
Abstract
Type 1 equilibrative nucleoside transporter (ENT1) promotes glutamate release by inhibition of adenosine signaling. However, whether ENT1 plays a role in epileptic seizure that involves elevated glutamatergic neurotransmission is unknown. Here, we report that both seizure rats and patients show increased expression of ENT1. Intrahippocampal injection of a specific inhibitor of ENT1, nitrobenzylthioinosine (NBTI), attenuates seizure severity and prolongs onset latency. In order to examine whether NBTI would be effective as antiepileptic after peripheral application, we injected NBTI intraperitoneally, and the results were similar to those obtained after intrahippocampal injection. NBTI administration leads to suppressed neuronal firing in seizure rats. In addition, increased mEPSC in seizure are inhibited by NBTI. Finally, NBTI results in deactivation of phosphorylated cAMP-response element-binding protein in the seizure rats. These results indicate that ENT1 plays an important role in the development of seizure. Inhibition of ENT1 might provide a novel therapeutic approach toward the control of epileptic seizure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epilepsy is one of the most common and disabling neurological disorder characterized by transient and unprovoked seizures. It affects about 50 million people worldwide of all ages and often requires lifelong medication (Xu et al. 2012; Maroso et al. 2010). Despite the use of anti-epileptic drugs (AEDs), about 20–40 % of patients remain refractory (French 2007).
Nucleoside transporters play important role in sleep, arousal, drug and alcohol addiction, nociception and analgesia. The therapeutic use of nucleoside transporter inhibitors in neoplastic, cardiovascular and infectious diseases has been described (King et al. 2006). There are two families of nucleoside transporters named concentrative and equilibrative in animals, which are encoded by the solute carrier families 28 and 29 (SLC28A and SLC29A), respectively (Baldwin et al. 2004). The SLC29A family encodes equilibrative nucleoside transporter (ENT) proteins including ENT1, ENT2, ENT3 and ENT4 (Gray et al. 2004). Both ENT1 and ENT2 mediate adenosine influx or efflux, in a direction dictated by the relative intracellular and extracellular concentrations of adenosine. ENT4 is known as plasma membrane monoamine transporter and play only minor role in adenosine signaling. In contrast to ENT4, the functional properties of ENT3 have not yet been described. It is suggested ENT3 might function as an intracellular rather than as a cell surface transporter (Baldwin et al. 2005; Barnes et al. 2006; Parkinson et al. 2011).
It is known that glutamate levels are elevated in epileptic brains and may directly trigger seizures by increasing neuronal excitability or promote epileptogenesis by inducing neuronal death (During and Spencer 1993). ENT1 that regulates extracellular adenosine concentrations across the plasma membrane appears to be present on both neurons and glia (Parkinson et al. 2005; Dunwiddie and Masino 2001). Study has shown that adenosine is an inhibitor of neuronal activity by suppressing synchronous discharges and epileptic seizures (Huber et al. 2001; Dale and Frenguelli 2009), which is associated with A1 adenosine receptor (Sebastiao 2010; Fredholm et al. 2005). Adenosine acts via A1 receptors presynaptically and inhibits glutamatergic synaptic transmission in the hippocampus (Lupica et al. 1992; Honore et al. 1998). Therefore, we hypothesize that inhibition of ENT1 by nitrobenzylthioinosine (NBTI), a specific inhibitor of ENT1, may attenuate seizure severity by glutamergic neurotransmission mechanism. We first measured neuronal ENT1 expression in patients with epilepsy and an epileptic seizure animal model. Then, we tested whether ENT1 inhibitor is effective in controlling seizure attacks, and the underlying mechanisms were investigated as well.
Materials and Methods
Experimental Model of Epileptic Seizure
All experimental procedures were approved by the Commission of Chongqing Medical University for the Ethics of Experiments on Animals in accordance with China Animal Welfare Legislation standards. The adult male Sprague–Dawley (SD) rats weighing 180–220 g were obtained from the Experimental Animal Center of Chongqing Medical University, China. The rats were maintained in a temperature controlled room (22–24 °C) with a 12-h light and 12-h dark cycle as well as free access to food and water. The rat model of epileptic seizure was produced as reported previously (Curia et al. 2008). Briefly, male rats were injected lithium chloride (127 mg/kg, i.p., Sigma-Aldrich Co., St. Louis, MO, USA) 20 h prior to the administration of pilocarpine. Furthermore, the rats were pretreated with atropine sulfate (1 mg/kg) 30 min prior to pilocarpine administration (30 mg/kg, i.p., Sigma-Aldrich, USA). The rats received repeated injections of pilocarpine (10 mg/kg, i.p.) every 30 min until they developed seizures. The seizures were evaluated by Racine score, and the latency to the first onset of seizure was measured as behavioral assessment. Only convulsive (motor) seizures were considered as the onset of seizure such as stage 4, rearing, and stage 5, rearing plus loss of balance and falling accompanied by generalized clonic seizures (Racine 1972; Xu et al. 2011). All rats were under video monitoring continuously starting immediately after pilocarpine injection until killed. The seizure rats were further studied randomly at four time points, 6, 24, 72 h, 1 week, following the onset of seizure (five rats for each time point). In addition, normal control rats received the same treatment with lithium chloride and atropine sulfate, but we used saline instead of pilocarpine.
Microinjection and Intraperitoneal Injection of NBTI
To examine whether inhibition of ENT1 in the hippocampus alters seizure behaviors and the downstream gene expression, we microinjected male SD rats with the ENT1-specific inhibitor, nitrobenzylthioinosine (NBTI). Rats were anesthetized with 3.5 % chloral hydrate (1 ml/100 g, i.p.) and placed in a stereotaxia frame (Stoelting Co., Ltd., USA). After the skull was exposed, 0.5-mm holes were drilled for placement of stainless steel guide cannulas aimed to the dorsal hippocampus (anterior–posterior −3.3 mm, medial–lateral ±1.8 mm, dorsal–ventral −2.6 mm). The guide cannulas were fixed by dental cement followed by insertion of dummy cannulas to prevent clogging. The rats were allowed to recover for 1 h and were left undisturbed in their cages for 7 days prior to study. Microinjections were given 15 min before pilocarpine injecting. NBTI [50 μM, dissolved in saline containing 2 % dimethyl sulphoxide (DMSO), Sigma-Aldrich Co.], 2 % DMSO (vehicle control) and artificial cerebrospinal fluid (ACSF, normal control) were infused using an infusion pump at 0.3 μl/min in a volume of 0.5 μl per side, respectively. The infusion cannulas were left in place for 1 min to allow for diffusion (Chen et al. 2007). In addition, to examine whether NBTI would be effective as antiepileptic after peripheral application, we injected male SD rats intraperitoneally with NBTI as described previously (Anderson et al. 1996). Intraperitoneal injections of NBTI (15 mg/kg) were given 45 min before pilocarpine injecting. Furthermore, the grouping method was the same as microinjection.
Selection of Brain Tissues and Tissue Preparation
Fifteen patients undergoing surgery for drug-resistant temporal lobe epilepsy (TLE), and 15 control samples from traumatic brain without epilepsy were included in this study. All brain tissues were chosen randomly from our epilepsy brain bank, which was reported in our previous study (Wang et al. 2014; Zhang et al. 2014). Before surgery, informed written consent form was obtained from the patients or their lineal relatives for the use of data and brain specimen. Experimental protocols complied with the guidelines for the conduct of research involving human subjects by National Institutes of Health of China and the Committee on Human Research at Chongqing Medical University, Chongqing, China. The diagnosis of epilepsy was made according to the International Classification of Epilepsy Seizures of the International League Against Epilepsy (1981). Presurgical assessment included detailed medical history, a neurological examination, interictal and ictal EEG studies, neuropsychological testing and neuroradiological studies. The epileptic lesion was localized in all patients by brain magnetic resonance imaging (MRI) or CT and 24-h EEG or video EEG. Sphenoidal electrode monitoring and intraoperative electrocorticography (ECOG) were performed to localize the epileptic lesion before resection in all patients. All patients were resistant to the maximal doses of three or more AEDs, including phenytoin (PHT), valproic acid (VPA), carbamazepine (CBZ), phenobabital (PB), topiramate (TPM) or lamotrigine (LTG). Cranial MRI scans found no progressive lesions in the central nervous system (CNS). In this epilepsy group, surgical removal of the epileptic discharge zone in the temporal neocortex was for strictly therapeutic purposes. After resection, intraoperative electrocorticography was carried out to ensure that the lesion was resected completely. Table 1 summarizes the clinical features of TLE patients. There are no significant differences in age and gender of the studied tissues between TLE and control tissues, which are shown in Table 2.
For both rat and human brain tissues, one portion of the samples obtained from each subjects was immediately placed in a cryovial, which had been soaked in buffered diethylpyrocarbonate (1:1,000) for 24 h and then stored in liquid nitrogen for western blotting study. Another portion of rat brain tissues was fixed with 4 % paraformaldehyde overnight at 4 °C and then embedded in paraffin, 5-μm-thick sections were cut and processed for immunohistochemistry and 10-μm-thick sections were cut for immunofluorescence.
Western Blotting
All samples were homogenized in 30 mM Tris–HCl and 100 mM phenylmethylsulfonyl fluoride. The protein concentration in the supernatant was measured by the Enhanced BCA Protein Assay Kit according to the manufacturer’s instructions (Beyotime, Haimen, China). Proteins (50 μg per lane) were separated by SDS-PAGE (5 % spacer gel; 10 % separation gel) and then transferred to polyvinylidene fluoride (PVDF) membranes (250 mA, 60 min). The PVDF membranes were blocked in 5 % skim milk for 1 h at room temperature and then incubated at 4 °C overnight with primary antibodies (rabbit anti-ENT1 1:1,000 and mouse anti-β-actin at 1:500, Santa Cruz Biotechnology, CA; rabbit anti-p-CREB 1:500, Cell signal, USA). The blots were washed 4 × 10 min each with PBS plus Tween-20 (0.1 %) and incubated with the appropriate diluted HRP-tagged secondary antibody (1:1,000, Santa Cruz Biotechnology, CA) for 1 h at room temperature. Blots were developed according to the manufacturer’s instructions with Super Signal West Pico Chemiluminescent HRP substrate (Rockford, IL, USA). Blots intensities were calculated with the Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
Immunohistochemistry
Paraffin embedded sections were deparaffinized, hydrated through graded alcohols and incubated in 0.3 % H2O2 for 15 min. The sections were heated (92–98 °C) in 10 mmol/l boiling sodium citrate buffer at pH 6.0 for 20 min and then blocked in normal goat serum (1:10) for 10 min (Zhongshan Golden Bridge Inc., Beijing, China). Sections were then incubated with the primary rabbit polyclonal anti-ENT1 antibody (1:50, Santa Cruz Biotechnology, CA) at 37 °C for 2 h then washed 4 times with PBS. Sections were then incubated with biotinylated goat anti-rabbit secondary antibody (streptavidin–peroxidase kits, Zhongshan Golden Bridge, Inc., Beijing, China) for 30 min and washed. Sections were then treated with ABC solution at 37 °C for 30 min, washed with PBS and incubated with DAB (3,3′-diaminobenzidine, Zhongshan Golden Bridge, Inc., Beijing, China) for 3 min. Counterstaining was carried out with Harris hematoxylin. For a negative control, primary and secondary antibodies were replaced with PBS. The OLYMPUS PM20 automatic microscope (Olympus, Osaka, Japan) and TC-FY-2050 pathology system (Yuancheng Inc., Beijing, China) were used to collect the images. Cell with buffy staining in cytoplasm was considered positive. Ten random visual field images at 400× magnification for each sample were selected for analysis. HPIAS 1000 high definition image analysis system (1,000 s of screen image engineering firm, Wuhan, China) was used to count the ENT1 positive cells.
Double-Labeling Immunofluorescence
To confirm the location of ENT1 in neurons and astrocytes in seizure hippocampus, immunoreactivity for ENT1 was observed by double-label immunofluorescence in rats 24 h after the onset of seizure. Paraffin sections were deparaffinized in xylene and rehydrated in a graded series of ethanol. The sections were heated in 0.01 M citric acid (pH 6.0) for 20 min at 92–98 °C for antigen recovery. Then, the tissues were permeabilized with 0.5 % Triton X-100 and were incubated in normal goat serum (Zhongshan Golden Bridge, Inc., Beijing, China) for 30 min. Sections were incubated with rabbit anti ENT1 (1:100; Santa Cruz Biotechnology, USA) and mouse anti-GFAP antibody (1:100; Zhongshan Golden Bridge, Inc., Beijing, China) overnight at 4 °C. After washing with PBS, the sections were incubated in the dark with anti-rabbit-FITC (green) and anti-mouse-TRITC (red) (Zhongshan Golden Bridge, Inc., Beijing, China) at a dilution of 1:100 for 60 min and 50 % glycerol and 50 % PBS mounted sections. The fluorescence was detected by laser scanning confocal microscopy (Leica, Heidelberg) on an Olympus IX70 inverted microscope (Olympus, Tokyo, Japan).
Electrophysiology
The rat was anesthetized with 3.5 % chloral hydrate (1 ml/100 g, i.p.). After cardiac perfusion by 0 °C slice solution (containing in mM: KCL, 2.5; NaH2PO4·2H2O, 1.25; MgCl2·H2O, 6; CaCl2. 1; NaHCO3, 26; sucrose, 220; glucose, 10), the whole brain was removed and placed in 0 °C slice solution for 3 min, then chopped into 350 μm thickness using an oscillating tissue slicer (Campden, NVSLM1). The brain slices were recovered in artificial cerebrospinal fluid (ACSF) at 37 °C (in mM: NaCl, 124; KCl, 3; NaH2PO4·2H2O, 1.23; NaHCO3, 26; CaCl2, 2; MgCl2, 2; glucose, 10, buffered with 95 % O2–5 % CO2) for 1 h and then transferred to ACSF (23 °C) for 30 min the following recordings were performed at 23 °C unless otherwise stated.
For spontaneous action potential, recording was conducted in gapfree mode. The average values of resting membrane potential were about −58 mV. After stable spontaneous action potentials were obtained, 100 nM NBTI was added in perfusion solution. To further illustrate the effect of NBTI on the action potential of epileptic rat, spikes were evoked by a 500 ms current pulse (10, 30, 50, 70 and 90 pA) in hippocampal CA1 pyramidal neurons. The adding mode of NBTI was the same as before. Patch pipette (2–4 ΩM) was filled with internal solution (mM): 60 K2SO4, 60 N-methyl-d-glucamine (NMG), 40 HEPES, 4 MgCl2, 0.5 BAPTA, 12 phosphocreatine, 2 Na2ATP, 0.2 Na3GTP, pH 7.2–7.3 (265–270 mOsm/l). ACSF was used as external solution. The signal was amplified using a Multiclamp 700B amplifier (Axon, USA) and digitized with a Digidata 1322A. Data were filtered at 10 kHz and then recorded with pClamp 9.2 software (Molecular Devices, Sunnyvale, CA, USA).
For the recording of excitatory postsynaptic currents, a patch pipette (2–4 ΩM) was filled with intracellular solution containing 130 mM Cs-methanesulfonate, 10 HEPES, 10 CsCl, 4 NaCl, 1 MgCl2, 1 EGTA, 5 NMG, 5 MgATP, and 0.5 Na2GTP and 12 phosphocreatine, pH 7.2, 275–290 mOsm. Bicuculline (10 μM) and TTX (1 µM) were added in ACSF to record mEPSC.
Statistical Analysis
Data were expressed as mean ± SEM. Statistical comparisons were made by one-way ANOVA or Student’s t test with SPSS (version 11.5) software. P < 0.05 was considered significance.
Results
The Expression of ENT1 in Patients with Epilepsy and a Pilocarpine-Induced Rat epileptic Seizure Model
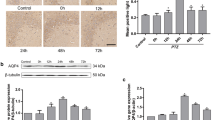
As shown in Fig. 1a, b (n = 15), ENT1 was significantly increased in patients with epilepsy. To test whether ENT1 was also altered in animal seizure model, we measured dynamic changes of ENT1 in pilocarpine-induced seizure in rats. As shown in Fig. 1c, d, increased expression of ENT1 was peaked at 24 h after seizure and remained significantly higher at 72 h and 1 week (n = 5 in each group).

Western blot analysis for ENT1 in the brain tissues of humans and rats. a Proteins from individual temporal neocortex of patients with TLE (epilepsy, EP) and nonepileptic patients (control, Con). The immunoreactive stainings of ENT1 in EP group are stronger than those in controls. b A comparison of the ratio of optical density of ENT1 over β-actin indicates significantly higher expression of ENT1 in patients with TLE than in controls (*P < 0.01, n = 15). c Hippocampal ENT1 blots in epileptic seizure rats are stronger than those in controls (Con) at 6, 24, 72 h and 1 week after the onset of seizures. d A comparison of the mean intensity ratio (ENT1/β-actin) indicates significantly higher expression of ENT1 in EP than in controls (*P < 0.01; **P < 0.05, n = 5) (ANOVA, F = 180.9)
In control hippocampus, ENT1 was presented mostly in cell membrane and cytoplasm of the pyramidal neurons and granule cells of the dentate gyrus (Fig. 2a, c). In seizure hippocampus, immunoreactivity was found in pyramidal neurons, granule cells and astrocytes (Fig. 2b, d). Significant increase of ENT1 positive cells was found in seizure group compared with that in control group (Fig. 2e, n = 5). Similarly, immunofluorescence study showed abundant ENT1 expression 24 h after seizure onset in CA1 pyramidal layers and DG granule layers (Fig. 3a, d). In addition, GFAP positive astrocytes also expressed ENT1 in the various hippocampal subfields including CA1 and DG (Fig. 3c, f). These results indicate that ENT1 is significantly increased after recurrent neuronal discharges.

Immunohistochemistry analysis for ENT1 in the hippocampus of control (Con) and epileptic seizure (EP) rats. In the control group (a, c), ENT1 immunoreactivity is located in neuronal membrane and cytoplasm (CA1 and dentate gyrus, DG); in the seizure rats 24 h after the onset of seizures (b, d), ENT1 immunoreactivity is located in neuronal and astrocytic membrane and cytoplasm (CA1 and DG). e The number of ENT1 positive cells is significantly lower in control (con) than in the EP group (*P < 0.01, n = 5). Scale bar 200 μm. Black arrows ENT1 positive neurons were enlarged above the top right corner. Red arrows ENT1 positive astrocytes were enlarged below the top right corner (Color figure online)

Double-labeled immunofluorescence analysis for ENT1 in the hippocampus of epileptic seizure rats 24 h after seizures in CA1 and dentate gyrus (DG). The yellow arrows signify co-expression of ENT1 (green a, d) with GFAP (red b, e). The white arrows signify ENT1 and GFAP without co-expression. c, f Merged images of ENT1 with GFAP. Scale bar 75 μm (Color figure online)
ENT1 Inhibitor NBTI Suppresses Epileptic Seizure Susceptibility
To determine whether ENT1 selective inhibitor NBTI may have impact on epileptic seizure behavior, we injected NBTI into the CA1 region of the hippocampus bilaterally. As shown in Fig. 4a, b, rats treated with NTB1 exhibited significantly longer onset latency and less seizure severity (P < 0.01, n = 9 in each group). In order to examine whether NBTI would be effective as antiepileptic after peripheral application, we injected NBTI intraperitoneally. The results (Fig. 4c, d, P < 0.01, n = 9 in each group) were similar to those obtained after intrahippocampal injection. These results indicate that NBTI possesses a strong inhibitory effect on seizure.

NBTI reduces seizure severity. In the microinjection study, the latency (min) to the first seizure (level 4 of Racine score) of rats undergone hippocampal injection of ACSF (Control, Con), DMSO (Vehicle) and NBTI (a). Racine score at different time points after pilocarpine injection (b). The same phenomenon is detected in the intraperitoneal injection (c, d). Significance is seen at every time point in NBTI treated rats compared with control or vehicle. *P < 0.01; # P > 0.05 (n = 9 in each group)
NBTI Decreases Neuronal Excitability in CA1 Pyramidal Neurons of Epileptic Seizure Rats
To validate the behavioral findings, we examined the firing properties of hippocampal CA1 pyramidal neurons in acutely isolated slices from seizure rats 24 h after the onset of seizures. Bath application of NBTI (100 nM) did not affect normal neurons (Fig. 5a), but significantly decreased the numbers of APs in seizure neurons (Fig. 5b, d, n = 5). The effect of NBTI on AP occurred in neurons treated with different current injections (Fig. 5c, e, n = 5 in each group, P < 0.01).

The effect of NBTI on the excitability in CA1 pyramidal neurons of epileptic seizure rats. a NBTI does not affect the action potential (AP) of normal neurons. b NBTI decreases epileptic spikes that recover after washout. c NBTI (100 nM) effect on APs in neurons injected with different currents. d Summary (taking from a) of AP numbers per minute in slices treated with ACSF, ACSF + NBTI and washout (n = 5). e Comparison of AP numbers between ACSF and ACSF + NBTI after neuron is injected with different currents (taking from c, n = 5 in each group). *P < 0.01, paired-sample t test
NBTI Inhibits Excitatory Neurotransmission
To test whether NBTI may inhibit excitatory neurotransmission, we measured the effect of NBTI on mEPSC. As shown in Fig. 6a–c, application of NBTI significantly reduced frequency, but not amplitude of mEPSC in CA1 neurons 24 h after the onset of seizures (n = 5).

Effect of NBTI on miniature EPSCs (mEPSCs) in CA1 pyramidal neurons. a Representative traces of mEPSCs in slices from control (left normal rat), epilepsy seizure rats without treatment (seizure without NBTI, middle) and seizure rats treated with NBTI (NBTI, 100 nM, right). b Cumulative fraction of mEPSC amplitude. c Cumulative fraction of inter-event intervals. NBTI decreases the frequency of mEPSCs in seizure neurons but not the amplitude. n = 5 in each group. Kolmogorov–Smirnov test
NBTI Inhibits the CREB Activation
It has been found that glutamate could stimulate phosphorylation of CREB (Lonze and Ginty 2002), we then tested whether NBTI may regulate CREB function. As shown in Fig. 7a, b, p-CREB was significantly increased 24 h after seizure, while NBTI injection in the hippocampus led to significantly decreased p-CREB (n = 5). The similar expression trend of CREB was detected in those groups (Fig. 7c, d).

Western blotting analysis of p-CREB in the hippocampus of rat. a Representative immunoblots showing p-CREB immunoreactivity in rats after hippocampal injection of vehicle (vehicle, epileptic seizure rats), saline (control rats without seizure, Con), NBTI (NBTI, epileptic rats) or without injection (EP, epileptic seizure rats). b Bar plot summary showing that p-CREB is increased in the hippocampus of seizure rats (EP) compared with control (Con) or seizure rats treated with NBTI. *P < 0.01; # P > 0.05, ANOVA (F = 71.2). The similar expression trend of CREB was detected in those groups (c, d). *P < 0.01; # P > 0.05, ANOVA (F = 52.51). n = 5 in each group
Discussion
The major findings of this study are that ENT1 is significantly increased in patients with epilepsy and an epileptic seizure animal model. ENT1 inhibitor NBTI attenuates seizure severity and prolongs onset latency. This effect is mediated by adenosine A1 receptor and may be attributed to decreased neuronal firing and glutamatergic neurotransmission, as well as deactivation of CREB. These results suggest that NBTI possesses a potential role in the treatment of seizure.
It is known that ENT1 mRNA is expressed in pyramidal neurons of the hippocampus and granule neurons of the dentate gyrus in adult human brain (Anderson et al. 1999). In spinal cord, ENT1 immunofluorescence is located in cell bodies of either neurons or glia (Governo et al. 2005). Consistently, the immunoreactive ENT1 exists in both pyramidal and dentate layers in the hippocampus in our study. Evidence has shown that ENT1 plays an important role in regulating brain function by modulating endogenous adenosine concentrations in CNS (Jennings et al. 2001) and is associated with sleep, arousal, drug and alcohol addiction, nociception and analgesia (King et al. 2006). However, whether ENT1 is associated with epileptic seizures remains unknown. Both Western blotting and immunohistochemical studies reveal that ENT1 expression is significantly increased in patients with epilepsy and pilocarpine-induced epileptic seizure model. Limited by the ethics, normal human brain is impossible to get, so we can only choose traumatic patients brain tissue as a control group. Hippocampal lesion is the most critical factor of epilepsy, however, the hippocampus of patients with epilepsy and nonepilepsy is not readily available. So, we choose the temporal cortex of patients and animal hippocampus in experiments. Thus, the present study provides direct evidence that links ENT1 with recurrent neuronal discharges.
Studies suggest that adenosine and overexpression of adenosine kinase (ADK) are pathological hallmarks of epileptic brain. The latter has emerged as a promising target for the prevention of epilepsy (Boison 2012). By analogy, ENT1 that controls extracellular concentration of adenosine may also regulate pathophysiology of seizure. It is known that ENT1 exerts a functional role in drug addiction and associated behaviors (Choi et al. 2004; Levran et al. 2009). Decreased ethanol sensitivity or increased ethanol drinking have been found in ENT1 null mice (Chen et al. 2010; Nam et al. 2010). Conversely, increased expression of ENT1 is associated with enhanced response to ethanol (Parkinson et al. 2009). Interestingly, microinjecting of NBTI into the amygdala reduces anxiety-like behavior of mice (Chen et al. 2007). The current study has demonstrated that microinjection of NBTI in the hippocampus significantly attenuates seizure severity and prolongs seizure latencies. Therefore, NBTI may serve as a potential therapeutic drug for treating epileptic seizure.
Our study demonstrates that consistent with behavioral findings, NBTI inhibits neuronal hyper-excitability as measured by AP spikes. However, it is relatively unclear how ENT1 may regulate neuronal excitability. One study reports that ENT1 controls glutamatergic neurotransmission. In rat spinal dorsal horn, ENT1 inhibitor reduces evoked EPSC in lamina II neurons in a dose-dependent manner (Ackley et al. 2003). Also, neuronal over expression ENT1 reduced extracellular adenosine levels induced by hypoxia and ischemia (Zhang et al. 2011), suggesting that ENT1 regulates neuronal activity though adenosine signaling. Limited by the experiment condition, we do not directly detect the adenosine concentration by microdialysis measurements at each time point.
In the current sudy, increased seizure intensity and decreased seizure latency are paralleled with an increase in hippocampus p-CREB in seizure rats. The CREB transcription factor is also recognized as a modulator of seizure. Significantly increased CREB phosphorylation is directly linked to CREB dependent gene transcription involved in neuronal excitability of epilepsy (Beaumont et al. 2012). A recent report shows that mice with decreased CREB levels exhibit about 50 % reduction in spontaneous seizures that requires more stimulation to reach kindling threshold (Zhu et al. 2012). The previous study has demonstrated that direct application of glutamate to the suprachiasmatic nucleus leads to increased p-CREB (Ding et al. 1997). Together with behavioral and electrophysiological findings, we assume that inhibition of ENT1 by NBTI results in activation of adenosine A1 receptor, inhibition of glutamatergic neurotransmission and attenuation of p-CREB levels, which contribute to suppressed seizure susceptibility.
In conclusion, the significant increase of ENT1 in hippocampus of epileptic rats and in brain specimens from epileptic patients suggests that this molecule might play an important role in epilepsy. NBTI suppression of seizure susceptibility implies that ENT1 might be potential target for prevention and control of epilepsy.
References
Ackley, M. A., Governo, R. J., Cass, C. E., Young, J. D., Baldwin, S. A., & King, A. E. (2003). Control of glutamatergic neurotransmission in the rat spinal dorsal horn by the nucleoside transporter ENT1. Journal of Physiology, 548(Pt 2), 507–517. doi:10.1113/jphysiol.2002.038091.
Anderson, C. M., Sitar, D. S., & Parkinson, F. E. (1996). Ability of nitrobenzylthioinosine to cross the blood–brain barrier in rats. Neuroscience Letters, 219(3), 191–194.
Anderson, C. M., Xiong, W., Geiger, J. D., Young, J. D., Cass, C. E., Baldwin, S. A., et al. (1999). Distribution of equilibrative, nitrobenzylthioinosine-sensitive nucleoside transporters (ENT1) in brain. Journal of Neurochemistry, 73(2), 867–873.
Baldwin, S. A., Beal, P. R., Yao, S. Y., King, A. E., Cass, C. E., & Young, J. D. (2004). The equilibrative nucleoside transporter family, SLC29. Pflügers Archiv European Journal of Physiology, 447(5), 735–743. doi:10.1007/s00424-003-1103-2.
Baldwin, S. A., Yao, S. Y., Hyde, R. J., Ng, A. M., Foppolo, S., Barnes, K., et al. (2005). Functional characterization of novel human and mouse equilibrative nucleoside transporters (hENT3 and mENT3) located in intracellular membranes. Journal of Biological Chemistry, 280(16), 15880–15887. doi:10.1074/jbc.M414337200.
Barnes, K., Dobrzynski, H., Foppolo, S., Beal, P. R., Ismat, F., Scullion, E. R., et al. (2006). Distribution and functional characterization of equilibrative nucleoside transporter-4, a novel cardiac adenosine transporter activated at acidic pH. Circulation Research, 99(5), 510–519. doi:10.1161/01.RES.0000238359.18495.42.
Beaumont, T. L., Yao, B., Shah, A., Kapatos, G., & Loeb, J. A. (2012). Layer-specific CREB target gene induction in human neocortical epilepsy. Journal of Neuroscience, 32(41), 14389–14401. doi:10.1523/JNEUROSCI.3408-12.2012.
Boison, D. (2012). Adenosine dysfunction in epilepsy. Glia, 60(8), 1234–1243. doi:10.1002/glia.22285.
Chen, J., Nam, H. W., Lee, M. R., Hinton, D. J., Choi, S., Kim, T., et al. (2010). Altered glutamatergic neurotransmission in the striatum regulates ethanol sensitivity and intake in mice lacking ENT1. Behavioural Brain Research, 208(2), 636–642. doi:10.1016/j.bbr.2010.01.011.
Chen, J., Rinaldo, L., Lim, S. J., Young, H., Messing, R. O., & Choi, D. S. (2007). The type 1 equilibrative nucleoside transporter regulates anxiety-like behavior in mice. Genes, Brain Behavior, 6(8), 776–783. doi:10.1111/j.1601-183X.2007.00311.x.
Choi, D. S., Cascini, M. G., Mailliard, W., Young, H., Paredes, P., McMahon, T., et al. (2004). The type 1 equilibrative nucleoside transporter regulates ethanol intoxication and preference. Nature Neuroscience, 7(8), 855–861. doi:10.1038/nn1288.
Curia, G., Longo, D., Biagini, G., Jones, R. S., & Avoli, M. (2008). The pilocarpine model of temporal lobe epilepsy. Journal of Neuroscience Methods, 172(2), 143–157. doi:10.1016/j.jneumeth.2008.04.019.
Dale, N., & Frenguelli, B. G. (2009). Release of adenosine and ATP during ischemia and epilepsy. Current Neuropharmacology, 7(3), 160–179. doi:10.2174/157015909789152146.
Ding, J. M., Faiman, L. E., Hurst, W. J., Kuriashkina, L. R., & Gillette, M. U. (1997). Resetting the biological clock: Mediation of nocturnal CREB phosphorylation via light, glutamate, and nitric oxide. Journal of Neuroscience, 17(2), 667–675.
Dunwiddie, T. V., & Masino, S. A. (2001). The role and regulation of adenosine in the central nervous system. Annual Review of Neuroscience, 24, 31–55. doi:10.1146/annurev.neuro.24.1.31.
During, M. J., & Spencer, D. D. (1993). Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet, 341(8861), 1607–1610.
Fredholm, B. B., Chen, J. F., Masino, S. A., & Vaugeois, J. M. (2005). Actions of adenosine at its receptors in the CNS: Insights from knockouts and drugs. Annual Review of Pharmacological Toxicology, 45, 385–412. doi:10.1146/annurev.pharmtox.45.120403.095731.
French, J. A. (2007). Can evidence-based guidelines and clinical trials tell us how to treat patients? Epilepsia, 48(7), 1264–1267. doi:10.1111/j.1528-1167.2007.01123.x.
Governo, R. J., Deuchars, J., Baldwin, S. A., & King, A. E. (2005). Localization of the NBMPR-sensitive equilibrative nucleoside transporter, ENT1, in the rat dorsal root ganglion and lumbar spinal cord. Brain Research, 1059(2), 129–138. doi:10.1016/j.brainres.2005.08.017.
Gray, J. H., Owen, R. P., & Giacomini, K. M. (2004). The concentrative nucleoside transporter family, SLC28. Pflügers Archiv European Journal of Physiology, 447(5), 728–734. doi:10.1007/s00424-003-1107-y.
Honore, P., Buritova, J., Chapman, V., & Besson, J. M. (1998). UP 202-56, an adenosine analogue, selectively acts via A1 receptors to significantly decrease noxiously-evoked spinal c-Fos protein expression. Pain, 75(2–3), 281–293.
Huber, A., Padrun, V., Deglon, N., Aebischer, P., Mohler, H., & Boison, D. (2001). Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. Proceedings of the National Academy of Sciences of the United States of America, 98(13), 7611–7616. doi:10.1073/pnas.131102898.
Jennings, L. L., Hao, C., Cabrita, M. A., Vickers, M. F., Baldwin, S. A., Young, J. D., et al. (2001). Distinct regional distribution of human equilibrative nucleoside transporter proteins 1 and 2 (hENT1 and hENT2) in the central nervous system. Neuropharmacology, 40(5), 722–731.
King, A. E., Ackley, M. A., Cass, C. E., Young, J. D., & Baldwin, S. A. (2006). Nucleoside transporters: From scavengers to novel therapeutic targets. Trends in Pharmacological Sciences, 27(8), 416–425. doi:10.1016/j.tips.2006.06.004.
Levran, O., Londono, D., O’Hara, K., Randesi, M., Rotrosen, J., Casadonte, P., et al. (2009). Heroin addiction in African Americans: A hypothesis-driven association study. Genes, Brain and Behavior, 8(5), 531–540. doi:10.1111/j.1601-183X.2009.00501.x.
Lonze, B. E., & Ginty, D. D. (2002). Function and regulation of CREB family transcription factors in the nervous system. Neuron, 35(4), 605–623.
Lupica, C. R., Proctor, W. R., & Dunwiddie, T. V. (1992). Presynaptic inhibition of excitatory synaptic transmission by adenosine in rat hippocampus: Analysis of unitary EPSP variance measured by whole-cell recording. Journal of Neuroscience, 12(10), 3753–3764.
Maroso, M., Balosso, S., Ravizza, T., Liu, J., Aronica, E., Iyer, A. M., et al. (2010). Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nature Medicine, 16(4), 413–419. doi:10.1038/nm.2127.
Nam, H. W., Lee, M. R., Hinton, D. J., & Choi, D. S. (2010). Reduced effect of NMDA glutamate receptor antagonist on ethanol-induced ataxia and striatal glutamate levels in mice lacking ENT1. Neuroscience Letters, 479(3), 277–281. doi:10.1016/j.neulet.2010.05.079.
Parkinson, F. E., Damaraju, V. L., Graham, K., Yao, S. Y., Baldwin, S. A., Cass, C. E., et al. (2011). Molecular biology of nucleoside transporters and their distributions and functions in the brain. Current Topics in Medicinal Chemistry, 11(8), 948–972.
Parkinson, F. E., Xiong, W., & Zamzow, C. R. (2005). Astrocytes and neurons: Different roles in regulating adenosine levels. Neurological Research, 27(2), 153–160. doi:10.1179/016164105X21878.
Parkinson, F. E., Xiong, W., Zamzow, C. R., Chestley, T., Mizuno, T., & Duckworth, M. L. (2009). Transgenic expression of human equilibrative nucleoside transporter 1 in mouse neurons. Journal of Neurochemistry, 109(2), 562–572. doi:10.1111/j.1471-4159.2009.05991.x.
Racine, R. J. (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalography and Clinical Neurophysiology, 32(3), 281–294.
Sebastiao, A. M. (2010). Adenosine and epilepsy-thinking beyond A(1) receptors. Purinergic Signal, 6(1), 1–2. doi:10.1007/s11302-010-9179-6.
Wang, L., Lv, Y., Deng, W., Peng, X., Xiao, Z., Xi, Z., et al. (2014). 5-HT6 receptor recruitment of mTOR modulates seizure activity in epilepsy. Molecular Neurobiology. doi:10.1007/s12035-014-8806-6.
Xu, Z., Xue, T., Zhang, Z., Wang, X., Xu, P., Zhang, J., et al. (2011). Role of signal transducer and activator of transcription-3 in up-regulation of GFAP after epilepsy. Neurochemical Research, 36(12), 2208–2215. doi:10.1007/s11064-011-0576-1.
Xu, Y., Zeng, K., Han, Y., Wang, L., Chen, D., Xi, Z., et al. (2012). Altered expression of CX3CL1 in patients with epilepsy and in a rat model. American Journal of Pathology, 180(5), 1950–1962. doi:10.1016/j.ajpath.2012.01.024.
Zhang, X., Chen, G., Lu, Y., Liu, J., Fang, M., Luo, J., et al. (2014). Association of mitochondrial letm1 with epileptic seizures. Cerebral Cortex, 24(10), 2533–2540. doi:10.1093/cercor/bht118.
Zhang, D., Xiong, W., Albensi, B. C., & Parkinson, F. E. (2011). Expression of human equilibrative nucleoside transporter 1 in mouse neurons regulates adenosine levels in physiological and hypoxic-ischemic conditions. Journal of Neurochemistry, 118(1), 4–11. doi:10.1111/j.1471-4159.2011.07242.x.
Zhu, X., Han, X., Blendy, J. A., & Porter, B. E. (2012). Decreased CREB levels suppress epilepsy. Neurobiology of Diseases, 45(1), 253–263. doi:10.1016/j.nbd.2011.08.009.
Acknowledgments
The author’s research was supported by National Natural Science Foundation of China (Nos. 81260201, 81271445 and 81201003) and Guizhou Provincial Science and Technology Foundation (No. [2013] 2327).
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Zucai Xu, Ping Xu and Yalan Chen have contributed equally to this article.
The First Affiliated Hospital of Chongqing Medical University and The Affiliated Hospital of Zunyi Medical College are contributed equally to this article.
Rights and permissions
About this article

Cite this article
Xu, Z., Xu, P., Chen, Y. et al. ENT1 Inhibition Attenuates Epileptic Seizure Severity Via Regulation of Glutamatergic Neurotransmission. Neuromol Med 17, 1–11 (2015). https://doi.org/10.1007/s12017-014-8338-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-014-8338-2