
Abstract
Recent findings indicate an isoform-specific role for apolipoprotein E (apoE) in the elimination of beta-amyloid (Aβ) from the brain. ApoE is closely associated with various lipoprotein receptors, which contribute to Aβ brain removal via metabolic clearance or transit across the blood–brain barrier (BBB). These receptors are subject to ectodomain shedding at the cell surface, which alters endocytic transport and mitigates Aβ elimination. To further understand the manner in which apoE influences Aβ brain clearance, these studies investigated the effect of apoE on lipoprotein receptor shedding. Consistent with prior reports, we observed an increased shedding of the low-density lipoprotein receptor (LDLR) and the LDLR-related protein 1 (LRP1) following Aβ exposure in human brain endothelial cells. When Aβ was co-treated with each apoE isoform, there was a reduction in Aβ-induced shedding with apoE2 and apoE3, while lipoprotein receptor shedding in the presence of apoE4 remained increased. Likewise, intracranial administration of Aβ to apoE-targeted replacement mice (expressing the human apoE isoforms) resulted in an isoform-dependent effect on lipoprotein receptor shedding in the brain (apoE4 > apoE3 > apoE2). Moreover, these results show a strong inverse correlation with our prior work in apoE transgenic mice in which apoE4 animals showed reduced Aβ clearance across the BBB compared to apoE3 animals. Based on these results, apoE4 appears less efficient than other apoE isoforms in regulating lipoprotein receptor shedding, which may explain the differential effects of these isoforms in removing Aβ from the brain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is an age-related condition, which affects approximately 36 million people worldwide (Gilbert 2013). This neurodegenerative process is characterized by a progressive deterioration in memory, executive function, and behavior (Reitz 2012) accompanied by selective neuronal degeneration and synaptic loss in the hippocampus, amygdala, and temporal neocortex (Serrano-Pozo et al. 2011). The key pathological hallmarks of AD include the formation of neurofibrillary tangles and the deposition of beta-amyloid proteins (Aβ) in the brain and cerebrovasculature (Citron 2010). While the exact pathogenesis is unknown, the major toxic agent in AD is thought to be Aβ (Gilbert 2013), which accumulates in the brain and leads to neuronal cell death and ultimately dementia (Armstrong 2009; Reitz 2012). Mounting evidence now suggests the excessive accumulation of Aβ in AD is the result of impaired Aβ clearance from the brain (Castellano et al. 2011; Mawuenyega et al. 2010). Furthermore, studies in mouse models of AD have indicated that lowering Aβ levels in the brain can minimize neurodegeneration and slow cognitive decline (Boche et al. 2005). Thus, targeting clearance-related pathways may prove most effective in attenuating Aβ accumulation in the AD brain.
One explanation for the attenuated clearance in AD is dysfunctional Aβ transport at the blood–brain barrier (BBB). The low-density lipoprotein receptor (LDLR) and the LDLR-related protein 1 (LRP1) are two BBB receptors that contribute to the brain-to-blood elimination of Aβ (Castellano et al. 2012; Deane et al. 2009). In addition to the transmembrane protein that transports molecules across the brain endothelium, these lipoprotein receptors also exist in a soluble form (Rebeck et al. 2006). The soluble receptor is generated via proteolytic cleavage at an extracellular region close to the cell surface, a process called ectodomain shedding (Begg et al. 2004; Etique et al. 2013; Selvais et al. 2010). When the soluble receptor is released from the cellular membrane, it retains the ability to bind ligands in the extracellular space (Grimsley et al. 1998; Quinn et al. 1997), but loses its functional capacity to internalize or transcytose ligands intracellularly (Rebeck et al. 2006; Selvais et al. 2010). It is believed the soluble receptor operates in a dominant negative fashion by attenuating the interaction between ligands and the membrane-associated receptor, thereby modulating endocytic activity and cell signaling (Etique et al. 2013; Rebeck et al. 2006).
While lipoprotein receptors interact with an array of ligands, one of the more closely associated is apolipoprotein E (apoE), which exists as three isoforms in humans (apoE2, apoE3, and apoE4). Numerous studies have acknowledged that possession of the apoE4 allele represents the strongest genetic risk factor for late-onset AD (Kim et al. 2009; Zhong and Weisgraber 2009). Our prior studies (Bachmeier et al. 2013) and the work of others indicate that when apoE is bound to Aβ, the BBB transport of Aβ is dramatically attenuated (Bell et al. 2007; Deane et al. 2008; Martel et al. 1997). However, when apoE is not bound to Aβ, apoE appears to have a supportive role in Aβ BBB clearance that is isoform-specific (Bachmeier et al. 2013). Along these lines, recent findings have suggested that apoE3 may promote Aβ clearance across the blood–cerebrospinal fluid barrier via the choroid plexus (Ruzali et al. 2012). As lipoprotein receptor shedding in the brain (and the BBB in particular) can be a major determinant in Aβ elimination, these studies investigated the influence of apoE on lipoprotein receptor shedding to further elucidate the role of apoE in Aβ removal from the brain.
Methods
Materials
Primary human brain microvascular endothelial cells (HBMEC) and associated culture reagents were purchased from Sciencell Research Laboratories (Carlsbad, CA, USA). Fibronectin, dextran (64,000–76,000 mol wt), and Hanks’ balanced salt solution (HBSS) were purchased from Sigma Chemical Co (St. Louis, MO, USA). DMEM/F-12 (Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12) and unlabeled human Aβ(1–42) were purchased from Invitrogen Corp (Carlsbad, CA, USA). The enzyme-linked immunosorbent assay (ELISA) kits for LRP1 and LDLR were purchased from Cedarlane Labs (Burlington, NC, USA). The ELISA for human apoE was purchased from MBL International (Woburn, MA, USA).
Animals
All animals (male mice 4–6 months of age) were purchased from Taconic Farms (Germantown, NY, USA) and allowed to adapt to the vivarium for 2 weeks prior to any experimental procedures. Mice were singly housed in a temperature and humidity-controlled room on a 12-h light/dark cycle with free access to food and water. The apoE-targeted replacement (apoE-TR) mice were created by gene targeting and carry one of the three human alleles (APOE2, APOE3, or APOE4) in place of the endogenous murine apoE gene (Sullivan et al. 1997). These mice retain the endogenous regulatory sequences required for apoE production and express the human apoE protein at physiological levels. The apoE knockout (apoE KO) mice were generated through disruption of the murine apoE gene, which results in a complete absence of the endogenous mouse apoE protein (Piedrahita et al. 1992). The wild-type mice were of the same background (C57BL/6) as the transgenic apoE animals described above. All experimental protocols involving animals were approved by the Institutional Animal Care and Use Committee of the Roskamp Institute, Inc.
Aβ Peptides
Using a standard process to limit aggregation, the Aβ peptides used in each of the studies were solubilized in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) to acquire a monomeric/dimeric sample and minimize the formation of β-sheet structures as we previously described (Bachmeier et al. 2010).
ApoE Isoforms
Dr. Mary Jo LaDu (University of Illinois at Chicago) kindly provided the mixed glial cultures. Cortical glial cultures were prepared from apoE-TR mice (apoE2-TR, apoE3-TR, or apoE4-TR) as previously described (Manelli et al. 2007). Briefly, dissected cortices from 1- to 2-day-old neonatal apoE-TR pups were dissociated by trypsinization and filtered sequentially through 100- and 40-μm cell strainers. Cells were plated in 150-cm2 flasks (~1½ brains per flask), and the medium (DMEM/F12 containing 10 % fetal bovine serum, 2 mM l-glutamine, and 1 % penicillin/streptomycin) was changed every 3–5 days (Fan et al. 2011; Manelli et al. 2007). On day 10, confluent cultures were trypsinized and passaged into 75-cm2 flasks (1 × 150 cm2 flask into 4 × 75 cm2 flasks). Upon confluency, cells were washed with serum-free media and incubated with serum-free media for 72 h (Fan et al. 2011). Glial-conditioned media (GCM) were collected and centrifuged at 1,000g for 3 min to remove any residual cells. The GCM were concentrated (10×) using the Vivaspin-15 centrifugal concentrator with a molecular weight cutoff of 10,000 kDa (Sartorius Mechatronics Corp., Bohemia, NY, USA). The resulting concentrate was analyzed for apoE content using a human apoE ELISA as per the manufacturer’s instructions and stored at −20 °C until further use.
Antibodies
Polyclonal rabbit LRP1 antibody recognizing the 85 kDa C-terminal subunit (LRP-85) and polyclonal rabbit laminin (Sigma Chemical Co., St. Louis, MO, USA), monoclonal rabbit synaptophysin (Cell Signaling Technology, Inc., Danvers, MA, USA), and mouse monoclonal anti-actin, clone C4 (EMD Millipore Corp., Billerica, MA, USA).
Human Brain Endothelial Cell Culture
HBMEC were seeded at 50,000 cells/cm2 onto fibronectin-coated 6-well plates as previously described (Bachmeier et al. 2010). At approximately 80 % confluency, the cells were treated with various concentrations (0.1, 0.2, 1, 2, and 10 µM) of Aβ(1–42) and incubated for 48 h at 37 °C. Similarly, for the apoE studies, HBMEC cells were treated with each apoE isoform (25 ng/ml) in the presence or absence of 2 µM Aβ(1–42) and incubated for 48 h at 37 °C. It was recently determined that the average concentration of apoE found in the brain interstitial fluid (ISF) of the same apoE transgenic animals used in the current study is 25 ng/ml (Ulrich et al. 2013). While the concentration of apoE is reported to be much greater in cerebrospinal fluid (5–10 µg/ml) (Bekris et al. 2008; Wahrle et al. 2007; Yamauchi et al. 1999), these studies used the concentration found in the ISF, as this is most relevant in studying the brain microvasculature. Following each treatment period, the extracellular media were collected and the cell monolayer was washed with ice-cold PBS. Cell lysates were collected using lysis buffer consisting of M-PER reagent (Pierce Biotechnology, Rockford, IL, USA) supplemented with phenylmethanesulfonyl fluoride (1 mM) and Halt protease and phosphatase inhibitor cocktail (Thermo Scientific, Waltham, MA, USA). Cellular toxicity in the HBMEC was assessed via the extracellular media using a lactate dehydrogenase (LDH) detection assay (Roche Applied Science, Indianapolis, IN, USA).
Intracerebral Aβ(1–42) Injections
Stereotaxic intracranial injections of Aβ were performed as previously described (Paris et al. 2011). Briefly, male mice (4–6 months of age) were anesthetized via inhalation using a 4 % isoflurane/oxygen mix. While under anesthesia, the mice were injected bilaterally with 3 μl of vehicle (DMSO) or 1 mM human Aβ(1–42) into the caudate putamen of each hemisphere of the brain (0.5 mm anterior to the bregma, 2 mm lateral to the midline, and 3 mm below the surface of the skull). Ten minutes after the second intracerebral injection, the mice were euthanatized. In addition, to determine the effect, if any, of the vehicle or the intracranial injection itself on lipoprotein receptor levels, we examined a group of age-matched naïve mice (i.e., no intracerebral injection). Upon killing, all mouse brains were collected (minus the cerebellum) and the outer vessels and meninges were removed using a dry cotton swab (Coisne et al. 2005). All tissue samples were immediately snap frozen in liquid nitrogen and stored at −80 °C.
Isolation of Brain Fractions
The cerebrovasculature and parenchyma from mouse brain tissue were isolated using a modified protocol (Triguero et al. 1990). As above, fresh mouse brains were collected (minus the cerebellum) and the outer vessels and meninges were removed using a dry cotton swab (Coisne et al. 2005). The mouse brains were pooled and minced with a blade prior to being ground with 6–8 passes of a Teflon pestle in a glass Dounce homogenizer (Erickson et al. 2012). Brain material was homogenized in fivefold excess of ice-cold HBSS containing 10 mM HEPES (Coisne et al. 2005). A sample of the brain homogenate was collected as a representation of the whole brain (Mitchell et al. 2011). An equal volume of 40 % dextran solution was added to the brain homogenate for a final concentration of 20 % dextran (Erickson et al. 2012) and immediately centrifuged at 6,000g for 15 min at 4 °C (Fryer et al. 2003). This procedure results in a pellet at the bottom of the container (cerebrovasculature) and a compact mass at the top of the solution (parenchyma) separated by a clear dextran interface (soluble fraction). The cerebrovascular pellet was washed with ice-cold HBSS and resuspended in lysis buffer. The parenchyma was collected in HBSS, centrifuged at 6,000g for 10 min at 4 °C, and the resulting pellet resuspended in lysis buffer. Finally, the dextran supernatant was added to an equal volume of HBSS and centrifuged at 6,000g for 5 min at 4 °C to pellet any remaining cellular material. The supernatant was collected, and all samples were stored at −80 °C until analysis.
Lipoprotein Receptor Analysis
For the in vitro studies, extracellular media samples and cell lysates were analyzed by ELISA for human LRP1 and human LDLR as per the manufacturer’s instructions. Protein expression levels in the extracellular media were expressed as ng of LRP1 or LDLR per ml of media. For the in vivo samples, the cerebrovasculature, parenchyma, and soluble brain fraction were analyzed by ELISA for mouse LRP1 and mouse LDLR as per the manufacturer’s instructions and normalized to total protein content using the bicinchoninic acid (BCA) protein assay (Thermo Scientific, Waltham, MA, USA). Protein expression levels were expressed as ng of LRP1 or LDLR per mg protein for brain tissue and ng/ml for the soluble brain fraction.
Immunoblotting
The efficiency of the cerebrovascular isolation was assessed by light microscopy and immunoblotting using LRP-85 (marker for the membrane-bound subunit of LRP1), laminin (brain blood vessel marker), and synaptophysin (neuronal marker). Samples were examined for total protein content using the BCA protein assay. Brain supernatants were denatured by boiling in Laemmli Buffer (Bio-Rad, Hercules, CA, USA) and loaded (100 µg of total protein) onto a Criterion 4–20 % Tris–HCl gradient gel (Bio-Rad, Hercules, CA, USA). Migration transpired in 10× Tris/Glycine/SDS (Bio-Rad, Hercules, CA, USA) electrophoresis buffer diluted in deionized water using 50–130 V over a 2-h period. Following migration, electrotransfer of 10× Tris/Glycine (Bio-Rad, Hercules, CA, USA) electrophoresis buffer and 20 % HPLC grade methanol in deionized water to an Immun-Blot PVDF (polyvinylidene fluoride) membrane occurred overnight at 4 °C and 90 mA. The membrane was blocked in 5 % Blotting-Grade Blocker (nonfat dry milk) for 1 h (Bio-Rad, Hercules, CA, USA) and then immunoprobed with antibodies for LRP-85 (1:500), laminin (1:800), synaptophysin (1:2,000), and the housekeeping protein actin (1:1,000) in 5 % Blotting-Grade Blocker overnight. The membrane was washed with deionized water and exposed to HRP-linked secondary (1:1,000) antibody (Cell Signaling Technology, Inc., Danvers, MA, USA) for 1 h. Following a 30-min wash in deionized water, the membrane was revealed using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Waltham, MA, USA) and exposed with a Bio-Rad ChemiDoc XRS molecular imager (Bio-Rad, Hercules, CA, USA).
Statistics
Statistical analyses were performed using an ANOVA and Bonferroni post hoc test.
Results
Lipoprotein Receptor Shedding in Human Brain Endothelial Cells
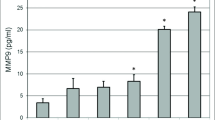
To determine the effect of Aβ exposure on lipoprotein receptor shedding in the BBB, human brain endothelial cells were treated with 2 µM Aβ(1–42) for 48 h and the extracellular media subsequently probed for soluble LRP1 and LDLR. A concentration-dependent increase in the appearance of LRP1 and LDLR in the media was observed upon exposure to Aβ(1–42). Moreover, Aβ concentrations ≥2 µM resulted in a statistically significant increase (approximately twofold at 2 µM) in lipoprotein receptor shedding compared to control conditions (Fig. 1). Additionally, cellular toxicity was monitored using a LDH detection assay, and there was no difference in LDH levels between control and Aβ-treated conditions (data not shown). The influence of apoE on lipoprotein receptor shedding was also examined in brain endothelial cells. For the most part, treatment with each apoE isoform alone demonstrated a modest increase in extracellular lipoprotein receptor levels compared to control conditions, though these values did not reach statistical significance and this effect was only around half of that produced with Aβ alone (Fig. 2). For LDLR specifically, the differences between apoE isoforms were obvious, with a rank order of apoE2 < apoE3 < apoE4 (Fig. 2b). Of these, only apoE4 treatment significantly altered LDLR levels (2.5-fold) from those observed under control conditions (Fig. 2b). For the combination studies, co-treatment of apoE2 or apoE3 with Aβ mitigated the effect of Aβ on lipoprotein receptor shedding, reducing the levels in the media (at least for LRP1) to the baseline observed with each isoform alone (Fig. 2a). In contrast, apoE4 treatment did not alter Aβ-induced lipoprotein receptor shedding as soluble LRP1 (Fig. 2a) and soluble LDLR (Fig. 2b) levels in the extracellular media were the same as those seen with Aβ insult alone. Importantly, for both lipoprotein receptors, the shedding levels in the media were significantly lower when apoE2 or apoE3 was administered with Aβ in comparison with apoE4 with Aβ (approximately a 1.6-fold difference). In addition, lipoprotein receptor expression was examined in the cell lysates, and no statistically significant difference was observed between any of the treatment groups (data not shown).

Appearance of extracellular soluble. a LRP1 or b LDLR in human brain endothelial cells (HBMECs) upon treatment with Aβ(1–42). HBMEC were exposed to various concentrations (0.1, 0.2, 1, 2, and 10 µM) of human Aβ(1–42) for 48 h at 37 °C. Following the treatment period, the extracellular media were collected and analyzed for LRP1 or LDLR content by ELISA. Values represent mean ± SEM (n = 3) and are expressed as ng of LRP1 or LDLR per ml of media. *P < 0.05 compared to control as determined by ANOVA and Bonferroni post hoc test

Appearance of extracellular soluble. a LRP1 or b LDLR in human brain endothelial cells (HBMEC) in the presence of Aβ(1–42), apoE isoforms, or combinations thereof. HBMEC were treated with human Aβ(1–42) (2 µM) and/or each apoE isoform (25 ng/ml) for 48 h at 37 °C. Following the treatment period, the extracellular media were collected and analyzed for LRP1 or LDLR content by ELISA. Values represent mean ± SEM (n = 3) and are expressed as ng of LRP1 or LDLR per ml of media. *P < 0.05 compared to control. *P < 0.05 comparing apoE4 in the presence of Aβ versus apoE2 or apoE3 in the presence of Aβ, as indicated on the graph. Statistics determined by ANOVA and Bonferroni post hoc test
Lipoprotein Receptor Levels In Vivo
As a complement to the in vitro studies, brain lipoprotein receptor levels were evaluated in various brain fractions of apoE transgenic animals following acute intracerebral Aβ insult. While subtle variations in soluble LRP1 and LDLR levels in the parenchyma (data not shown) and cerebrovasculature (Fig. 3) between apoE genotypes were apparent, there was no obvious trend and none of the values in these brain fractions were statistically different. While we did not observe demonstrable differences in lipoprotein receptor expression levels in the parenchyma and cerebrovasculature, the soluble brain fraction revealed both Aβ treatment- and apoE genotype-dependent differences in lipoprotein receptor levels. Under control conditions, the soluble brain levels of both LRP1 (Fig. 4a) and LDLR (Fig. 4b) varied across apoE genotype with a rank order of apoE2 < apoE3 < apoE4. Specifically, soluble lipoprotein receptor levels in apoE4 brains were significantly greater (twofold) than that observed in apoE2 or apoE3 animals. Moreover, in every genotype, soluble brain lipoprotein receptor levels increased upon Aβ insult (especially for LRP1) compared to vehicle, with a rank order of apoE2 < apoE3 < apoE4 < apoE KO = wild-type (Fig. 4). The most dramatic change was observed in the wild-type mice where soluble brain lipoprotein receptor levels were approximately sixfold higher in the Aβ-treated animals compared to vehicle (Fig. 4). Additionally, soluble brain lipoprotein receptor levels were examined in naïve mice (i.e., no intracerebral injection), and no significant differences were observed in these animals compared to the intracerebral vehicle-injected group (data not shown), indicating exposure to the vehicle or the intracranial injection itself does not appreciably impact the soluble levels of these proteins.

Expression of a LRP1 and b LDLR in cerebrovasculature isolated from apoE transgenic mice. Human Aβ(1–42) or vehicle was intracranially administered to male mice (4–6-month old). Ten minutes after the intracerebral injection, the brains were collected and various brain fractions were isolated. LRP1 or LDLR levels in the cerebrovasculature were determined using an ELISA and normalized to total protein content using the BCA protein assay. Values represent mean ± SEM (n = 6 animals) and are expressed as ng of LRP1 or LDLR per mg protein. No comparisons reached statistical significance as determined by ANOVA and Bonferroni post hoc test

Levels of a LRP1 and b LDLR in the soluble brain fraction of apoE transgenic mice. Human Aβ(1–42) or vehicle was intracranially administered to male mice (4–6-month old). Ten minutes after the intracerebral injection, the brains were collected and various brain fractions were isolated. LRP1 or LDLR levels in the soluble brain fraction were determined using an ELISA. Values represent mean ± SEM (n = 6 animals) and are expressed as ng of LRP1 or LDLR per ml of soluble brain material. *P < 0.05 comparing apoE4 to apoE2 or apoE3 for the respective control and Aβ(1–42) groups as determined by ANOVA and Bonferroni post hoc test
Brain Fraction Isolation
To assess the efficiency of our method to isolate various fractions of the brain, each of the resulting brain fractions was probed with specific protein markers (Fig. 5). Since lipoprotein receptors are the focus of these studies, the presence of the C-terminal subunit of LRP1 (LRP-85) was examined in each brain fraction. LRP-85 contains the transmembrane and cytoplasmic domains of LRP1 and is associated with the cellular membrane prior to and following ectodomain shedding of the soluble receptor. The membranous LRP-85 was present in each of the cellular brain fractions (homogenate, parenchyma, and vasculature), which is expected as LRP1 is expressed in neurons (Bu et al. 1994) and cerebrovascular cells such as pericytes (Wilhelmus et al. 2007) and brain endothelia (Shibata et al. 2000). Furthermore, LRP-85 was not detected in the soluble protein fraction, indicating the soluble layer is devoid of cell-associated material. A neuronal marker (synaptophysin) and a blood vessel marker (laminin) were also examined in each brain fraction. Synaptophysin was present in each of the cellular fractions and not in the soluble preparation. While there were detectable levels of synaptophysin in the vasculature, these levels were considerably lower than those found in the parenchymal fraction. Laminin, on the other hand, was only detected in the vascular preparation, indicating all of the blood vessel components of the brain were confined to this fraction during the isolation process. Lastly, we employed a prototypical housekeeping protein (actin) and found similar levels in each fraction with the exception of the soluble layer, which is expected as this fraction consists only of soluble (noncell-associated) protein and would thus contain lower actin levels than the cell-based preparations (i.e., homogenate, parenchyma, and vasculature).

Western blot analysis of brain fractions isolated from wild-type mice. Brain fractions from wild-type male mice (4–6-month old) were examined for the presence of LRP-85 (marker for the membrane-bound subunit of LRP1), laminin (brain blood vessel marker), synaptophysin (neuronal marker), and the housekeeping protein actin. Samples were collected from the brain fractions of three naïve (i.e., no intracerebral injection) wild-type mice and loaded into separate lanes of the gel
Discussion
Prior reporting has indicated Aβ clearance from the brain is differentially regulated by the type of apoE isoform expressed (Castellano et al. 2011). Multiple studies, including our own (Bachmeier et al. 2013), have demonstrated an isoform-specific disruption of Aβ transit across the BBB when Aβ is complexed with apoE (Bell et al. 2007; Deane et al. 2008; Martel et al. 1997). In addition, it has been proposed that soluble apoE (i.e., not bound to Aβ) can support Aβ clearance across the BBB in an isoform-dependent manner (Bachmeier et al. 2013). In line with this, recent findings suggest apoE3 may promote Aβ clearance across the blood–cerebrospinal fluid barrier in the choroid plexus as well (Ruzali et al. 2012). Despite the close association of apoE with lipoprotein receptors (Bu 2009; Zaiou et al. 2000), the manner in which apoE isoforms influence lipoprotein receptors and impact Aβ clearance from the brain is not entirely understood. To determine whether the effects of apoE on brain Aβ removal are due to lipoprotein receptor quantity, we examined the expression of these receptors in both the parenchyma and cerebrovasculature isolated from the brains of apoE transgenic animals. While several studies have investigated the correlation between apoE and LRP1 expression in the brain (Akram et al. 2012; Arelin et al. 2002; Qiu et al. 2001; Shinohara et al. 2013), few have done so in relation to apoE genotype or isolated brain vasculature. Of those that have LRP1 mRNA levels in whole brain homogenate were found to be different between genotypes in apoE transgenic animals (Kajiwara et al. 2010). At the protein level, in control and AD human brains, apoE genotype was not associated with significant variations in LRP1 in the frontal and occipital cortices or the meningeal blood vessels (Ruzali et al. 2012). Likewise, in our examination of lipoprotein receptor levels in the parenchyma and isolated brain vasculature of apoE transgenic mice, we did not observe substantial differences between apoE genotypes in LRP1 or LDLR protein expression. Thus, at least at the protein level in these apoE transgenic animals, it does not appear that alterations in lipoprotein expression are driving the isoform-specific effects of apoE on Aβ elimination from the brain.
Having observed a lack of variation in the lipoprotein expression among the apoE genotypes, we investigated the role of apoE in lipoprotein receptor processing, i.e., ectodomain shedding. When the soluble receptor is released from the cell following proteolysis, it is no longer involved in endocytic cellular transport (Rebeck et al. 2006; Selvais et al. 2010), which impairs Aβ clearance across the BBB. Lipoprotein receptor shedding has been shown to be induced by inflammation (Begg et al. 2004; Gorovoy et al. 2010), acute respiratory distress (Wygrecka et al. 2011), and exposure to Aβ(1–42) (Liu et al. 2009). In the current studies, treatment with Aβ(1–42) resulted in a dose-dependent increase of lipoprotein receptor shedding in brain endothelial cells. In these same cells, we also examined the effect of each apoE isoform on lipoprotein receptor shedding and observed a modest increase upon apoE treatment. However, only apoE4 treatment resulted in shedding levels that were significantly greater than control (at least for LDLR), and the extent of shedding in the presence of apoE, regardless of isoform, was lower than that observed for Aβ. Importantly, there were no differences in lipoprotein receptor levels in the cell lysates between treatment groups indicating lipoprotein receptor expression changes are not driving the observed effects of Aβ and apoE in vitro, which is consistent with the in vivo findings discussed above. Our observation of increased lipoprotein receptor shedding in the presence of apoE is similar to prior work in which apoE binding to apoE receptors caused an increase in the release of the extracellular domain (Hoe and Rebeck 2005). Also, proteolysis of apoE receptors has been shown to be promoted by other ligands such as α2-macroglobulin and reelin (Hoe and Rebeck 2005), indicating some degree of receptor shedding is common with many ligands upon lipoprotein receptor binding, though the degree to which this occurs may vary between ligands. In the current studies, when the apoE isoforms were co-treated with Aβ in brain endothelial cells, Aβ-induced shedding in the presence of apoE2 and apoE3 was significantly lower than with apoE4. While apoE appears to play a role in the lipoprotein receptor shedding process (one that is isoform-specific), it is unclear whether apoE is simply less able to induce receptor shedding than other ligands (e.g., Aβ), or if apoE (apoE2 and apoE3 in particular) is meant to provide some protection to the lipoprotein receptor under certain conditions. Of note, a general protective role for apoE has been reported using a variety of experimental paradigms, and like our observations, these studies found apoE4 to be less adept than apoE3 in exerting a protective function (Buttini et al. 1999; Hayashi et al. 2007; Sen et al. 2012).
To complement our in vitro findings, we investigated lipoprotein receptor shedding in vivo by examining the soluble brain fraction in apoE transgenic animals. As prior reporting (Liu et al. 2009) and our in vitro studies demonstrate lipoprotein receptor shedding is induced by Aβ exposure, these in vivo studies also included an Aβ paradigm by administering Aβ via stereotaxic intracranial injection. Examination of the soluble fraction of the brain revealed substantial differences in lipoprotein receptor levels across apoE genotypes after Aβ insult. For mice administered vehicle intracerebrally, we observed differences in lipoprotein receptor shedding between apoE isoforms, similar to that found in vitro for apoE treatment alone (i.e., no Aβ exposure). Thus, even in the absence of Aβ insult, the degree of receptor shedding at baseline appears higher for apoE4 versus apoE2 or apoE3. In comparing the wild-type mice and the apoE KO animals, murine apoE does not appear to have a role in LRP1 shedding at baseline or in response to Aβ, while at the same time suppressing basal LDLR shedding levels. Consistent with the above in vitro studies, intracranial exposure to Aβ exacerbated receptor shedding in all genotypes with a rank order of wt = apoE KO > apoE4 > apoE3 > apoE2. Not only were the baseline shedding levels for apoE2 and apoE3 significantly lower than apoE4, but these two isoforms appear to offer more protection against Aβ insult as the degree of receptor shedding following intracerebral Aβ exposure was not nearly as extensive as that observed in the other genotypes (apoE4, apoE KO, and wt), especially for LDLR.
As mentioned above, the greatest degree of shedding was observed in the wild-type and apoE knockout animals, suggesting the presence of apoE (in particular human apoE) provides some protection to lipoprotein receptors from Aβ-induced shedding. This apparent protection provides rationale for reports that an absence of apoE altogether leads to reduced Aβ brain clearance (Shibata et al. 2000) and increased Aβ levels in the brain (DeMattos et al. 2004; Dodart et al. 2002). In the in vitro studies, apoE4 did not attenuate Aβ-induced lipoprotein receptor shedding as it did in the in vivo studies. This may be due to differences in cell type. The in vitro studies investigated lipoprotein receptor shedding exclusively in brain endothelial cells, while the in vivo studies examined lipoprotein receptor shedding in the soluble fraction of the entire brain. Thus, any lipoprotein receptor-expressing cell in the brain could have contributed to the soluble receptor levels we observed. This would include cells such as neurons (Kanekiyo et al. 2013), pericytes (Wilhelmus et al. 2007), smooth muscle cells (Kanekiyo et al. 2012), astrocytes (Basak et al. 2012; Koistinaho et al. 2004), and microglia (Lee and Landreth 2010) involved in metabolic Aβ clearance. The dynamics of apoE4 in these cells may differ from brain endothelial cells, which might explain the discrepancy between the in vitro and in vivo studies. Moreover, it is important to note that altered lipoprotein receptor shedding by apoE in any of these cell types would likely contribute to Aβ accumulation in the AD brain. Overall, it is clear from both the in vitro and in vivo studies that an apoE isoform-specific effect exists such that lipoprotein receptor shedding is more prevalent in the presence of apoE4 than with apoE2 or apoE3.
In correlating lipoprotein receptor shedding (in the current studies) with BBB-mediated Aβ clearance (from our prior work) (Bachmeier et al. 2013), there is a strong inverse relationship (P < 0.05, Pearson’s correlation) for both lipoprotein receptors (LRP1 and LDLR) that is apoE genotype-specific (Fig. 6). Based on these data, Aβ clearance across the BBB appears to be at least partially mediated by lipoprotein receptor shedding, a process that is differentially regulated by the apoE isoforms. Receptor shedding not only depletes the population of endocytic transporters available for BBB clearance, but increases the concentration of soluble receptors in the extracellular space, which can bind Aβ (among other ligands) and extend its half-life in the brain. At this stage, it is uncertain how apoE can alter Aβ-induced shedding. One explanation is that apoE directly binds to Aβ, preventing Aβ from accessing the lipoprotein receptors. However, a recent study determined apoE does not readily bind soluble Aβ in physiological fluids, but instead acts through a shared receptor (e.g., LRP1) to influence Aβ brain removal (Verghese et al. 2013), a revelation that coincides with the findings of the current study and our prior work (Bachmeier et al. 2013). Alternatively, upon interacting with the lipoprotein receptor, apoE may promote Aβ endocytosis through cooperative binding or by inducing a conformational change in the receptor. Further exploration is necessary to understand the nature and consequence of these interactions.

Correlation between brain lipoprotein receptor shedding and Aβ clearance across the BBB in apoE transgenic mice. LRP1 and LDLR levels in the soluble fraction of the brain were plotted versus the appearance of Aβ in the plasma (i.e., Aβ BBB clearance) following intracerebral Aβ administration (LRP1, R 2 = 0.94; LDLR, R 2 = 0.96). The Aβ BBB clearance data were derived from our previously published work (Bachmeier et al. 2013). Values represent the mean of six animals for each genotype. P < 0.05 for both LRP1 and LDLR as determined by Pearson’s correlation
Conclusion
Our previous findings and the work of others indicate an isoform-specific role for apoE in the elimination of Aβ from the brain (Bachmeier et al. 2013; Castellano et al. 2011). The present studies indicate that apoE influences lipoprotein receptor shedding, a process that may explain the impact of apoE on Aβ brain BBB clearance, as increased shedding is associated with a loss of endocytic transport function (Rebeck et al. 2006; Selvais et al. 2010). Furthermore, the observed effect on shedding is apoE isoform-specific as both our in vitro and in vivo studies showed increased lipoprotein receptor shedding in the presence of apoE4, compared with apoE2 or apoE3, under basal conditions and following Aβ insult. Thus, apoE4 appears less efficient than other apoE isoforms in regulating lipoprotein receptor shedding, which culminates in reduced Aβ elimination from the brain. These studies further our understanding of the relationship between apoE and lipoprotein receptors and provide rationale for the increased Aβ brain burden in apoE4 transgenic animals (Bales et al. 2009; Holtzman et al. 2000) and AD patients carrying the apoE4 allele (Bogdanovic et al. 2002; Schmechel et al. 1993). Moving forward, as our group (Kennelly et al. 2012) and others (Risner et al. 2006; Salloway et al. 2009) have observed that apoE4 carriers are often less responsive to therapeutic intervention than apoE4 noncarriers, new AD treatment modalities targeting this process could be particularly beneficial to individuals with this genotype.
References
Akram, A., Schmeidler, J., Katsel, P., Hof, P. R., & Haroutunian, V. (2012). Association of ApoE and LRP mRNA levels with dementia and AD neuropathology. Neurobiology of Aging, 33(3), e1–e14.
Arelin, K., Kinoshita, A., Whelan, C. M., Irizarry, M. C., Rebeck, G. W., Strickland, D. K., et al. (2002). LRP and senile plaques in Alzheimer’s disease: Colocalization with apolipoprotein E and with activated astrocytes. Molecular Brain Research, 104(1), 38–46.
Armstrong, R. A. (2009). The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathologica, 47(4), 289–299.
Bachmeier, C., Mullan, M., & Paris, D. (2010). Characterization and use of human brain microvascular endothelial cells to examine beta-amyloid exchange in the blood–brain barrier. Cytotechnology, 62(6), 519–529.
Bachmeier, C., Paris, D., Beaulieu-Abdelahad, D., Mouzon, B., Mullan, M., & Crawford, F. (2013). A multifaceted role for apoE in the clearance of beta-amyloid across the blood–brain barrier. Neurodegenerative Diseases, 11(1), 13–21.
Bales, K. R., Liu, F., Wu, S., Lin, S., Koger, D., DeLong, C., et al. (2009). Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. Journal of Neuroscience, 29(21), 6771–6779.
Basak, J. M., Verghese, P. B., Yoon, H., Kim, J., & Holtzman, D. M. (2012). Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Abeta uptake and degradation by astrocytes. Journal of Biological Chemistry, 287(17), 13959–13971.
Begg, M. J., Sturrock, E. D., & van der Westhuyzen, D. R. (2004). Soluble LDL-R are formed by cell surface cleavage in response to phorbol esters. European Journal of Biochemistry, 271(3), 524–533.
Bekris, L. M., Millard, S. P., Galloway, N. M., Vuletic, S., Albers, J. J., Li, G., et al. (2008). Multiple SNPs within and surrounding the apolipoprotein E gene influence cerebrospinal fluid apolipoprotein E protein levels. Journal of Alzheimer’s Disease, 13(3), 255–266.
Bell, R. D., Sagare, A. P., Friedman, A. E., Bedi, G. S., Holtzman, D. M., Deane, R., et al. (2007). Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. Journal of Cerebral Blood Flow and Metabolism, 27(5), 909–918.
Boche, D., Nicoll, J. A., & Weller, R. O. (2005). Immunotherapy for Alzheimer’s disease and other dementias. Current Opinion in Neurology, 18(6), 720–725.
Bogdanovic, N., Corder, E., Lannfelt, L., & Winblad, B. (2002). APOE polymorphism and clinical duration determine regional neuropathology in Swedish APP(670, 671) mutation carriers: Implications for late-onset Alzheimer’s disease. Journal of Cellular and Molecular Medicine, 6(2), 199–214.
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nature Reviews Neuroscience, 10(5), 333–344.
Bu, G., Maksymovitch, E. A., Nerbonne, J. M., & Schwartz, A. L. (1994). Expression and function of the low density lipoprotein receptor-related protein (LRP) in mammalian central neurons. Journal of Biological Chemistry, 269(28), 18521–18528.
Buttini, M., Orth, M., Bellosta, S., Akeefe, H., Pitas, R. E., Wyss-Coray, T., et al. (1999). Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: Isoform-specific effects on neurodegeneration. Journal of Neuroscience, 19(12), 4867–4880.
Castellano, J. M., Deane, R., Gottesdiener, A. J., Verghese, P. B., Stewart, F. R., West, T., et al. (2012). Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Abeta clearance in a mouse model of beta-amyloidosis. Proc Natl Acad Sci USA, 109(38), 15502–15507.
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., DeMattos, R. B., Patterson, B. W., et al. (2011). Human apoE isoforms differentially regulate brain amyloid–beta peptide clearance. Science Translational Medicine, 3(89), 57–67.
Citron, M. (2010). Alzheimer’s disease: Strategies for disease modification. Nature Reviews Drug Discovery, 9(5), 387–398.
Coisne, C., Dehouck, L., Faveeuw, C., Delplace, Y., Miller, F., Landry, C., et al. (2005). Mouse syngenic in vitro blood–brain barrier model: A new tool to examine inflammatory events in cerebral endothelium. Laboratory Investigation, 85(6), 734–746.
Deane, R., Bell, R. D., Sagare, A., & Zlokovic, B. V. (2009). Clearance of amyloid–beta peptide across the blood–brain barrier: Implication for therapies in Alzheimer’s disease. CNS and Neurological Disorders: Drug Targets, 8(1), 16–30.
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., et al. (2008). apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. The Journal of Clinical Investigation, 118(12), 4002–4013.
DeMattos, R. B., Cirrito, J. R., Parsadanian, M., May, P. C., O’Dell, M. A., Taylor, J. W., et al. (2004). ApoE and clusterin cooperatively suppress Abeta levels and deposition: Evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron, 41(2), 193–202.
Dodart, J. C., Bales, K. R., Johnstone, E. M., Little, S. P., & Paul, S. M. (2002). Apolipoprotein E alters the processing of the beta-amyloid precursor protein in APP(V717F) transgenic mice. Brain Research, 955(1–2), 191–199.
Erickson, M. A., Hartvigson, P. E., Morofuji, Y., Owen, J. B., Butterfield, D. A., & Banks, W. A. (2012). Lipopolysaccharide impairs amyloid beta efflux from brain: Altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood–brain barrier. Journal of Neuroinflammation, 9, 150–164.
Etique, N., Verzeaux, L., Dedieu, S., & Emonard, H. (2013). LRP-1: A checkpoint for the extracellular matrix proteolysis. BioMed Research International, 2013, 1–7.
Fan, J., Stukas, S., Wong, C., Chan, J., May, S., DeValle, N., et al. (2011). An ABCA1-independent pathway for recycling a poorly lipidated 8.1 nm apolipoprotein E particle from glia. Journal of Lipid Research, 52(9), 1605–1616.
Fryer, J. D., Taylor, J. W., DeMattos, R. B., Bales, K. R., Paul, S. M., Parsadanian, M., et al. (2003). Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. Journal of Neuroscience, 23(21), 7889–7896.
Gilbert, B. J. (2013). The role of amyloid beta in the pathogenesis of Alzheimer’s disease. Journal of Clinical Pathology, 66(5), 362–366.
Gorovoy, M., Gaultier, A., Campana, W. M., Firestein, G. S., & Gonias, S. L. (2010). Inflammatory mediators promote production of shed LRP1/CD91, which regulates cell signaling and cytokine expression by macrophages. Journal of Leukocyte Biology, 88(4), 769–778.
Grimsley, P. G., Quinn, K. A., & Owensby, D. A. (1998). Soluble low-density lipoprotein receptor-related protein. Trends in Cardiovascular Medicine, 8(8), 363–368.
Hayashi, H., Campenot, R. B., Vance, D. E., & Vance, J. E. (2007). Apolipoprotein E-containing lipoproteins protect neurons from apoptosis via a signaling pathway involving low-density lipoprotein receptor-related protein-1. Journal of Neuroscience, 27(8), 1933–1941.
Hoe, H. S., & Rebeck, G. W. (2005). Regulation of ApoE receptor proteolysis by ligand binding. Molecular Brain Research, 137(1–2), 31–39.
Holtzman, D. M., Bales, K. R., Tenkova, T., Fagan, A. M., Parsadanian, M., Sartorius, L. J., et al. (2000). Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA, 97(6), 2892–2897.
Kajiwara, Y., Franciosi, S., Takahashi, N., Krug, L., Schmeidler, J., Taddei, K., et al. (2010). Extensive proteomic screening identifies the obesity-related NYGGF4 protein as a novel LRP1-interactor, showing reduced expression in early Alzheimer’s disease. Molecular Neurodegeneration, 5(1), 1–11.
Kanekiyo, T., Cirrito, J. R., Liu, C. C., Shinohara, M., Li, J., Schuler, D. R., et al. (2013). Neuronal clearance of amyloid-beta by endocytic receptor LRP1. Journal of Neuroscience, 33(49), 19276–19283.
Kanekiyo, T., Liu, C. C., Shinohara, M., Li, J., & Bu, G. (2012). LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-beta. Journal of Neuroscience, 32(46), 16458–16465.
Kennelly, S., Abdullah, L., Kenny, R. A., Mathura, V., Luis, C. A., Mouzon, B., et al. (2012). Apolipoprotein E genotype-specific short-term cognitive benefits of treatment with the antihypertensive nilvadipine in Alzheimer’s patients—An open-label trial. International Journal of Geriatric Psychiatry, 27(4), 415–422.
Kim, J., Basak, J. M., & Holtzman, D. M. (2009). The role of apolipoprotein E in Alzheimer’s disease. Neuron, 63(3), 287–303.
Koistinaho, M., Lin, S., Wu, X., Esterman, M., Koger, D., Hanson, J., et al. (2004). Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nature Medicine, 10(7), 719–726.
Lee, C. Y., & Landreth, G. E. (2010). The role of microglia in amyloid clearance from the AD brain. Journal of Neural Transmission, 117(8), 949–960.
Liu, Q., Zhang, J., Tran, H., Verbeek, M. M., Reiss, K., Estus, S., et al. (2009). LRP1 shedding in human brain: Roles of ADAM10 and ADAM17. Molecular Neurodegeneration, 4, 17–23.
Manelli, A. M., Bulfinch, L. C., Sullivan, P. M., & LaDu, M. J. (2007). Abeta42 neurotoxicity in primary co-cultures: Effect of apoE isoform and Abeta conformation. Neurobiology of Aging, 28(8), 1139–1147.
Martel, C. L., Mackic, J. B., Matsubara, E., Governale, S., Miguel, C., Miao, W., et al. (1997). Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood–brain barrier transport of circulating Alzheimer’s amyloid beta. Journal of Neurochemistry, 69(5), 1995–2004.
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science, 330(6012), 1774.
Mitchell, R. W., On, N. H., Del Bigio, M. R., Miller, D. W., & Hatch, G. M. (2011). Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. Journal of Neurochemistry, 117(4), 735–746.
Paris, D., Bachmeier, C., Patel, N., Quadros, A., Volmar, C. H., Laporte, V., et al. (2011). Selective antihypertensive dihydropyridines lower Abeta accumulation by targeting both the production and the clearance of Abeta across the blood–brain barrier. Molecular Medicine, 17(3–4), 149–162.
Piedrahita, J. A., Zhang, S. H., Hagaman, J. R., Oliver, P. M., & Maeda, N. (1992). Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA, 89(10), 4471–4475.
Qiu, Z., Strickland, D. K., Hyman, B. T., & Rebeck, G. W. (2001). Elevation of LDL receptor-related protein levels via ligand interactions in Alzheimer disease and in vitro. Journal of Neuropathology and Experimental Neurology, 60(5), 430–440.
Quinn, K. A., Grimsley, P. G., Dai, Y. P., Tapner, M., Chesterman, C. N., & Owensby, D. A. (1997). Soluble low density lipoprotein receptor-related protein (LRP) circulates in human plasma. Journal of Biological Chemistry, 272(38), 23946–23951.
Rebeck, G. W., LaDu, M. J., Estus, S., Bu, G., & Weeber, E. J. (2006). The generation and function of soluble apoE receptors in the CNS. Molecular Neurodegeneration, 1, 15–27.
Reitz, C. (2012). Alzheimer’s disease and the amyloid cascade hypothesis: A critical review. International Journal of Alzheimers Disease, 2012, 1–11.
Risner, M. E., Saunders, A. M., Altman, J. F., Ormandy, G. C., Craft, S., Foley, I. M., et al. (2006). Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics Journal, 6(4), 246–254.
Ruzali, W. A., Kehoe, P. G., & Love, S. (2012). LRP1 expression in cerebral cortex, choroid plexus and meningeal blood vessels: Relationship to cerebral amyloid angiopathy and APOE status. Neuroscience Letters, 525(2), 123–128.
Salloway, S., Sperling, R., Gilman, S., Fox, N. C., Blennow, K., Raskind, M., et al. (2009). A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology, 73(24), 2061–2070.
Schmechel, D. E., Saunders, A. M., Strittmatter, W. J., Crain, B. J., Hulette, C. M., Joo, S. H., et al. (1993). Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA, 90(20), 9649–9653.
Selvais, C., Dedieu, S., Hornebeck, W., & Emonard, H. (2010). Post-translational proteolytic events influence LRP-1 functions. BioMedical Materials and Engineering, 20(3), 203–207.
Sen, A., Alkon, D. L., & Nelson, T. J. (2012). Apolipoprotein E3 (ApoE3) but not ApoE4 protects against synaptic loss through increased expression of protein kinase C epsilon. Journal of Biological Chemistry, 287(19), 15947–15958.
Serrano-Pozo, A., Frosch, M. P., Masliah, E., & Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harbor Perspectives in Medicine, 1(1), 1–23.
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood–brain barrier. The Journal of Clinical Investigation, 106(12), 1489–1499.
Shinohara, M., Petersen, R. C., Dickson, D. W., & Bu, G. (2013). Brain regional correlation of amyloid-beta with synapses and apolipoprotein E in non-demented individuals: Potential mechanisms underlying regional vulnerability to amyloid-beta accumulation. Acta Neuropathologica, 125(4), 535–547.
Sullivan, P. M., Mezdour, H., Aratani, Y., Knouff, C., Najib, J., Reddick, R. L., et al. (1997). Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. Journal of Biological Chemistry, 272(29), 17972–17980.
Triguero, D., Buciak, J., & Pardridge, W. M. (1990). Capillary depletion method for quantification of blood–brain barrier transport of circulating peptides and plasma proteins. Journal of Neurochemistry, 54(6), 1882–1888.
Ulrich, J. D., Burchett, J. M., Restivo, J. L., Schuler, D. R., Verghese, P. B., Mahan, T. E., et al. (2013). In vivo measurement of apolipoprotein E from the brain interstitial fluid using microdialysis. Molecular Neurodegeneration, 8(13), 1–7.
Verghese, P. B., Castellano, J. M., Garai, K., Wang, Y., Jiang, H., Shah, A., et al. (2013). ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci USA, 110(19), E1807–E1816.
Wahrle, S. E., Shah, A. R., Fagan, A. M., Smemo, S., Kauwe, J. S., Grupe, A., et al. (2007). Apolipoprotein E levels in cerebrospinal fluid and the effects of ABCA1 polymorphisms. Molecular Neurodegeneration, 2, 1–9.
Wilhelmus, M. M., Otte-Holler, I., van Triel, J. J., Veerhuis, R., Maat-Schieman, M. L., Bu, G., et al. (2007). Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. American Journal of Pathology, 171(6), 1989–1999.
Wygrecka, M., Wilhelm, J., Jablonska, E., Zakrzewicz, D., Preissner, K. T., Seeger, W., et al. (2011). Shedding of low-density lipoprotein receptor-related protein-1 in acute respiratory distress syndrome. American Journal of Respiratory and Critical Care Medicine, 184(4), 438–448.
Yamauchi, K., Tozuka, M., Nakabayashi, T., Sugano, M., Hidaka, H., Kondo, Y., et al. (1999). Apolipoprotein E in cerebrospinal fluid: Relation to phenotype and plasma apolipoprotein E concentrations. Clinical Chemistry, 45(4), 497–504.
Zaiou, M., Arnold, K. S., Newhouse, Y. M., Innerarity, T. L., Weisgraber, K. H., Segall, M. L., et al. (2000). Apolipoprotein E–low density lipoprotein receptor interaction. Influences of basic residue and amphipathic α–helix organization in the ligand. Journal of Lipid Research, 41(7), 1087–1095.
Zhong, N., & Weisgraber, K. H. (2009). Understanding the basis for the association of apoE4 with Alzheimer’s disease: Opening the door for therapeutic approaches. Current Alzheimer Research, 6(5), 415–418.
Acknowledgments
We would like to thank the laboratory of Dr. Mary Jo LaDu (University of Illinois at Chicago) including Dr. Leon Tai and Susan Wohlgenant for isolating and preparing the mixed glial cultures. Research was supported by the National Institute on Aging of the National Institutes of Health under award number R01AG041971. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Ethical standards
All experimental protocols involving animals were approved by the Institutional Animal Care and Use Committee of the Roskamp Institute, Inc. The manuscript does not contain clinical studies or patient data.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bachmeier, C., Shackleton, B., Ojo, J. et al. Apolipoprotein E Isoform-Specific Effects on Lipoprotein Receptor Processing. Neuromol Med 16, 686–696 (2014). https://doi.org/10.1007/s12017-014-8318-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-014-8318-6