
Abstract
For many years, adipose tissues have been considered a lipid storage organ, with a vital role limited to energy balance. However, since the discovery of leptin 20 years ago, this view changed radically. Now, it is well recognized that white adipose tissue is an endocrine organ able to secrete a wide variety of factors called adipokines. These hormones have been demonstrated to play relevant roles in metabolism, immunity, inflammation and also in bone metabolism. Thus, this review summarizes the recent findings in basic research about the involvement of several adipokines in bone metabolism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Before the discovery of leptin 20 years ago, adipose tissue was considered a mere fat depot. However, in the last years, this view totally changed, and now it is well recognized that white adipose tissue (WAT) is also an endocrine organ able to produce and secrete a wide variety of factors termed adipokines [1, 2]. These hormones have been demonstrated to be involved in several biological processes such as metabolism, immunity and inflammation [3–6]. More recently, there is a growing interest about the role of adipokines in the regulation of bone metabolism [7–11].
Alterations at bone level are common features of different diseases such as osteoarthritis (OA) and especially osteoporosis. Then, the study of the complex network that regulates bone biology, in physiological and pathological conditions, represents an interesting field aimed to achieve new therapeutic targets for bone-associated disorders. Recently, many data showed the participation of adipokines in the regulation of critical processes as bone formation and resorption [12, 13]. These data suggest that adipose tissue-derived hormones could impact bone physiology, highlighting a promising research field, in which adipose tissue, through the action of adipokines, may regulate certain disorders related with a dysregulation of bone metabolism.
Thus, in this, we provide an overview of recent advances on basic aspects of bone metabolism.
Leptin
Leptin is a 16 kDa non-glycosylated protein encoded by the ob gene, the murine homologue of the human gene LEP, cloned in 1994 [1]. Leptin is a member of the superfamily of adipose tissue-derived hormones, which have been termed collectively “adipokines.” It is mainly produced by adipocytes, and in physiological conditions, leptin circulating levels correlate positively with WAT mass. This hormone acts in the brain as a regulating factor that induces a decrease in food intake and an increase in energy consumption by inducing anorexigenic factors (e.g., CART [cocaine- and amphetamine-regulated transcript], POMC [pro-opiomelanocortin]) and suppressing orexigenic neuropeptides (e.g., NPY [neuropeptide Y], AgRP [agouti-related peptide] and orexin) [4]. Its own synthesis is mainly regulated by food intake, but also depends on energy status, sex hormones (being inhibited by testosterone and increased by ovarian sex steroids) and a wide range of inflammation mediators, [14, 15] being increased or suppressed by pro-inflammatory cytokines depending on whether their action is acute or chronic. Through the mediation of these latter agents, leptin synthesis is increased also by acute infection and sepsis [16]. As a result of the effects of sex hormones, leptin levels are higher in women than in men even when adjusted for BMI, which may be relevant to the influence of sex on the development or frequency of certain diseases [17], such as OA or osteoporosis (Table 1).
Leptin and Bone
The first evidence of the possible involvement of leptin in controlling bone metabolism was published by Thomas et al. These authors demonstrated that leptin increased osteoblast differentiation and inhibited differentiation of adipocytes in conditionally immortalized human marrow stromal cells [18]. Few years later, other authors also reported an increased proliferation and decreased apoptosis of osteoblasts after leptin exposure [19]. In addition, this effect was accompanied by increased mineralization and expression of different markers of osteoblast differentiation [19]. More recently, it was published that disruption of the long form of leptin receptor in primary one marrow stromal cells resulted in decreased mineralization and increased adipogenesis [20].
However, some of the effects of leptin on bone marrow stromal cells, previously published, are not in agreement with the above mentioned findings. It has been reported that leptin increased human bone marrow stromal cells apoptosis, and this process seems to occur through ERK/cPLA2/cytochrome c pathway and the activation of the caspases 3 and 9 [21]. Moreover, it was also showed that leptin signaling blockade increased osteoblast differentiation [22]. Very recently, it was also postulated that leptin may participate in the osteoblastic differentiation of vascular smooth muscle cells [23, 24]; this is a complex process resulting in arterial calcification. Two studies demonstrated that leptin was able to regulate this differentiation process via GSK-3β and RANKL/BMP4 pathways [23, 24].
Studies aimed to define the regulation of osteoclastogenesis by leptin presented controversial results. Some authors did not find any difference in osteoclast differentiation and function in cell cultures obtained from leptin-deficient mice in comparison to those obtained from wild-type mice [25]. On the other hand, it was reported that leptin inhibited osteoclastogenesis in mouse bone marrow cultures [26]. In line with this, Holloway et al. [27] showed that leptin decreased osteoclast generation in cultures of human peripheral blood mononuclear cells in presence of hM-CSF and RANKL.
In the past years, some studies demonstrated that osteoblasts could also participate in joint destruction in several diseases as rheumatoid arthritis or OA [28]. Leptin was able to increase the expression of oncostatin M by downregulating the microRNA miR93 and Akt signaling [29]. Oncostatin M is a cytokine of the interleukin-6 family, which is able to produce certain chemotactic proteins as monocyte chemoattractant protein-1 (MCP-1) or metalloproteinases as MMP-1 in human synovial fibroblasts [30]. MCP-1 has been demonstrated to participate in joint destruction in adjuvant-induced arthritis model [31]. This study revealed that MCP-1 inhibition decreased macrophage accumulation and pro-inflammatory cytokines production in the joint. Moreover, these authors observed less radiographic joint damage in animals treated with MCP-1 inhibitor [31]. Similarly, MMP-1 is well known for its prominent role in collagen network remodeling and degradation, which confers to this proteinase a relevant role in tissue destruction during joint degenerative diseases [32].
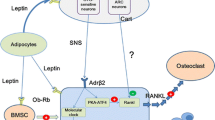
In a similar way, in vivo studies showed contradictory results and the effect of leptin on bone metabolism is still under debate. Nevertheless, most works suggest a negative role for leptin by enhancing sympathetic output to bone from the hypothalamus by suppressing serotonin system in the brainstem [25, 33]. According to these data, leptin, secreted from adipose tissue, crosses the blood brain barrier and acts through the leptin receptor to inhibit serotonin (5HT) production in containing neurons in the brainstem [25, 34]. Normally, serotonin would be secreted from these nerve terminals in the ventromedial hypothalamus to suppress sympathetic activity to bone. However, under leptin-induced inhibition of serotonin synthesis, the sympathetic nervous system signals to osteoblasts by releasing norepinephrine onto β2 adrenergic receptors. This, in turn, suppresses bone formation and increases resorption through increased RANK ligand expression [7].
Suppression of bone formation by leptin was demonstrated in leptin-deficient mice [25], but also in a leptin gain-of-function model [35]. These authors showed that this mouse model presented a low bone mass phenotype, without any alteration in appetite or energy expenditure. These authors, through mouse genetic studies causing a deletion of the leptin receptor in neurons, demonstrated an increase in bone formation and bone resorption, resulting in a high bone mass as seen in leptin-deficient mice. On the other hand, the same deletion in osteoblasts does not influence bone remodeling [35] (Fig. 1).

Schematic representation of the effect of leptin on bone metabolism
Adiponectin
Adiponectin, also known as GBP28, apM1, Acrp30 or AdipoQ, is a 244-residue protein with structural homology to types VIII and X collagen and complement factor C1q that is prevalently synthesized by adipose tissue. Adiponectin circulates in the blood in large amounts and constitutes approximately 0.01 % of the total plasma proteins and can be found as different molecular forms (trimers, hexamers and also 12–18-monomer forms) [36, 37]. The gene that encodes for human adiponectin is located on chromosome 3q27, a locus linked with susceptibility to diabetes and cardiovascular diseases [38]. Ablation of the adiponectin gene has no dramatic effect on knockout mice on a normal diet, but when placed on a high fat/sucrose diet, animals develop severe insulin resistance and exhibit lipid accumulation in muscles [39]. Circulating adiponectin levels tend to be low in morbidly obese patients and increase with weight loss and with the use of thiazolidinediones (PPAR agonists) which enhance sensitivity to insulin [36, 40]. Adiponectin decreases insulin resistance by stimulating glucose uptake, by increasing fatty acid oxidation and reducing the synthesis of glucose in the liver and other tissues [36].
Adiponectin acts via two receptors, one (AdipoR1) found predominantly in skeletal muscle and the other (AdipoR2) in liver. Transduction of the adiponectin signal by AdipoR1 and AdipoR2 involves the activation of AMPK, PPAR-α, PPAR-γ and other signaling molecules [36]. To note, targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and all its metabolic actions [41].
Adiponectin and Bone
Several studies [13, 42] have supported a functional role for adiponectin in bone biology, demonstrating that this adipokine and its receptors are expressed in osteoblasts [43], which also differentiate in response to this hormone. In fact, adiponectin was able to stimulate the proliferation and mineralization of human osteoblasts via the p38 mitogen-activated protein kinase (MAPK) signaling pathway in autocrine and/or paracrine manners [44, 45]. Moreover, this adipokine enhanced the expression of bone morphogenetic protein 2 (BMP-2), which plays a fundamental role in osteoblast differentiation and bone formation, in cultured osteoblastic cells [46]. Adiponectin inhibited differentiation of bone marrow macrophages and CD14+ mononuclear cells into osteoclasts [42]. In contrast, adiponectin indirectly activates osteoclasts by stimulating RANKL and inhibiting OPG (osteoprotegerin) production in osteoblasts [8]. In vitro studies also showed that adiponectin was able to stimulate the production of VEGF, MMP-1, MMP-13, IL-6 and IL-8 in cultured osteoblasts [47], suggesting an important role for adiponectin in joint destruction by inducing catabolic and pro-inflammatory mediators in osteoblasts.
In vivo studies highlighted interesting but controversial results. It has been demonstrated that mice treated with adiponectin showed increased trabecular bone mass and decreased number of osteoclasts [42]. In line with this, it was also reported that adiponectin produced an increase in bone mass in mice and this effect occurred through an adipokine-mediated decrease in the sympathetic tone, by acting on neurons of locus coeruleus [48]. However, these authors also showed that adiponectin inhibits osteoblast differentiation and promotes their apoptosis [48]. These results demonstrated that adiponectin could act in two different opposite manners depending on the site of action. In contrast, another study revealed that adiponectin knockout mice displayed increased bone mass [49]. Moreover, adiponectin deficiency can protect against osteoporosis in ovariectomized mice [50]. All together, these findings suggest that adiponectin participates in the balance of bone formation and bone resorption. However, more studies will be necessary to clarify the exact role of this adipokine in bone metabolism.
Visfatin
Visfatin, also called PBEF (pre-B cell colony-enhancing factor) and Nampt (nicotinamide phosphoribosyltransferase), is a protein of approximately 471 amino acids and 52 kDa [51]. It is a hormone that originally was discovered in liver, bone marrow and muscle, but it is also secreted by visceral fat [51, 52].
It has been reported that visfatin levels are increased in obesity. Moreover, leukocytes from obese patients produce higher amounts of visfatin compared with lean subjects, and specifically, granulocytes and monocytes are the major producing cells [53, 54]. Macrophages have been also described as a source for visfatin production [55].
It is supposed that visfatin has insulin mimetic properties, but the role of this adipokine in glucose metabolism is still unclear [52, 56]. Visfatin is upregulated in models of acute injury and sepsis [57], and its synthesis is regulated by factors such as glucocorticoids, TNF-α, IL-6 and growth hormone. Moreover, visfatin has been shown to induce chemotaxis and the production of IL-1β, TNF-α and IL-6 in lymphocytes [58].
Visfatin and Bone
Several studies demonstrated the involvement of visfatin in bone biology. In fact, it was reported that this adipokine stimulated the proliferation of cultured osteoblasts [59]. In addition, visfatin was able to promote glucose transport and type I collagen production in the same cell type being these effects appear dependent on insulin receptor transduction pathway [59]. In line with this, it was also observed that visfatin levels increased during osteogenic differentiation, and this increase was associated with higher nicotinamide adenine dinucleotide (NAD) levels [60]. Interestingly, visfatin knockdown or inhibition caused a decrease in NAD concentration and osteogenesis [60]. These results suggested that osteogenic differentiation depends on NAD levels, and in this scenario, visfatin could play a regulatory role.
Regarding the effect of visfatin on osteoclastogenesis, contradictory results have been published. On one hand, it was reported that visfatin inhibited osteoclastogenesis [61], but on the contrary, it was also showed that visfatin knockdown decreased osteoclast formation [62].
Very recently, it was demonstrated that OA subchondral bone released visfatin, and in vitro experiments showed that the administration of this adipokine induced the expression of IL6 and MCP-1 in osteoblasts [63]. Thus, visfatin could participate in the activation of this cell type during OA.
Taken together, these data suggested that adipose tissue could influence bone biology through the action of adipokines such as visfatin. Very recently, it was proposed that visfatin could exert positive effects on fetal/neonatal bone metabolism [64]. On the other side, senile osteoporosis is related with a progressive decrease in bone mass and higher accumulation of marrow fat. The marrow adipocytes and osteoblasts share the same cellular progenitor and recent in vitro studies showed that visfatin activity could influence the differentiation of mesenchymal stem cells to adipocytes or osteoblasts [65], revealing a potential role for visfatin in the development of senile osteoporosis.
Resistin
Resistin, also called adipocyte-secreted factor or found in inflammatory zone 3 (FIZZ3), was discovered in 2001 and it is a 12.5 kDa protein, which is constituted by 108 amino acids in human and 114 amino acids in mice. Resistin belongs to FIZZ family (also known RELMs, resistin-like molecules) [66, 67]. The gene that codes for human resistin is located on chromosome 19p13.2. Mouse and human resistin genomic DNA are of 46.7 % similarly, and the proteins are 59 % [67]. The major source of resistin in mice is WAT [66], whereas in humans, it is predominantly expressed in macrophages [68]. Thus, in human adipose tissue, resistin is mainly produced by non-adipocyte resident inflammatory cells [69].
Resistin acts as a circulating polypeptide, and it participates in many different processes. Multiple tissues have been described to be responsive to resistin, and the mechanism of action might be both endocrine and paracrine, but the resistin receptor remains still unidentified.
Resistin and Bone
There are few evidences of the implication of resistin in bone biology. Resistin expression has been reported in osteoblasts and osteoclasts [70]. Interestingly, the production of this adipokine increased during osteoclasts differentiation and appeared to be regulated by PKC and PKA signaling pathways [70]. In line with this, recombinant resistin stimulates osteoclasts differentiation, suggesting a role for resistin in osteoclastogenesis. In addition, this adipokine also induced osteoblasts proliferation [70]. These results suggest that resistin could contribute to bone metabolism and remodeling via two main ways: enhancement of osteoclasts differentiation and the recruitment of osteoblasts.
Other Adipokines
Lipocalin-2
Lipocalin-2, also known as siderocalin, 24p3, uterocalin and neutrophil gelatinase-associated lipocalin (NGAL), is constitutively expressed in myelocytes and stored in neutrophil specific granules [71, 72]. LCN2 expression was also found in chondrocytes [73], although WAT is thought to be the main source [74]. NGAL can exist as a monomer, as a disulfide-linked homodimer, or as a disulfide-linked heterodimer in a complex with MMP-9 [75, 76]. Human NGAL consists of a single disulfide-bridged polypeptide chain of 178 amino acid residues with a calculated molecular weight of 22 kDa. The molecular weight of NGAL increases to 25 kDa when the molecule is glycosylated [77]. LCN2 is involved in a series of processes such as apoptosis of haematopoietic cells [78], transport of fatty acids and iron [79] and modulation of inflammation [80]. It was shown that LCN2 binds iron and delivers it to the cells through a small molecular weight siderophore [81, 82]. Particularly, LCN2 recognized a transmembrane receptor, recently cloned and named megalin (GP330), which is internalized in the cell by endocytosis [83, 84].
LCN2 synthesis has been described in bone [85]. Recently, Costa et al. demonstrated that LCN2 could modulate the bone marrow microenvironment by increasing the expression of one of the most important bone niche factors, the SDF-1 (stromal derived factor 1), a chemokine involved in the recruitment of hemopoietic precursors. This result suggests that this adipokine could play a major role in both tissue repair and maintenance of the bone marrow microenvironment [86]. The same authors also reported that LCN2 expression increased during osteoblast differentiation, and interestingly, transgenic mice over-expressing LCN2 presented bone microarchitectural changes [87], specifically, this transgenic mouse showed reduced trabecular number and bone mass, growth plate alterations, decreased bone formation rate and higher bone resorption [87]. Also, very recently was determined that LCN2 is a novel mechanoresponding gene in osteoblasts [88].
Chemerin
Chemerin, also known as tazarotene-induced gene 2 and retinoic acid receptor responder 2 (RARRES2), is an adipokine with chemoattractant activity [89]. It is secreted as an 18 kDa inactive proprotein, and it is activated by posttranslational C-terminal cleavage [89]. Chemerin acts via the G-protein-coupled receptor chemokine-like receptor 1 (CMKLR1 or ChemR23) [89]. CMKLR1 gene was first cloned in 1996 as a gene encoding a putative 371 amino acid receptor containing seven transmembrane domains [90]. The downstream signaling involves different pathways, including ERK1/2 and Akt pathways [91].
Chemerin and its receptor are mainly, but not exclusively, expressed in adipose tissue [92], and, for instance, dendritic cells, and macrophages express chemerin receptor [93]. Chemerin is also expressed in preosteoblastic cells and it seems to be involved in osteoblast differentiation [94]. Moreover, very recently, it was demonstrated that chemerin neutralization blocked osteoclast differentiation of hematopoietic stem cells [95].
Apelin
Apelin is a peptide, recently identified as the ligand of the orphan G-protein-coupled receptor APJ [96]. Several active apelin forms exist such as apelin-36, apelin-17, apelin-13 and pyroglutamated form of apelin-13. Apelin has been detected in adipose tissue and secreted by adipocytes [96, 97].
Recently, it has been demonstrated an increase in bone mass in mice lacking apelin [98]; in fact, these animals presented increased rates of bone formation and accelerated osteoblast formation and differentiation [98].
Vaspin
Vaspin is a serpin (serine protease inhibitor) that is produced in the visceral adipose tissue [99]. Human vaspin gene contains 1,245 nucleotides encoding a putative protein with 415 amino acids [99].
It has been reported that vaspin attenuates RANKL-induced osteoclastogenesis in RAW264.7 cell line [100]. In addition, it was also showed that vaspin was able to reduce osteoblast apoptosis induced by serum deprivation and this affect could be mediated by the regulation of ERK signaling [101].
Conclusions
In this review, we attempt to summarize the current understanding on adipokines biology in skeletal system. A lot of works has been made in the past decade on this topic, clearly showing the importance of the close interactions existing between adipose tissue and bone, via activation of specific pathways elicited by adipokines. Although a wealth of literature published in the last years certainly contributed to enlighten the role of adipokines in bone metabolism, several aspects are still inconsistent. In particular, incongruous results exist between in vivo and in vitro experimental systems.
It is evident that we are on the right way and future data, coming from a range of interrelated models, will provide new insights into the relationships existing between adipokines and bone metabolism. In particular, we believe that future studies, using unbiased all-encompassing systems (metabolomic/proteomics), will develop new avenues for novel therapeutical approaches that will impact on skeletal diseases.
References
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91.
Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol. 2005;115:911–9 quiz 920.
Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–2.
Busso N, So A, Chobaz-Peclat V, Morard C, Martinez-Soria E, Talabot-Ayer D, et al. Leptin signaling deficiency impairs humoral and cellular immune responses and attenuates experimental arthritis. J Immunol. 2002;168:875–82.
Lago F, Dieguez C, Gomez-Reino J, Gualillo O. The emerging role of adipokines as mediators of inflammation and immune responses. Cytokine Growth Factor Rev. 2007;18:313–25.
Elefteriou F, Ahn JD, Takeda S, Starbuck M, Yang X, Liu X, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434:514–20.
Luo XH, Guo LJ, Xie H, Yuan LQ, Wu XP, Zhou HD, et al. Adiponectin stimulates RANKL and inhibits OPG expression in human osteoblasts through the MAPK signaling pathway. J Bone Miner Res. 2006;21:1648–56.
Scotece M, Conde J, Abella V, Lopez V, Pino J, Lago F, et al. Bone metabolism and adipokines: Are there perspectives for bone diseases drug discovery? Expert Opin Drug Discov. 2014;9:945–57.
Scotece M, Conde J, Vuolteenaho K, Koskinen A, Lopez V, Gomez-Reino J, et al. Adipokines as drug targets in joint and bone disease. Drug Discov Today. 2014;19:241–58.
Yadav VK, Karsenty G. Leptin-dependent co-regulation of bone and energy metabolism. Aging (Albany NY). 2009;1:954–6.
Motyl KJ, Rosen CJ. Understanding leptin-dependent regulation of skeletal homeostasis. Biochimie. 2012;94:2089–96.
Shinoda Y, Yamaguchi M, Ogata N, Akune T, Kubota N, Yamauchi T, et al. Regulation of bone formation by adiponectin through autocrine/paracrine and endocrine pathways. J Cell Biochem. 2006;99:196–208.
Sarraf P, Frederich RC, Turner EM, Ma G, Jaskowiak NT, Rivet DJ 3rd, et al. Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med. 1997;185:171–5.
Gualillo O, Eiras S, Lago F, Dieguez C, Casanueva FF. Elevated serum leptin concentrations induced by experimental acute inflammation. Life Sci. 2000;67:2433–41.
Tschop J, Dattilo JR, Prakash PS, Kasten KR, Tschop MH, Caldwell CC. The leptin system: a potential target for sepsis induced immune suppression. Endocr Metab Immune Disord Drug Targets. 2010;10:336–47.
Blum WF, Englaro P, Hanitsch S, Juul A, Hertel NT, Muller J, et al. Plasma leptin levels in healthy children and adolescents: dependence on body mass index, body fat mass, gender, pubertal stage, and testosterone. J Clin Endocrinol Metab. 1997;82:2904–10.
Thomas T, Gori F, Khosla S, Jensen MD, Burguera B, Riggs BL. Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology. 1999;140:1630–8.
Gordeladze JO, Drevon CA, Syversen U, Reseland JE. Leptin stimulates human osteoblastic cell proliferation, de novo collagen synthesis, and mineralization: Impact on differentiation markers, apoptosis, and osteoclastic signaling. J Cell Biochem. 2002;85:825–36.
Scheller EL, Song J, Dishowitz MI, Soki FN, Hankenson KD, Krebsbach PH. Leptin functions peripherally to regulate differentiation of mesenchymal progenitor cells. Stem Cells. 2010;28:1071–80.
Kim GS, Hong JS, Kim SW, Koh JM, An CS, Choi JY, et al. Leptin induces apoptosis via ERK/cPLA2/cytochrome c pathway in human bone marrow stromal cells. J Biol Chem. 2003;278:21920–9.
Zhang J, Li T, Xu L, Li W, Cheng M, Zhuang J, et al. Leptin promotes ossification through multiple ways of bone metabolism in osteoblast: a pilot study. Gynecol Endocrinol. 2013;29:758–62.
Zeadin MG, Butcher MK, Shaughnessy SG, Werstuck GH. Leptin promotes osteoblast differentiation and mineralization of primary cultures of vascular smooth muscle cells by inhibiting glycogen synthase kinase (GSK)-3beta. Biochem Biophys Res Commun. 2012;425:924–30.
Liu GY, Liang QH, Cui RR, Liu Y, Wu SS, Shan PF, et al. Leptin promotes the osteoblastic differentiation of vascular smooth muscle cells from female mice by increasing RANKL expression. Endocrinology. 2014;155:558–67.
Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100:197–207.
Cornish J, Callon KE, Bava U, Lin C, Naot D, Hill BL, et al. Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J Endocrinol. 2002;175:405–15.
Holloway WR, Collier FM, Aitken CJ, Myers DE, Hodge JM, Malakellis M, et al. Leptin inhibits osteoclast generation. J Bone Miner Res. 2002;17:200–9.
Lisignoli G, Piacentini A, Toneguzzi S, Grassi F, Cocchini B, Ferruzzi A, et al. Osteoblasts and stromal cells isolated from femora in rheumatoid arthritis (RA) and osteoarthritis (OA) patients express IL-11, leukaemia inhibitory factor and oncostatin M. Clin Exp Immunol. 2000;119:346–53.
Yang WH, Tsai CH, Fong YC, Huang YL, Wang SJ, Chang YS, et al. Leptin induces oncostatin M production in osteoblasts by downregulating miR-93 through the Akt signaling pathway. Int J Mol Sci. 2014;15:15778–90.
Langdon C, Leith J, Smith F, Richards CD. Oncostatin M stimulates monocyte chemoattractant protein-1- and interleukin-1-induced matrix metalloproteinase-1 production by human synovial fibroblasts in vitro. Arthritis Rheum. 1997;40:2139–46.
Shahrara S, Proudfoot AE, Park CC, Volin MV, Haines GK, Woods JM, et al. Inhibition of monocyte chemoattractant protein-1 ameliorates rat adjuvant-induced arthritis. J Immunol. 2008;180:3447–56.
Martel-Pelletier J, Pelletier JP. Wanted—the collagenase responsible for the destruction of the collagen network in human cartilage! Br J Rheumatol. 1996;35:818–20.
Karsenty G, Oury F. The central regulation of bone mass, the first link between bone remodeling and energy metabolism. J Clin Endocrinol Metab. 2010;95:4795–801.
Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111:305–17.
Shi Y, Yadav VK, Suda N, Liu XS, Guo XE, Myers MG Jr, et al. Dissociation of the neuronal regulation of bone mass and energy metabolism by leptin in vivo. Proc Natl Acad Sci USA. 2008;105:20529–33.
Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–51.
Oh DK, Ciaraldi T, Henry RR. Adiponectin in health and disease. Diabetes Obes Metab. 2007;9:282–9.
Stumvoll M, Tschritter O, Fritsche A, Staiger H, Renn W, Weisser M, et al. Association of the T–G polymorphism in adiponectin (exon 2) with obesity and insulin sensitivity: interaction with family history of type 2 diabetes. Diabetes. 2002;51:37–41.
Whitehead JP, Richards AA, Hickman IJ, Macdonald GA, Prins JB. Adiponectin—a key adipokine in the metabolic syndrome. Diabetes Obes Metab. 2006;8:264–80.
Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50:2094–9.
Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13:332–9.
Oshima K, Nampei A, Matsuda M, Iwaki M, Fukuhara A, Hashimoto J, et al. Adiponectin increases bone mass by suppressing osteoclast and activating osteoblast. Biochem Biophys Res Commun. 2005;331:520–6.
Berner HS, Lyngstadaas SP, Spahr A, Monjo M, Thommesen L, Drevon CA, et al. Adiponectin and its receptors are expressed in bone-forming cells. Bone. 2004;35:842–9.
Luo XH, Guo LJ, Yuan LQ, Xie H, Zhou HD, Wu XP, et al. Adiponectin stimulates human osteoblasts proliferation and differentiation via the MAPK signaling pathway. Exp Cell Res. 2005;309:99–109.
Kanazawa I. Adiponectin in metabolic bone disease. Curr Med Chem. 2012;19:5481–92.
Huang CY, Lee CY, Chen MY, Tsai HC, Hsu HC, Tang CH. Adiponectin increases BMP-2 expression in osteoblasts via AdipoR receptor signaling pathway. J Cell Physiol. 2010;224:475–83.
Lee YA, Ji HI, Lee SH, Hong SJ, Yang HI, Chul Yoo M, et al. The role of adiponectin in the production of IL-6, IL-8, VEGF and MMPs in human endothelial cells and osteoblasts: implications for arthritic joints. Exp Mol Med. 2014;46:e72.
Kajimura D, Lee HW, Riley KJ, Arteaga-Solis E, Ferron M, Zhou B, et al. Adiponectin regulates bone mass via opposite central and peripheral mechanisms through FoxO1. Cell Metab. 2013;17:901–15.
Williams GA, Wang Y, Callon KE, Watson M, Lin JM, Lam JB, et al. In vitro and in vivo effects of adiponectin on bone. Endocrinology. 2009;150:3603–10.
Wang F, Wang PX, Wu XL, Dang SY, Chen Y, Ni YY, et al. Deficiency of adiponectin protects against ovariectomy-induced osteoporosis in mice. PLoS One. 2013;8:e68497.
Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–7.
Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426–30.
Friebe D, Neef M, Kratzsch J, Erbs S, Dittrich K, Garten A, et al. Leucocytes are a major source of circulating nicotinamide phosphoribosyltransferase (NAMPT)/pre-B cell colony (PBEF)/visfatin linking obesity and inflammation in humans. Diabetologia. 2011;54:1200–11.
Catalan V, Gomez-Ambrosi J, Rodriguez A, Ramirez B, Silva C, Rotellar F, et al. Association of increased visfatin/PBEF/NAMPT circulating concentrations and gene expression levels in peripheral blood cells with lipid metabolism and fatty liver in human morbid obesity. Nutr Metab Cardiovasc Dis. 2011;21:245–53.
Curat CA, Wegner V, Sengenes C, Miranville A, Tonus C, Busse R, et al. Macrophages in human visceral adipose tissue: increased accumulation in obesity and a source of resistin and visfatin. Diabetologia. 2006;49:744–7.
Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, et al. Retraction. Science. 2007;318:565.
Jia SH, Li Y, Parodo J, Kapus A, Fan L, Rotstein OD, et al. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J Clin Invest. 2004;113:1318–27.
Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007;178:1748–58.
Xie H, Tang SY, Luo XH, Huang J, Cui RR, Yuan LQ, et al. Insulin-like effects of visfatin on human osteoblasts. Calcif Tissue Int. 2007;80:201–10.
Li Y, He J, He X, Lindgren U. Nampt expression increases during osteogenic differentiation of multi- and omnipotent progenitors. Biochem Biophys Res Commun. 2013;434:117–23.
Moschen AR, Geiger S, Gerner R, Tilg H. Pre-B cell colony enhancing factor/NAMPT/visfatin and its role in inflammation-related bone disease. Mutat Res. 2010;690:95–101.
Venkateshaiah SU, Khan S, Ling W, Bam R, Li X, van Rhee F, et al. NAMPT/PBEF1 enzymatic activity is indispensable for myeloma cell growth and osteoclast activity. Exp Hematol. 2013;41(547–557):e2.
Laiguillon MC, Houard X, Bougault C, Gosset M, Nourissat G, Sautet A, et al. Expression and function of visfatin (Nampt), an adipokine-enzyme involved in inflammatory pathways of osteoarthritis. Arthritis Res Ther. 2014;16:R38.
Briana DD, Boutsikou M, Boutsikou T, Malamitsi-Puchner A. Associations of novel adipocytokines with bone biomarkers in intra uterine growth-restricted fetuses/neonates at term. J Matern Fetal Neonatal Med. 2014;27:984–8.
Li Y, He X, He J, Anderstam B, Andersson G, Lindgren U. Nicotinamide phosphoribosyltransferase (Nampt) affects the lineage fate determination of mesenchymal stem cells: a possible cause for reduced osteogenesis and increased adipogenesis in older individuals. J Bone Miner Res. 2011;26:2656–64.
Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–12.
Ghosh S, Singh AK, Aruna B, Mukhopadhyay S, Ehtesham NZ. The genomic organization of mouse resistin reveals major differences from the human resistin: functional implications. Gene. 2003;305:27–34.
Patel L, Buckels AC, Kinghorn IJ, Murdock PR, Holbrook JD, Plumpton C, et al. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem Biophys Res Commun. 2003;300:472–6.
Fain JN, Cheema PS, Bahouth SW, Lloyd Hiler M. Resistin release by human adipose tissue explants in primary culture. Biochem Biophys Res Commun. 2003;300:674–8.
Thommesen L, Stunes AK, Monjo M, Grosvik K, Tamburstuen MV, Kjobli E, et al. Expression and regulation of resistin in osteoblasts and osteoclasts indicate a role in bone metabolism. J Cell Biochem. 2006;99:824–34.
Borregaard N, Cowland JB. Neutrophil gelatinase-associated lipocalin, a siderophore-binding eukaryotic protein. Biometals. 2006;19:211–5.
Chakraborty S, Kaur S, Guha S, Batra SK. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta. 2012;1826:129–69.
Conde J, Gomez R, Bianco G, Scotece M, Lear P, Dieguez C, et al. Expanding the adipokine network in cartilage: identification and regulation of novel factors in human and murine chondrocytes. Ann Rheum Dis. 2011;70:551–9.
Triebel S, Blaser J, Reinke H, Tschesche H. A 25 kDa alpha 2-microglobulin-related protein is a component of the 125 kDa form of human gelatinase. FEBS Lett. 1992;314:386–8.
Kjeldsen L, Bainton DF, Sengelov H, Borregaard N. Identification of neutrophil gelatinase-associated lipocalin as a novel matrix protein of specific granules in human neutrophils. Blood. 1994;83:799–807.
Kjeldsen L, Cowland JB, Borregaard N. Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochim Biophys Acta. 2000;1482:272–83.
Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268:10425–32.
Devireddy LR, Teodoro JG, Richard FA, Green MR. Induction of apoptosis by a secreted lipocalin that is transcriptionally regulated by IL-3 deprivation. Science. 2001;293:829–34.
Chu ST, Lin HJ, Huang HL, Chen YH. The hydrophobic pocket of 24p3 protein from mouse uterine luminal fluid: fatty acid and retinol binding activity and predicted structural similarity to lipocalins. J Pept Res. 1998;52:390–7.
Cowland JB, Borregaard N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from humans. Genomics. 1997;45:17–23.
Yang J, Goetz D, Li JY, Wang W, Mori K, Setlik D, et al. An iron delivery pathway mediated by a lipocalin. Mol Cell. 2002;10:1045–56.
Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033–43.
Richardson DR. 24p3 and its receptor: Dawn of a new iron age? Cell. 2005;123:1175–7.
Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123:1293–305.
Bartsch S, Tschesche H. Cloning and expression of human neutrophil lipocalin cDNA derived from bone marrow and ovarian cancer cells. FEBS Lett. 1995;357:255–9.
Costa D, Biticchi R, Negrini S, Tasso R, Cancedda R, Descalzi F, et al. Lipocalin-2 controls the expression of SDF-1 and the number of responsive cells in bone. Cytokine. 2010;51:47–52.
Costa D, Lazzarini E, Canciani B, Giuliani A, Spano R, Marozzi K, et al. Altered bone development and turnover in transgenic mice over-expressing lipocalin-2 in bone. J Cell Physiol. 2013;228:2210–21.
Rucci N, Capulli M, Piperni SG, Cappariello A, Lau P, Frings-Meuthen P, et al. Lipocalin 2: a new mechanoresponding gene regulating bone homeostasis. J Bone Miner Res. 2014;. doi:10.1002/jbmr.2341.
Wittamer V, Franssen JD, Vulcano M, Mirjolet JF, Le Poul E, Migeotte I, et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med. 2003;198:977–85.
Gantz I, Konda Y, Yang YK, Miller DE, Dierick HA, Yamada T. Molecular cloning of a novel receptor (CMKLR1) with homology to the chemotactic factor receptors. Cytogenet Cell Genet. 1996;74:286–90.
Berg V, Sveinbjornsson B, Bendiksen S, Brox J, Meknas K, Figenschau Y. Human articular chondrocytes express ChemR23 and chemerin; ChemR23 promotes inflammatory signalling upon binding the ligand chemerin (21–157). Arthritis Res Ther. 2010;12:R228.
Bozaoglu K, Bolton K, McMillan J, Zimmet P, Jowett J, Collier G, et al. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology. 2007;148:4687–94.
Luangsay S, Wittamer V, Bondue B, De Henau O, Rouger L, Brait M, et al. Mouse ChemR23 is expressed in dendritic cell subsets and macrophages, and mediates an anti-inflammatory activity of chemerin in a lung disease model. J Immunol. 2009;183:6489–99.
Muruganandan S, Roman AA, Sinal CJ. Role of chemerin/CMKLR1 signaling in adipogenesis and osteoblastogenesis of bone marrow stem cells. J Bone Miner Res. 2010;25:222–34.
Muruganandan S, Dranse HJ, Rourke JL, McMullen NM, Sinal CJ. Chemerin neutralization blocks hematopoietic stem cell osteoclastogenesis. Stem Cells. 2013;31:2172–82.
Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251:471–6.
Boucher J, Masri B, Daviaud D, Gesta S, Guigne C, Mazzucotelli A, et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology. 2005;146:1764–71.
Wattanachanya L, Lu WD, Kundu RK, Wang L, Abbott MJ, O’Carroll D, et al. Increased bone mass in mice lacking the adipokine apelin. Endocrinology. 2013;154:2069–80.
Hida K, Wada J, Eguchi J, Zhang H, Baba M, Seida A, et al. Visceral adipose tissue-derived serine protease inhibitor: a unique insulin-sensitizing adipocytokine in obesity. Proc Natl Acad Sci USA. 2005;102:10610–5.
Kamio N, Kawato T, Tanabe N, Kitami S, Morita T, Ochiai K, et al. Vaspin attenuates RANKL-induced osteoclast formation in RAW264.7 cells. Connect Tissue Res. 2013;54:147–52.
Zhu X, Jiang Y, Shan PF, Shen J, Liang QH, Cui RR, et al. Vaspin attenuates the apoptosis of human osteoblasts through ERK signaling pathway. Amino Acids. 2013;44:961–8.
Disclosures
Conflict of interest
Javier Conde, Morena Scotece, Vanessa Abella, Verónica López, Jesús Pino, Juan Jesús Gómez-Reino and Oreste Gualillo declare that they have no conflict of interest. Oreste Gualillo is Staff Personnel of Xunta de Galicia (SERGAS) through a research-staff stabilization contract (ISCIII/SERGAS). Oreste Gualillo is supported by Instituto de Salud Carlos III (PI11/01073). Oreste Gualillo is a member of RETICS Programme, RD12/0009/0008 (RIER:Red de Investigación en Inflamación y Enfermedades Reumáticas) via Instituto de Salud Carlos III (ISCIII). Morena Scotece is a recipient of the “FPU” programme of the Spanish Ministry of Education. Javier Conde is a recipìent of a predoctoral program of Fundación Ramón Domínguez. Vanessa Abella is a predoctoral recipient of Xunta de Galicia. Veronica Lopez is a recipient of a grant from ISCIII.
Animal/Human studies
This article does not include any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Conde, J., Scotece, M., Abella, V. et al. Basic Aspects of Adipokines in Bone Metabolism. Clinic Rev Bone Miner Metab 13, 11–19 (2015). https://doi.org/10.1007/s12018-014-9175-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12018-014-9175-4