in pancreatic duct cells [2]. Inter alia, the relatively high concentrations of HCO3 − (alkaline pH) and of Na+ (about 36 mM) in islet cells exposed to 16.7 mM d-glucose could, however, allow NBCe1-B to cotranport one Na+ and two HCO3 − ions from inside the islet cells into the extracellular fluid. This would generate a net inward depolarizing current, consistent with the hyperpolarization accompanying inhibition of the transporter by tenidap.
In conclusion, the present study is compatible with the view that in rat pancreatic islet cells, variants of NBCe1 Na+–HCO3 − cotransporter contribute to the efflux of HCO3 − generated at the intervention of carbonic anhydrase V from CO2 itself produced through the oxidative catabolism of nutrient insulin secretagogues. Such a proposal does not rule out, however, the participation of other modalities of HCO3 − outflow from islet cells, e.g., through other NBC isoforms or through volume-sensitive anion channels, in a fashion similar to that recently proposed for the exit of phosphate ions during the early so-called phosphate flush provoked by d-glucose and other nutrient secretagogues in pancreatic islets [11]. Furthermore, the postulated cotransport of Na+ and HCO3 − may well participate in the increase of 22Na+ outflow from prelabeled islets provoked by raising extracellular d-glucose concentration [12, 13].
Research design and methods
Animals
Wistar rats (Iffa Credo; L’Arbresle, France) were sacrificed under CO2 anesthesia. The pancreas and kidney were quickly excised and immediately frozen in liquid nitrogen or processed for microscopy or for islet isolation.
Reverse transcription-polymerase chain reaction
Total RNA, extracted from frozen rat tissues using AURUMTM total RNA fatty acid and tissue kit (Biorad, Hercules, CA, USA) was quantified on a nanodrop spectrophotometer (Nanodrop Technologies, Inc., Wilmington, DE, USA) and its quality was checked using an experion automated Electrophoresis system (Bio-Rad, Hercules, CA, USA). Total RNA was reversed transcribed from 1 μg of total RNA in a final volume reaction of 20 μl using Revert AidTm first strand cDNA synthesis kit (Fermentas, St Leon Rot, Germany). The resulting cDNA was subjected to independent PCR amplification with primer sets corresponding to NBCe1 variants, NBCe1-A (accession n° AF027362, amplicon: 344pb), NBCe1-B (accession n° AF254802, amplicon: 473pb) and a universal (uNBCe1) primer set amplifying all variants [14, 15].
RT-PCR reactions were carried out in a final volume of 20 μl, containing 1 μl of cDNA added to 4 μl of GO Taq Green 5X buffer, 0.2 mM dNTP, 0.5 μm of each primer, 0.5 U of GO TaqTM DNA polymerase (Promega, Madison, WI, USA). Amplification was performed in a thermocycler system (i-Cycler, Bio-rad Laboratories, Hercules, CA, USA). Thermocycling conditions for variant A and variant B were: denaturation at 95°C for 90 s, followed by 33 cycles of (95°C for 30 s, 55°C for 30 s, 72°C for 60 s), and terminated by a final elongation at 72°C for 5 min. For the amplification using the universal primer set uNBCe1, the conditions were adapted: denaturation at 95°C for 90 s, followed by 35 cycles of (95°C for 20 s, 61°C for 20 s, 72°C for 30 s), and terminated by a final elongation at 72°C for 5 min. For β-actin amplification, 30 cycles were performed at 95°C for 30 s, 55°C for 30 s, 72°C for 60 s. Ten microliters of each PCR product were submitted to 1.2% agarose gel electrophoresis in TAE buffer (40 mM Tris, 40 mM acetate, 1 mM EDTA) in the presence of 0.5 μg/ml of ethidium bromide. Gels were visualized by UV translumination using a GelDoc apparatus (Bio-Rad Laboratories, Hercules, CA, USA).
Western blot analysis
Crude membranes from rat kidney, pancreas, and isolated pancreatic islets were prepared as described previously [16] except that a membrane pellet of 17,000g centrifugation was used for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Rat pancreatic plasma membranes were prepared exactly as described elsewhere [15]. The samples were analyzed by using 7.5% polyacrylamide gels. Proteins were then transferred to polyvinylidene difluoride membranes and immunolabeled using the anti-NBC1 antibody (AB3204, Chemicon, Temecula, CA, USA) or using antibodies specific to NBCe1-A and NBC1e-B according to a procedure described elsewhere [15, 17]. The bound antibodies were then detected using the enhanced chemiluminescence method (ECL kit, Amersham, Piscataway, USA).
Tissue preparation for immunocytochemistry
Tissues were fixed for 24 h, at room temperature, in formalin 10% v/v diluted in PBS (NaCl 145 mM, NaH2PO4/Na2HPO4 10 mM, pH 7.2). After automated dehydration (Leica Microsystems, Belgium), tissues were embedded in paraffin. Sections sized 5 μm were cut with a conventional microtome (Reichert-Jung, Germany) and placed onto Superfrost slides (International Medical Products, Belgium).
Immunohistochemistry ABC-DAB
Immunodetection followed the standard ABC-DAB technique [18] using the avidin–biotin blocking kit (Vector Laboratories, Belgium). The slides were incubated overnight at 4°C with the first antibody: anti-NBC-1 (AB-3204, Chemicon, Temecula, CA, USA), a rabbit polyclonal, affinity purified, antibody raised against sequence 990–1035 of rat Na+–HCO3 − cotransporter. The primary antibody was used at dilutions ranging from 1/100 to 1/1000 in PBS with appropriate blocking serum at a dilution of 1/20. Purified immunoglobulins (IgG) (Sigma-Aldrich, Belgium) from non-immunized rabbit were used as negative controls. The slides were further incubated with the secondary biotinylated antibody [goat anti-rabbit IgG (H + L)] (BA-1000, Vector Labs, Belgium) at a dilution of 1/300 in PBS for 30 min, at room temperature. The staining pattern was ascertained with an Axioplan and recorded with an Axiocam (Carl Zeiss, Germany).
Immunofluorescence labeling using universal anti-NBC1 antibody
Rehydrated paraffin sections were blocked for non-specific reactions with normal goat serum (Vector Labs, Belgium) and non-specific staining related to endogenous biotin was blocked with the avidin–biotin blocking kit (Vector Labs, Belgium). The slides were incubated with the primary anti-NBC1 antibody overnight at 4°C, at a concentration of 1/100 in normal goat serum (1/20 in PBS). The secondary antibody, a biotinylated goat anti-rabbit IgG (H + L) (BA-1000, Vector Labs, Belgium) was applied at a dilution of 1/300 for 30 min at room temperature. Further 30 min incubation with FITC-Avidin D (Vector Labs, Belgium) was used for revelation of the second antibody. The staining patterns were observed with an Axioplan and recorded with an Axiocam (Carl Zeiss, Germany). The slides were unmounted in PBS at 37°C. Anti-insulin (I2018, Sigma-Aldrich, Belgium) or anti-glucagon (G2654, Sigma-Aldrich, Belgium) was applied at a dilution of 1/3000 and 1/2000, respectively, overnight, at 4°C. The secondary antibody was a Rhodamine Red X-conjugated goat anti-mouse IgG (H + L) (115-295-146, Jackson ImmunoResearch Laboratories, USA) diluted at 1/100 in PBS/normal goat serum 1/20.
Immunofluorescence labeling using variant-specific anti-NBCe1-A and NBCe1-B antibodies
The pancreata were fixed in 4% paraformaldehyde. Immunofluorescence was performed in 10 μm fixed cryosections, as previously described [15]. Sections were treated with 1% SDS/PBS for 5 min, washed with PBS, and blocked with 1% BSA/PBS for 15 min. Subsequently, sections were incubated with rabbit polyclonal antibody raised against either NBCe1-A or NBCe1-B at a dilution of 1:100 overnight at 4°C, followed by incubation with goat-anti-rabbit IgG coupled to CY3, at a dilution of 1:600 for 1 h at room temperature. For double labeling of NBCe1-A and NBCe1-B with insulin or glucagon, sections were treated as above, followed by incubation with mouse monoclonal antibody against insulin at a dilution of 1:3,000 or glucagon at a dilution of 1:2,000 as second primary antibody. Finally, sections were incubated with goat anti-mouse IgG coupled to FITC as second secondary antibody at a dilution of 1:200 for 1 h at 20°C. After washing with PBS, sections were mounted with Vectashield and viewed with a ZEISS Apotome fluorescence microscope. The findings illustrated in Figs. 4, 5 are representative of several independent experiments.
Functional studies
The methods used for the isolation of rat pancreatic islets [19] and to measure insulin release [19] and d-glucose metabolism [20] were previously described in the cited references. Likewise, intracellular pH measurements and electrophysiological experiments, both carried out with dispersed rat islet cells, were performed as previously described [1, 21]. The membrane potential of insulin-producing cells was monitored with the perforated configuration of the patch clamp technique. Intracellular pH was assessed from the 440 nm to 480 nm fluorescence ratio in cells loaded with the pH-sensitive dye BCECF [21].
For the study of 22 Na+ net uptake by dispersed cells, groups of 5–10 × 104 dispersed islet cells were pre-incubated for 30 min at 37°C in 50 μl of a salt-balanced medium [22] equilibrated against O2/CO2 (95/5, v/v) and containing 5 mg/ml bovine serum albumin and 16.7 mM d-glucose, in the presence or absence of ouabain (2.0 mM). After addition of 50 μl of the same salt-balanced medium (except for the absence of ouabain) now containing a tracer amount of 22Na+ (3.1 nCi/μl) and, when required, 0.2 mM tenidap (Pfizer, Groton, CT, USA), the cells were further incubated for 5, 10, and 20 min at 37°C and eventually separated from the incubation medium as detailed elsewhere [23]. Their 22Na+ content was measured by liquid scintillation.
All results are presented as mean values ± SEM together with either the number of separate determinations (n) or degree of freedom (df). The statistical significance of differences between mean values was assessed by use of Student’s t-test.
References
A. Sener, H. Jijakli, S. Zahedi Asl, P. Courtois, A.P. Yates, S. Meuris, L.C. Best, W.J. Malaisse, Am. J. Physiol. 292, E1624 (2007)
M.D. Parker, W.F. Boron, in Seldin and Giebisch’s the Kidney: Physiology and Pathophyiology, ed. by R.J. Alpern, S.C. Hebert (Elsevier Academic Press, Amsterdam, 2008), p. 1481
W.F. Boron, E.L. Boulpaep, J. Gen. Physiol. 81, 53 (1983)
M.F. Romero, M.A. Hediger, E.L. Boulpaep, W.F. Boron, Nature 387, 409 (1997)
M.F. Romero, P. Fong, U.V. Berger, M.A. Hediger, W.F. Boron, Am. J. Physiol. 274, F425 (1998)
O. Ducoudret, A. Diakov, S. Müller-Berger, M.F. Romero, E. Frömter, Eur. J. Physiol. 442, 709 (2001)
J. Bondeson, Gen. Pharmacol. 27, 943 (1996). 27
H. Matsumoto, A. Fujii, J. Pharmacol. Exp. Ther. 300, 668 (2002)
A. Sener, W.J. Malaisse, Diabete Metab 6, 97 (1980)
W.J. Malaisse, K. Louchami, A. Sener, R. Beauwens, Diabetes 57(Suppl 1), A730 (2008)
K. Louchami, Y. Zhang, R. Beauwens, W.J. Malaisse, A. Sener, Endocrine 31, 1 (2007)
A.C. Boschero, W.J. Malaisse, Arch. Int. Physiol. Biochim. 86, 479 (1978)
S. Kawazu, A.C. Boschero, W.J. Malaisse, Pflügers Arch. 375, 197 (1978)
J. Praetorius, Y.H. Kim, E.V. Bouzinova, S. Friesche, A. Rojek, C. Aalkaer, S. Nielsen, Am. J. Physiol. 286, F903 (2004)
E. Roussa, W. Nastainczyk, F. Thévenod, Biochem. Biophys. Res. Commun. 314, 382 (2004)
M.S. Soyfoo, C. De Vriese, H. Debaix, M.D. Martin-Martinez, C. Mathieu, O. Devuyst, S.D. Steinfeld, C. Delporte, Arthritis Rheum. 56, 2566 (2007)
A. Brandes, O. Oehlke, A. Schümann, S. Heidrich, F. Thévenod, E. Roussa, Am. J. Physiol. 293, R2400 (2007)
S.M. Hsu, L. Raine, H. Fanger, J. Histochem. Cytochem. 29, 577 (1981)
F. Malaisse-Lagae, W.J. Malaisse, in Methods in Diabetes Research, vol. I, ed. by J. Larner, S. Pohl (Wiley, New York, 1984), p. 174
W.J. Malaisse, A. Sener, Biochim. Biophys. Acta 971, 246 (1988)
L. Best, A.C. Elliott, Mol. Cell. Endocrinol. 111, 191 (1995)
W.J. Malaisse, C. Maggetto, V. Leclercq-Meyer, A. Sener, J. Clin. Invest. 91, 432 (1993)
M.-H. Giroix, E. Agascioglu, B. Oguzhan, K. Louchami, Y. Zhang, P. Courtois, W.J. Malaisse, A. Sener, Biochim. Biophys. Acta 1757, 773 (2006)
Acknowledgments
We thank Y. Zhang, E. Hupkens, and C. Demesmaeker for scientific, technical, and secretarial help. This work was supported by grants from the Belgian Foundation for Scientific Medical Research (3.4520.07) and the Deutsche Forschungsgemeinschaft (F.T). M.S.S is recipient of a scholarship from the Erasme Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Soyfoo, M.S., Bulur, N., Virreira, M. et al. Expression of the electrogenic Na+–HCO3 −-cotransporters NBCe1-A and NBCe1-B in rat pancreatic islet cells. Endocr 35, 449–458 (2009). https://doi.org/10.1007/s12020-009-9175-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-009-9175-1