
Abstract
Neuronal ceroid lipofuscinoses (NCLs) are a group of neurodegenerative disorders caused by mutations in fourteen distinct ceroid lipofuscinoses, neuronal (CLN) genes described with various severe symptoms such as seizures, visual failure, motor decline, and progressive cognitive deterioration. The current research represents novel CLN5 (c.741G > A) and CLN8 (c.565delT) mutations in two different Iranian families with late-infantile NCL (LINCL) and their relatives by using whole-exome sequencing (WES). The first family had a 10-year-old male with consanguineous parents and severe NCL symptoms, including motor clumsiness, telangiectasia, and cerebellar atrophy. The second family with a child who suffered from nystagmus rotation, motor difficulties, and seizure was a 5-year-old male with consanguineous parent. WES of probands 1 and 2 revealed homozygotic mutations in exon 4 of CLN5 (c.741G > A, p.W247X) and deletion in exon 3 (c.565delT, p.F189fs) of CLN8, respectively. Both patients’ parents were heterozygous for these alterations. In concordance with previous studies, our results indicate that pathogenic mutations in CLN genes, especially CLN5 and 8, are a main cause of LINCL; these results also suggest that LINCL is not a regionally or nationally dependent disorder and can occur in any ethnic group despite the fact that some populations may be more at risk. Consequently, CLN gene screening for patients with typical signs of LINCL is recommended.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neuronal ceroid lipofuscinoses (NCLs) are a group of inherited progressive neurodegenerative disorders characterized by epileptic seizures, visual loss, progressive cognitive and motor decline, and often reduced life expectancy (Hall et al. 2001; Kollmann et al. 2013). Its estimated incidence range is 1:1,000,000 to 1:14,000 worldwide (Haltia 2003). NCLs are inherited primarily in an autosomal recessive manner. According to the primary onset of symptoms and clinical features, this group has been classified into four subtypes: infantile (INCL), late-infantile (LINCL), juvenile (JNCL), and adult (ANCL) (Katata et al. 2016; Marotta et al. 2017). Lysosomal accumulation of autofluorescent storage material in the central nervous system (CNS) is a crucial pathological finding of NCLs (Getty and Pearce 2011; Zelnik et al. 2007). So far, 13 candidate genes for NCLs with different roles have been identified (Table 1). As shown in Table 1, CLN9 has not yet been molecularly characterized (Kollmann et al. 2013; Schulz et al. 2004; Warrier et al. 2013; Williams and Mole 2012). To date, more than 440 mutations in more over 13 candidate genes have been reported in NCLs and registered in the NCL mutation database (http://www.ucl.ac.uk/ncl) (Beesley et al. 2017; Mole and Cotman 2015). Approximately 47 NCLs patients have been attributed to pathogenic alterations in the CLN8 gene (Gao et al. 2018). Clinical syndromes associated with CLN8 variants have been found to occur in two main Northern Finnish and Turkish phenotypes. The Northern Finnish phenotype, also known as Northern Epilepsy (NE) Syndrome, is a progressive epilepsy with mental retardation (EPMR) characterized by normal early development, slow progressive mental decline, and mildly reduced to normal visual acuity (Herva et al. 2000; Zelnik et al. 2007), whereas the Turkish phenotype shows a rapid disease progression with epilepsy, mental decline, ataxia, and visual impairment (Ranta et al. 2004; Reinhardt et al. 2010). Further studies described CLN8 mutations in Italian, Pakistani, and Israeli patients with the LINCL phenotype. These patients showed both rapid and slow clinical courses (Cannelli et al. 2006; Mahajnah and Zelnik 2012; Ranta et al. 2004; Vantaggiato et al. 2009; Zelnik et al. 2007). CLN8 is a non-glycosylated transmembrane protein with 286 amino acids belonging to the TLC superfamily which is involved in biosynthesis, sensing, and metabolism of lipids (Winter and Ponting 2002). In non-neuronal cells, this protein shuttles between the ER and the ER-Golgi intermediate complex (ERGIC), while in polarized cells, such as neurons, it takes place outside of the ER (Lonka et al. 2000, 2004).
A clinical syndrome associated with CLN5 that was first described in Finland has also been found in other ethnic groups, but is most common in Finland and northern Europe (Kousi et al. 2012). The variant late-infantile form is the most common form (VLINCL) of CLN5 and is characterized by motor clumsiness, attention disturbances, visual impairment, motor decline, ataxia, and mental deterioration (Uusi-Rauva et al. 2017). Other studies described CLN5 mutations in different nations such as Portugal, Pakistan, and Qatar [http://www.ucl.ac.uk]. CLN5 is an N-glycosylated protein with 407 amino acids secreted from lysosomes (Mancini et al. 2015; Mole and Cotman 2015) and plays a crucial role as a sensor in lysosomal traffic (Getty and Pearce 2011). This study examines the first Iranian cases of NCL patients with CLN5 and 8 using whole-exome sequencing (WES).
Materials and Methods
Participants
CLN5 Proband
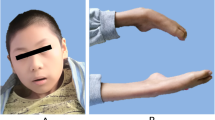
The subject is a 10-year-old Iranian Azeri Turkish boy whose parents are consanguineous who revealed several symptoms in early childhood, including gait ataxia, myoclonus, and motor clumsiness (Fig. 1). The prenatal period was unremarkable; he was born via normal delivery at term with a birth weight of 4200 g. Until the age of three, he was completely normal then gradually began to show symptoms of physical disability encompassing ataxia and myoclonus until he lost the ability to walk at the age of eight and developed motor clumsiness in his hands with motor loss at the age of seven. The patient became completely dependent for daily activities, and there are signs of telangiectasia in both eyes along with myoclonic seizures. Magnetic resonance imaging (MRI) revealed cerebellar atrophy (Fig. 2).

Family tree of case 1

Magnetic resonance imaging (MRI) findings in case 1 (a) and case 2 (b) both show cerebellar atrophy
CLN8 Proband
The subject is a 5-year-old Iranian Azeri Turkish male whose parents are consanguineous who was referred to our department because of frequent drop seizures and permanent loss of motor and psychological ability (Fig. 3). He was born via normal delivery at term with a birth weight of 4250 g. During his third year, he developed motor difficulties, seizures, and slowing cognitive skills. Initially, the seizures occurred 3–4 times every month; later they began to take place every 2–3 months. After treatment with medicine, the patient had no seizures for 1 year. For the first 3 years, he had normal development and appeared to be healthy. At the age of four, he exhibited ataxic gait, nystagmus rotation (visual problems), and gastrointestinal problems, including dysphagia, nausea, and diarrhea. On admission at age five, he had significant difficulties with communication and was unable to stand independently. His MRI indicated cerebellar atrophy (Fig. 2).

Family tree of case 2
Whole-Exon Sequencing (WES)
Genomic DNA was extracted from peripheral blood of the patients, their parents, and five members of their relatives according to standard protocols (GeneAll Exgene Blood SV Mini). WES was performed on individual samples and PCR amplification was designed for identification of variants on exon 4 of CLN5 and exon 3 of CLN8 by using specific primers 5′-TAGTTGGGTTTGGTGGCCA-3′, 5′-TGTTTCCTGTTGGCCCAAAA-3′ and 5′-ACT TCC CAG GGT CTA CGT ATG G-3′, 5′-AGA GCC AGT CCG ACA AGG AA-3′, respectively.
Results
MRI Findings
After patients underwent MRI, evaluation of several images in numerous locations indicated the MRI abnormalities. As shown in Fig. 2, the test results of patients display moderate cerebellar atrophy for CLN5 proband and partly extreme cerebellar atrophy for CLN8 proband.
Molecular Genetics Results
Although the patients and their family members had been observed clinically for years, clinical diagnosis failed to suggest an especial disease based on MRI findings. To identify the apparent causative variants, affected individuals were analyzed using WES. Through WES of the probands two novel mutations were identified. The first one is a nonsense mutation, CLN5. c.741G > A (p.Trp247*), which was seen in patient 1 (Fig. 4, left). The other one is deletion mutation, CLN8. c.565delT (p.Phe189fs) which was discovered in patient 2 (Fig. 4, right). The nucleotide 741 in exon 4 of the CLN5 gene and its corresponding amino acid tryptophan is partly conserved throughout a variety of species (up to Ptroglodytes and considering 6 spices). Also, MutationTaster software suggests a pathogenicity for this mutation. Deletion of one nucleotide (c.565delT) in exon 3 of CLN8 gene is predicted to cause a frameshift mutation and premature stop codon and results in 97 amino acids missing in a truncated protein. Protein structure prediction was performed for assessing mutant and normal forms of these proteins using SWISS-MODEL software (Fig. 5). As can be seen in Fig. 5, both variants lead to production of truncated proteins which may result in functional and structural changing. The co-segregation analysis of the variants in family members of the case 1 and case 2 who are closely related to the probands in pedigree was also performed by Sanger sequencing (Fig. 4). According to the genotype, patient 1 and patient 2’s parents are heterozygotes for the CLN5 and CLN8 mutations, respectively, whereas other members are homozygous for the wild-type allele and negative for neurologic dysfunction.

Sanger traces for PCR products. CLN5 (c.741G > A) left and CLN8 (c.565delT) right (Affected proband, father, and mother, respectively)

CLN5 (upper) CLN8 (down). A, normal forms and B, mutated forms
Systematic Literature Review
Several articles were used as collected specimens. Clinical presentation, examination results, and molecular analysis of LINCL patients similar to our affected individuals were collected. As a result, three patients with CLN5 mutations and 7 patients with CLN8 variations met the inclusion criteria and finally were picked from articles and compared with our patients (Tables 2, 3). In addition, all reports that had represented CLN5 and CLN8 mutations and their association with LINCL were screened. The summary of all published reports is given in Table 4 for further comparison of Iranian patients with patients of other races, especially in high incidence districts.
Discussion
In this study, we identified two novel mutations, CLN5, c.G741A and CLN8, c.565delT, through the investigation of two cases from different Iranian families. To our knowledge, these two variants have not been reported in ClinVar. The neuronal ceroid lipofuscinoses (NCLs) are a group of progressive neurodegenerative disorders with variable age of onset. These diseases are clinically and genetically heterogeneous and are characterized by lysosomal accumulations of autofluorescent storage material, neuroinflammation, especially in neurons, resulting in psychomotor deterioration, visual failure, and premature death (Getty and Pearce 2011; Katata et al. 2016; Kollmann et al. 2013; Kousi et al. 2012). One of the most genetically heterogeneous forms of NCL is late-infantile-onset (LINCL), with causative variations found in CLN1, 2, 5, 6, 7, 8, and 14 (Cannelli et al. 2006; Mole et al. 2005; Siintola et al. 2007). This type of NCL is inherited mostly in an autosomal recessive manner (Vantaggiato et al. 2009).
CLN5 (ceroid lipofuscinosis, neuronal, 5) pathogenic mutations lead to an LINCL phenotype (MIM#256731) with motor clumsiness as the most common presenting symptom and attention disturbances in the early years between four to seven, progressive visual failure, ataxia, mental decline, myoclonia accompanied by epilepsy; early death usually occurs between 10 and 30 years of age (Mancini et al. 2015; Setty et al. 2013). The brains of such patients show the earliest and more severe atrophy in the cerebellum followed by storage deposition, astrocytosis, hypomyelination, and destruction of cerebral neurons, suggesting a modifying role for the CLN5 protein itself (Tyynelä et al. 2004; Uusi-Rauva et al. 2017). The soluble lysosomal glycoprotein encoded by this gene is ubiquitously expressed in glia and neurons, but its function is still largely unknown (Holmberg et al. 2004; Kopra et al. 2004; Schmiedt et al. 2012, 2010; Vesa and Peltonen 2002; von Schantz et al. 2008). In vitro studies revealed that the CLN5 protein share a common interaction partner with CLN1 and plays a role in cholesterol transport through the plasma membrane (Martinez et al. 2003). Furthermore, this protein is implicated in the function of the sphingolipid transport and in Rab7-mediated endosomal sorting (Haddad et al. 2012; Mamo et al. 2012). Although the exact role of the CLN5 protein and the cellular consequence of this gene mutation is currently unknown, some research has informed and examined common variations in CLN5 gene. Kristiina et al. in 2017 studied the effects of p.Tyr392X SNP, and found alteration occurs in 94% of Finnish types in induced pluripotent stem cells (iPSCs) and that these cells accumulate autofluorescent storage material and mitochondrial ATP synthase’s subunit C, both demonstrating the hallmarks of many NCL forms, as well as CLN5 disease. They also found many intracellular organelles with abnormalities and neuronal sphingolipid transportation aberrations (Uusi-Rauva et al. 2017). In a study by Xin et al. (2010, p.Ser98LeufsX13), mutation was described as infantile onset disease and p.Tyr374Cys was identified as the adult form with mild impact on the protein (Xin et al. 2010). The majority of the pathogenic mutations constitute prematurely terminated transcripts that cause mRNA instability as well as the presence of subjects, degradation via nonsense-mediated decay (NMD), and reduced protein expression (Kousi et al. 2012). Here, we identified a novel c.G741A (NM_006493) mutation in an Iranian Azeri child with gait ataxia, myoclonus, and motor clumsiness. Clinical symptoms of case 1 and other patients with CLN5 pathogenic mutations are given in Table 1. A bioinformatics analysis showed that this pathogenic nonsense mutation is on exon 4 and strongly causes a truncated protein (p.W247X) (Fig. 5). Our patient for this mutation was homozygote and his parents were heterozygote for this SNP, suggesting that NCLs are wholly genetic-based disorders and other factors, such as environmental, are not associated with these diseases.
The deletion mutation c.565delT (NM_018941) on exon 3 was found in our case 2, the five-year-old boy with motor difficulties, seizures, ataxic gait, nystagmus rotation, gastrointestinal problems, and cognitive skills (Table 3), which is similar to other LINCL symptoms that have been reported in patients in Turkey, Italy, Israel, and Japan (Kousi et al. 2012). His parents were heterozygous for this variation; therefore, autosomal recessive inheritance is suggested. With respect to the clinical symptoms of the patient which are caused by the truncated protein p.F189fs due to this mutation (Fig. 5), the critical role of CLN8 protein becomes clear. CLN8 as a transmembrane protein has 286 amino acid residues with five hydrophilic regions, including a TLC (TRAM-LAG1-CLN8) domain. This TLC domain encompasses amino acids from 62 to 262, including five transmembrane helices (Winter and Ponting 2002). As seen in our mutation, other alterations that affect this domain lead to severe forms of NCL and may be due to complete loss of function and even protein location (Kousi et al. 2012). Even though p.(Gln194Arg) as a missense mutation at the TLC domain was reported in a patient with a slightly more severe disease course (Cannelli et al. 2006), p.(Gln256Glu) in affected siblings resulted in noticeably varying symptoms (Zelnik et al. 2007). CLN8 as a transmembrane protein is localized to the endoplasmic reticulum (ER) and recycles between this organelle and the ER-Golgi intermediate compartment (ERGIC). The exact function is unknown, but based on prior studies it involves metabolism, transport, detection, and biosynthesis of lipids (Jalanko and Braulke 2009) and also corrects growth and apoptosis (Haddad et al. 2012). Since molecular partners of CLN8 have roles in apoptosis, autophagy, lipid transport, and vesicular trafficking, it seems that CLN8 mutations induce apoptosis.
Considering the number of CLN5 and CLN8 mutations associated with LINCLs, it seems that the prevalence of LINCLs among Europe and the Middle East population is similar but slightly higher than seen in other countries. Turkish families are at more risk for these mutations in the Middle East, and in Europe, British and Italian populations are susceptible to CLN5 and CLN8 variations, respectively (Table 4). This assessment may vary among other subtypes of NCLs, so more prospective registered studies with a higher clinical value will help to confirm or refuse this evaluation.
To sum up, in line with previous reports, our findings confirm that the CLN5 and CLN8 mutations associated with LINCL can occur in many populations in spite of the fact that some populations may be more at risk. Therefore, we recommend that cases with typical signs of LINCL should be screened for CLN genes mutation especially for CLN5 and 8.
References
Beesley, C., Guerreiro, R. J., Bras, J. T., Williams, R. E., Taratuto, A. L., Eltze, C., & Mole, S. E. (2017). CLN 8 disease caused by large genomic deletions. Molecular Genetics & Genomic Medicine, 5(1), 85–91.
Cannelli, N., Cassandrini, D., Bertini, E., Striano, P., Fusco, L., Gaggero, R.,.. . Bruno, C. (2006). Novel mutations in CLN8 in Italian variant late infantile neuronal ceroid lipofuscinosis: Another genetic hit in the Mediterranean. Neurogenetics, 7(2), 111.
Gao, Z., Xie, H., Jiang, Q., Wu, N., Chen, X., & Chen, Q. (2018). Identification of two novel null variants in CLN8 by targeted next-generation sequencing: First report of a Chinese patient with neuronal ceroid lipofuscinosis due to CLN8 variants. BMC medical genetics, 19(1), 21.
Getty, A. L., & Pearce, D. A. (2011). Interactions of the proteins of neuronal ceroid lipofuscinosis: Clues to function. Cellular and molecular life sciences, 68(3), 453–474.
Haddad, S. E., Khoury, M., Daoud, M., Kantar, R., Harati, H., Mousallem, T.,.. . Boustany, R. M. (2012). CLN 5 and CLN 8 protein association with ceramide synthase: B iochemical and proteomic approaches. Electrophoresis, 33(24), 3798–3809.
Hall, J. C., Dunlap, J. C., Friedmann, T., Giannelli, F., Wisniewski, K. E., & Zhong, N. (2001). Batten disease: Diagnosis, treatment, and research (Vol. 45). Amsterdam: Elsevier.
Haltia, M. (2003). The neuronal ceroid-lipofuscinoses. Journal of Neuropathology & Experimental Neurology, 62(1), 1–13.
Herva, R., Tyynelä, J., Hirvasniemi, A., Syrjäkallio-Ylitalo, M., & Haltia, M. (2000). Northern epilepsy: A novel form of neuronal ceroid-lipofuscinosis. Brain Pathology, 10(2), 215–222.
Holmberg, V., Jalanko, A., Isosomppi, J., Fabritius, A.-L., Peltonen, L., & Kopra, O. (2004). The mouse ortholog of the neuronal ceroid lipofuscinosis CLN5 gene encodes a soluble lysosomal glycoprotein expressed in the developing brain. Neurobiology of Disease, 16(1), 29–40.
Jalanko, A., & Braulke, T. (2009). Neuronal ceroid lipofuscinoses. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 1793(4), 697–709.
Katata, Y., Uematsu, M., Sato, H., Suzuki, S., Nakayama, T., Kubota, Y.,.. . Kure, S. (2016). Novel missense mutation in CLN8 in late infantile neuronal ceroid lipofuscinosis: The first report of a CLN8 mutation in Japan. Brain and Development, 38(3), 341–345.
Kollmann, K., Uusi-Rauva, K., Scifo, E., Tyynelä, J., Jalanko, A., & Braulke, T. (2013). Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 1832(11), 1866–1881.
Kopra, O., Vesa, J., von Schantz, C., Manninen, T., Minye, H., Fabritius, A.-L.,.. . Jalanko, A. (2004). A mouse model for Finnish variant late infantile neuronal ceroid lipofuscinosis, CLN5, reveals neuropathology associated with early aging. Human Molecular Genetics, 13(23), 2893–2906.
Kousi, M., Lehesjoki, A. E., & Mole, S. E. (2012). Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Human Mutation, 33(1), 42–63.
Lonka, L., Kyttälä, A., Ranta, S., Jalanko, A., & Lehesjoki, A.-E. (2000). The neuronal ceroid lipofuscinosis CLN8 membrane protein is a resident of the endoplasmic reticulum. Human Molecular Genetics, 9(11), 1691–1697.
Lonka, L., Salonen, T., Siintola, E., Kopra, O., Lehesjoki, A. E., & Jalanko, A. (2004). Localization of wild-type and mutant neuronal ceroid lipofuscinosis CLN8 proteins in non-neuronal and neuronal cells. Journal of Neuroscience Research, 76(6), 862–871.
Mahajnah, M., & Zelnik, N. (2012). Phenotypic heterogeneity in consanguineous patients with a common CLN8 mutation. Pediatric Neurology, 47(4), 303–305.
Mamo, A., Jules, F., Dumaresq-Doiron, K., Costantino, S., & Lefrancois, S. (2012). The role of ceroid lipofuscinosis neuronal protein 5 (CLN5) in endosomal sorting. Molecular and Cellular Biology, 32(10), 1855–1866.
Mancini, C., Nassani, S., Guo, Y., Chen, Y., Giorgio, E., Brussino, A.,.. . Funaro, A. (2015). Adult-onset autosomal recessive ataxia associated with neuronal ceroid lipofuscinosis type 5 gene (CLN5) mutations. Journal of Neurology, 262(1), 173–178.
Marotta, D., Tinelli, E., & Mole, S. E. (2017). NCLs and ER: A stressful relationship. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 1863(6), 1273–1281.
Martinez, L. O., Jacquet, S., Esteve, J.-P., Rolland, C., Cabezón, E., Champagne, E.,.. . Tercé, F. (2003). Ectopic β-chain of ATP synthase is an apolipoprotein AI receptor in hepatic HDL endocytosis. Nature, 421(6918), 75.
Mole, S. E., & Cotman, S. L. (2015). Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 1852(10), 2237–2241.
Mole, S. E., Williams, R. E., & Goebel, H. H. (2005). Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics, 6(3), 107–126.
Ranta, S., Topcu, M., Tegelberg, S., Tan, H., Üstübütün, A., Saatci, I.,.. . Alembik, Y. (2004). Variant late infantile neuronal ceroid lipofuscinosis in a subset of Turkish patients is allelic to Northern epilepsy. Human Mutation, 23(4), 300–305.
Reinhardt, K., Grapp, M., Schlachter, K., Brück, W., Gärtner, J., & Steinfeld, R. (2010). Novel CLN8 mutations confirm the clinical and ethnic diversity of late infantile neuronal ceroid lipofuscinosis. Clinical Genetics, 77(1), 79–85.
Schmiedt, M. L., Bessa, C., Heine, C., Ribeiro, M. G., Jalanko, A., & Kyttälä, A. (2010). The neuronal ceroid lipofuscinosis protein CLN5: New insights into cellular maturation, transport, and consequences of mutations. Human Mutation, 31(3), 356–365.
Schmiedt, M.-L., Blom, T., Blom, T., Kopra, O., Wong, A., von Schantz-Fant, C.,.. . Cooper, J. D. (2012). Cln5-deficiency in mice leads to microglial activation, defective myelination and changes in lipid metabolism. Neurobiology of Disease, 46(1), 19–29.
Schulz, A., Dhar, S., Rylova, S., Dbaibo, G., Alroy, J., Hagel, C.,.. . Boustany, R. M. (2004). Impaired cell adhesion and apoptosis in a novel CLN9 Batten disease variant. Annals of Neurology, 56(3), 342–350.
Setty, G., Saleem, R., Khan, A., & Hussain, N. (2013). Atypical juvenile neuronal ceroid lipofuscinosis: A report of three cases. Journal of Pediatric Neurosciences, 8(2), 117.
Siintola, E., Topcu, M., Aula, N., Lohi, H., Minassian, B. A., Paterson, A. D.,.. . Anttonen, A.-K. (2007). The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. The American Journal of Human Genetics, 81(1), 136–146.
Tyynelä, J., Cooper, J. D., Khan, M. N., Shemilt, S. J., & Haltia, M. (2004). Hippocampal pathology in the human neuronal ceroid-lipofuscinoses: Distinct patterns of storage deposition, neurodegeneration and glial activation. Brain Pathology, 14(4), 349–357.
Uusi-Rauva, K., Blom, T., von Schantz-Fant, C., Blom, T., Jalanko, A., & Kyttälä, A. (2017). Induced pluripotent stem cells derived from a CLN5 patient manifest phenotypic characteristics of neuronal ceroid lipofuscinoses. International Journal of Molecular Sciences, 18(5), 955.
Vantaggiato, C., Redaelli, F., Falcone, S., Perrotta, C., Tonelli, A., Bondioni, S.,.. . Bonaglia, M. C. (2009). A novel CLN8 mutation in late-infantile-onset neuronal ceroid lipofuscinosis (LINCL) reveals aspects of CLN8 neurobiological function. Human Mutation, 30(7), 1104–1116.
Vesa, J., & Peltonen, L. (2002). Mutated genes in juvenile and variant late infantile neuronal ceroid lipofuscinoses encode lysosomal proteins. Current Molecular Medicine, 2(5), 439–444.
von Schantz, C., Saharinen, J., Kopra, O., Cooper, J. D., Gentile, M., Hovatta, I.,.. . Jalanko, A. (2008). Brain gene expression profiles of Cln1 and Cln5 deficient mice unravels common molecular pathways underlying neuronal degeneration in NCL diseases. BMC Genomics, 9(1), 146.
Warrier, V., Vieira, M., & Mole, S. E. (2013). Genetic basis and phenotypic correlations of the neuronal ceroid lipofusinoses. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 1832(11), 1827–1830.
Williams, R. E., & Mole, S. E. (2012). New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology, 79(2), 183–191.
Winter, E., & Ponting, C. P. (2002). TRAM, LAG1 and CLN8: members of a novel family of lipid-sensing domains? Trends in Biochemical Sciences, 27(8), 381–383.
Xin, W., Mullen, T., Kiely, R., Min, J., Feng, X., Cao, Y.,.. . Mole, S. (2010). CLN5 mutations are frequent in juvenile and late-onset non-Finnish patients with NCL. Neurology, 74(7), 565–571.
Zelnik, N., Mahajna, M., Iancu, T. C., Sharony, R., & Zeigler, M. (2007). A novel mutation of the CLN8 gene: is there a Mediterranean phenotype? Pediatric Neurology, 36(6), 411–413.
Acknowledgements
The authors deeply acknowledge all the study participants for their cooperation and contribution towards this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Parvin, S., Rezazadeh, M., Hosseinzadeh, H. et al. The Neuronal Ceroid Lipofuscinoses-Linked Loss of Function CLN5 and CLN8 Variants Disrupt Normal Lysosomal Function. Neuromol Med 21, 160–169 (2019). https://doi.org/10.1007/s12017-019-08529-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-019-08529-7