
Abstract
Rett syndrome (RTT) is a severe and progressive neurological disorder, which mainly affects young females. Mutations of the methyl-CpG binding protein 2 (MECP2) gene are the most prevalent cause of classical RTT cases. MECP2 mutations or altered expression are also associated with a spectrum of neurodevelopmental disorders such as autism spectrum disorders with recent links to fetal alcohol spectrum disorders. Collectively, MeCP2 relation to these neurodevelopmental disorders highlights the importance of understanding the molecular mechanisms by which MeCP2 impacts brain development, mental conditions, and compromised brain function. Since MECP2 mutations were discovered to be the primary cause of RTT, a significant progress has been made in the MeCP2 research, with respect to the expression, function and regulation of MeCP2 in the brain and its contribution in RTT pathogenesis. To date, there have been intensive efforts in designing effective therapeutic strategies for RTT benefiting from mouse models and cells collected from RTT patients. Despite significant progress in MeCP2 research over the last few decades, there is still a knowledge gap between the in vitro and in vivo research findings and translating these findings into effective therapeutic interventions in human RTT patients. In this review, we will provide a synopsis of Rett syndrome as a severe neurological disorder and will discuss the role of MeCP2 in RTT pathophysiology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rett syndrome (RTT) is a pervasive neurological disorder (Rett 1966; Hagberg et al. 1983), and it is characterized by compromised brain functions, severe mental retardation, language and learning disabilities, repetitive stereotyped hand movements and developmental regression. RTT is predominantly found in young females, with an incidence of 1:10,000–20,000 live births, with rare cases reported in males (Moog et al. 2003). RTT is considered to be a monogenic neurological disorder (Renieri et al. 2003; Amir et al. 1999), because approximately 90 % cases of classical RTT patients harbor loss-of-function mutations of the X-linked methyl-CpG binding protein 2 (MECP2) gene (Bienvenu et al. 2000). Rett syndrome is perhaps the best example for contribution of epigenetic mechanisms in disease pathology, mostly due to the involvement of MeCP2, a key epigenetic modulator in the brain. Epigenetic mechanisms include DNA methylation, histone posttranslational modifications (PTMs) and noncoding RNAs, which regulate gene expression without altering the corresponding DNA sequences (Delcuve et al. 2009). Epigenetic mechanisms are involved in controlling embryonic development, stem cell differentiation and have a high impact in human disease (Olynik and Rastegar 2012; Barber and Rastegar 2010). As a well-studied epigenetic factor, MeCP2 controls gene expression and modulates chromatin architecture through binding to methylated DNA (Zachariah and Rastegar 2012).
Considerable progress has been made during the last few decades to understand the role of MECP2 mutations in RTT pathogenesis, the functional role of MeCP2 PTMs, MECP2/Mecp2 isoforms, MeCP2 regulation and effective therapeutic strategies for RTT. The focus on this literature review is to discuss the advances in MeCP2 research work with regard to Rett syndrome.
Rett Syndrome
Rett Syndrome in Females and Males
Due to the X-linked nature of MECP2 gene, RTT is mainly found in females, and it is the leading cause of mental retardation in females (Jedele 2007; Shahbazian and Zoghbi 2002). RTT also affects males in rare cases, and the male RTT patients manifest a range of symptoms including severe encephalopathy, mild mental retardation and dystonia apraxia (Jan et al. 1999).
Causes of Rett Syndrome
The primary cause of Rett syndrome in approximately 90 % of patients is the mutations in the MECP2 gene (Amir et al. 1999). Studies have shown a genotype–phenotype relationship between RTT phenotypes and MECP2 mutations (Amir and Zoghbi 2000). For example, truncation mutations within the MECP2 gene show correlation with more severe RTT phenotypes (Weaving et al. 2003). Other than mutations in the MECP2 gene, mutations in CDKL5 and FOXG1 genes are also associated with approximately 10 % of RTT cases (Evans et al. 2005a; Philippe et al. 2010).
Moreover, since MECP2 is an X-linked gene, X-chromosome inactivation (XCI) has also been shown to impact the RTT pathology and clinical severity. The XCI causes uneven expression of wild type and mutant MECP2 alleles within the brain. Even though, the results observed by different groups seem to be complex, the fact that X-chromosome inactivation patterns (either skewed or random, whether the mutant allele is of paternal or maternal origin) are linked to the severity of RTT phenotypes has been established by these groups (Ishii et al. 2001; Gibson et al. 2005; Xinhua et al. 2008). Since the relationship between clinical severity of RTT and XCI is inconclusive, it has been postulated that other factors such as environmental modifiers including epigenetic mechanisms and other MeCP2-related genes might be contributing to RTT clinical severity (Hoffbuhr et al. 2002).
Multiple Stages of Rett Syndrome Progression
The standard criteria of RTT diagnosis describe four major stages of RTT pathogenesis, subsequent to a normal prenatal and perinatal development up to about 5 months of age (Hagberg 2002). Stage I of RTT is defined by an early onset of developmental stagnation, which starts approximately from 6 months to 1.5 years of age. During this period, major RTT phenotypes such as microcephaly (reduced brain/head size), reduced growth rate, loss of language and behavioral skills and seizures start to appear (Chahrour and Zoghbi 2007). Stage II is mainly defined by developmental regression with an onset of 1–4 years of age. Affected patients start losing the already acquired skills in communication and behavior and show symptoms of mental retardation (Lee et al. 2013). Stage III is usually referred to as pseudo-stationary period, which follows the stage II and approximately spans around 4–7 years. This period is also referred as wake up period because some patients are able to regain certain skills such as communication abilities. However, these patients would still suffer from respiratory problems, disturbed sleeping patterns, scoliosis (abnormal curvature of the spine), anxiety and hand apraxia/dyspraxia. Late motor deterioration is the Stage IV, during which patients lose their ability to walk and would become non-ambulatory or completely dependent on a wheelchair for mobility. In more severe cases of RTT, patients may develop Parkinsonian phenotypes at later stages of this period (Roze et al. 2007). The last stage of RTT may last from several years to decades.
Major Phenotypes of Rett Syndrome
As a neurological disorder, the majority of RTT phenotypes are associated with nervous system and brain. A major phenotype observed in brain is the altered size/volume of whole brain, usually reduced brain size, which is also referred to as microcephaly (Reiss et al. 1993; Subramaniam et al. 1997). Studies also show that the volume of specific brain regions, namely cerebellum and cortex is reduced in RTT patients (Reiss et al. 1993; Murakami et al. 1992). Rett syndrome patients generally share many phenotypes with autistic patients, such as impaired social interactions or social withdrawal. For the same reason, several RTT patients are frequently misdiagnosed as autistic patients (Young et al. 2008; Olsson and Rett 1987; Gillberg 1986). Another major feature of this devastating disorder is the repetitive stereotyped hand movements, such as clapping, wringing, squeezing, tapping and loss of purposeful hand movements (Carter et al. 2010). Impaired learning, language and communication skills are also frequently seen in RTT patients. These patients are typically unable to communicate properly and have disabilities in speaking. Some patients also experience sudden loss of speech. However, during stage III, they might regain some of the lost abilities to communicate (Woodyatt and Ozanne 1993).
Apart from major phenotypes associated with nervous system, both human RTT patients and RTT mouse models show phenotypes in other organs. One such phenotype is the breathing abnormalities (Stettner et al. 2008; Katz et al. 2009; Cirignotta et al. 1986). The underlying causes of these breathing abnormalities are linked to MeCP2 dysfunction in specific brain regions such as ventro-lateral medulla and pons (Ramirez et al. 2013). The reduced life span and higher rates of sudden deaths of RTT patients are associated with cardiac dysrhythmias (Byard 2006). Several impairments in heart function are also reported in RTT cases (Guideri and Acampa 2005; Guideri et al. 2004; Madan et al. 2004) and RTT mouse models (McCauley et al. 2011). The neurological contribution of the cardiac problems is demonstrated by the immature cardiorespiratory neurons (Julu et al. 1997) and impairments in cardiac autonomic nervous system (Acampa and Guideri 2006). Rett syndrome patients also suffer from several bone problems including, osteopenia and difficulties in limb movements, fragile bones, loss of muscle tone (hypotonia) and scoliosis (Budden and Gunness 2003; Heilstedt et al. 2002; Holm and King 1990). Finally, children suffering from Rett syndrome mostly have retarded growth due to the difficulties in feeding and malnutrition (Morton et al. 1997; Motil et al. 2012).
Neurodevelopmental Versus Neurological Disorders
Rett syndrome was primarily discovered as a progressive neurodevelopmental disorder (Rett 1966; Hagberg et al. 1983; Neul and Zoghbi 2004). The high prevalence of RTT in small children and the associated developmental regression support the fact that RTT is a neurodevelopmental disorder. However, recent studies in adult RTT mouse models suggest that it is rather a neurological, not a neurodevelopmental, disorder (McGraw et al. 2011; Guy et al. 2007). This concept was based on the neurological phenotypes caused by the loss of Mecp2 in adult mice.
MeCP2 Structure, Expression and Functions
MeCP2 Structure
Perhaps MeCP2 is the mostly studied protein of the Methyl-CpG Binding Protein family that binds to methylated DNA through their unique Methyl Binding Domain (MBD) (Hung and Shen 2003; Singh et al. 2008). MECP2/Mecp2 gene is an X-linked gene that spans ~76 kb in the long arm of the X-chromosome (Xq28). The MECP2/Mecp2 gene is located in between the Interleukin-1 receptor associated kinase gene (IRAK1) and the Red Opsin gene (RCP) (Fig. 1a). In both human and mouse, the MECP2/Mecp2 gene comprises four major exons (exon 1–4) and three introns (intron 1–3) (Fig. 1b-c). MeCP2 protein structure is composed of five major domains, N-terminal Domain (NTD), Methyl Binding Domain, Inter-Domain (ID), Transcription Repression Domain (TRD) and C-terminal Domain (CTD) and is approximately 53 kDa in size (Fig. 1d). Even though the theoretical molecular weight of MeCP2 protein is 53 kDa, it is usually detected at 75 kDa by Western blot analysis (Zachariah et al. 2012; Olson et al. 2014), probably due to its posttranslational modifications. The known tertiary structure of MeCP2 is composed of 4 % α-helices, 21 % β-sheets and 13 % β-turns. However, the majority (~60 %) of MeCP2 protein is unstructured. Therefore, MeCP2 is considered to be an intrinsically disordered protein (Adams et al. 2007). Among the aforementioned MeCP2 protein domains, MBD is the only domain that has a definite secondary structure, while the rest of MeCP2 protein domains are highly unstructured (Ghosh et al. 2010b; Hite et al. 2012).

The structure of the MECP2/Mecp2 gene and MeCP2 protein. a The MECP2/Mecp2 gene is located in X-chromosome (Xq28), flanked by the RCP and IRAK genes. b The schematic of the MECP2/Mecp2 gene is shown that is composed of four exons (exon 1–4) and three introns (intron 1–3). The gene has three polyadenylation sites at the 3′UTR. The sizes of each exonic and intronic region in human MECP2 and mouse Mecp2 genes are indicated. c The generation of two MeCP2 isoforms: MeCP2E1 and MeCP2E2. The translation start site (ATG) for each isoform is indicated by arrows. MeCP2E1 isoform is encoded by exons 1, 3 and 4. MeCP2E2 isoform is encoded by exons 2, 3 and 4. Red coding sequence. Green noncoding sequence. d Protein structure of human MeCP2E1 and MeCP2E2. The human MeCP2E1 and MeCP2E2 are 498 and 486 amino acids in size, respectively. In contrast, the mouse MeCP2E1 and MeCP2E2 are 501 and 484 amino acids in size, respectively. MBD methyl-CpG-binding domain, ID inter-domain, TRD transcriptional repression domain, CTD C-terminal domain, NTD N-terminal domain, AT-hook a domain found within the TRD which allows binding to adenine–thymine (AT) rich DNA
MeCP2 Expression
MeCP2 is widely expressed in many organs, and its highest expression is detected in brain, lung and spleen, compared with which the expression levels are lower in liver, heart, kidney and small intestines (Shahbazian et al. 2002b). However, the brain-specific expression of MeCP2 is extensively studied, as the majority of RTT phenotypes are neurological. Additionally, abolishing Mecp2 expression in the embryonic brain results in similar RTT phenotypes caused by Mecp2 null mutations affecting all murine tissues (Chen et al. 2001; Guy et al. 2001). Nonetheless, non-neuronal RTT symptoms such as scoliosis, breathing/respiratory abnormalities, cardiac problems, difficulty in feeding and limb movements indicate the importance of MeCP2 expression outside the central nervous system (Guideri and Acampa 2005; Ogier and Katz 2008; Nomura and Segawa 1992; Isaacs et al. 2003; Ezeonwuka and Rastegar 2014).
Within the brain, both the distribution and levels of MeCP2 have been shown to be different. For instance, we have recently demonstrated the distribution profile of MeCP2 in the adult murine brain regions, specifically, in the olfactory bulb, cortex, striatum, hippocampus, thalamus, cerebellum and brain stem (Zachariah et al. 2012; Olson et al. 2014). Analysis of whole cell extracts isolated from these brain regions indicated the highest MeCP2 expression in the cortex and cerebellum among the studied brain regions (Zachariah et al. 2012). In contrast, analysis of nuclear extracts from the same brain regions indicated relatively even levels of MeCP2E1 and differential levels of MeCP2E2 (Olson et al. 2014). Implicating the importance of brain region-specific MeCP2 expression in Rett syndrome pathogenesis, others have shown that the expression levels of MeCP2 in different mouse brain regions correlated with impaired behavioral phenotypes in a RTT mouse model (Wither et al. 2013). Several RTT mouse models have been generated by deleting the Mecp2 expression in specific brain regions and/or specific cell types within the brain regions that show varying degrees of RTT phenotypes. For example, loss of MeCP2 expression in the neurons in basolateral amygdala causes increased anxiety-like behavior and impaired cue-dependent fear learning (Adachi et al. 2009; Wu and Camarena 2009). Several studies have shown abnormal social behaviors, anxiety and also autistic features in RTT mouse models lacking MeCP2 expression in the neurons of forebrain (Gemelli et al. 2006; Chen et al. 2001; Chao et al. 2010). Moreover, MeCP2 deletion from hypothalamic neurons resulted in abnormal physiological stress response, hyper-aggressiveness and obesity (Fyffe et al. 2008).
Within the brain, cellular expression of MeCP2 is predominantly detected in neurons. Other than neurons, MeCP2 expression has also been demonstrated in astrocytes, oligodendrocytes and microglia (Zachariah et al. 2012; Ballas et al. 2009; Rastegar et al. 2009; Derecki et al. 2012; Liyanage et al. 2013; Olson et al. 2014). Our studies also show that MeCP2 is localized to chromocenters in neurons, astrocytes and oligodendrocytes (Zachariah et al. 2012; Rastegar et al. 2009; Liyanage et al. 2013; Olson et al. 2014).
MeCP2 in Neurons
We and others have shown that among the MeCP2-expressing cells, neurons show the highest MeCP2 expression, while lower MeCP2 levels are detected in glial cell types (Zachariah et al. 2012; Ballas et al. 2009). MeCP2 expression in neuronal nuclei is as high as the levels of histone proteins (Skene et al. 2010). For the same reasons, RTT phenotypes are thought to be majorly of neuronal origin (Chen et al. 2001; Guy et al. 2001). Proper MeCP2 expression in neurons is required for neuronal maturation (Kishi and Macklis 2004; Singleton et al. 2011; Thatcher and LaSalle 2006; Shahbazian et al. 2002b) and proper neuronal functions (Nguyen et al. 2012; Shahbazian and Zoghbi 2002). In a previous section (MeCP2 Expression), we described that loss of Mecp2 in neurons found within specific brain regions of mouse models leads to RTT phenotypes.
MeCP2 in Astrocytes
Even though, astrocytes express lower levels of MeCP2 relative to neurons (Zachariah et al. 2012; Ballas et al. 2009), proper expression of MeCP2 in astrocytes is essential for normal neuronal function. For example, abnormal neuronal morphology in primary neurons can be caused by Mecp2-deficiency in the neighboring astrocytes (Ballas et al. 2009; Maezawa et al. 2009). Furthermore, loss of MeCP2 in astrocytes has been shown to cause abnormalities in neurodevelopment and thereby contributing to RTT progression (Maezawa et al. 2009; Okabe et al. 2012; Lioy et al. 2011). A recent study also reported a set of MeCP2 targets in astrocytes which might be important in RTT pathology. The confirmed targets of MeCP2 in astrocytes include Apoc2, Cdon, Csrp and Nrep, which the authors suggest to be important for proper astrocytic functions (Yasui et al. 2013).
MeCP2 in Microglia
Microglia is a type of glial cells, which is mainly involved in neuro-inflammatory processes. Similar to other glial cell types, microglia can modulate neuronal morphology through secreting soluble factors. MeCP2-deficient microglia secrete higher levels of glutamate, leading to abnormal neuronal morphology, highlighting the potential role of MeCP2 in microglia that impacts RTT progression (Maezawa and Jin 2010). Further supporting the importance of microglial expression of MeCP2, re-expression of MeCP2 in microglia of Mecp2-null mouse model has been shown to restore several RTT phenotypes (Derecki et al. 2012).
MeCP2 in Oligodendrocytes
Mecp2-deficiency in the cortex and hippocampus of a RTT mouse model has been shown to cause reduced CNPase expression, a specific marker for oligodendrocytes (Wu et al. 2012). The role of MeCP2 in regulating myelin-related genes including myelin basic protein, proteolipid protein and myelin-associated glycoprotein has also been shown in a RTT mouse model (Vora et al. 2010). A recent report demonstrated RTT phenotypes such as hindlimb grasping in a mouse model lacking MeCP2 expression in oligodendrocyte lineage cells (Nguyen et al. 2013). Together, these reports suggest that MeCP2 expression is required for proper oligodendrocyte functions and RTT pathology.
MeCP2 Functions
MeCP2 is a multifunctional protein that is involved in transcriptional regulation as well as modulating chromatin structure. The different domains of MeCP2 have been assigned to facilitate multiple functions through direct DNA binding, interaction with protein partners or by recruiting other factors (Guy et al. 2011). Different MeCP2 functions are described in detail in the following sections.
MeCP2 in Transcriptional Regulation
The role of MeCP2 in transcriptional regulation has been described in several aspects of gene regulation, including transcriptional repression/silencing and activation. However, the precise role of MeCP2 as a transcriptional repressor (Nan et al. 1998) or transcriptional activator (Chahrour et al. 2008) is paradoxical. For example, brain-derived neurotrophic factor (BDNF) is one of the most studied MeCP2 target genes. Binding of MeCP2 to the Bdnf promoter has been shown to repress its expression, and the removal of MeCP2 from Bdnf promoter due to neuronal activity-dependent phosphorylation of MeCP2 caused the de-repression/activation of Bdnf expression (Chen et al. 2003; Martinowich et al. 2003; Zhou et al. 2006). In contradiction, the Mecp2-deficient mice demonstrated reduced levels of Bdnf expression, raising the question whether MeCP2 is a transcriptional activator of Bdnf (Abuhatzira et al. 2007).
The transcriptional regulatory role of MeCP2 seems to be dependent on its interacting protein partners. The association of MeCP2 with repressor complexes containing SIN3a and histone deacetylases (HDAC) leads to transcriptional repression. In contrast, MeCP2-mediated transcriptional activation of genes occurs in association with the activator complexes containing cAMP response element-binding protein (CREB) (Chahrour et al. 2008). MeCP2 recruitment to 5hmC-enriched active genomic loci (Mellen et al. 2012) and its association with TET1 protein (Cartron et al. 2013) further support the role of MeCP2 in gene activation. Even though numerous MeCP2 target genes have been reported in multiple cellular systems (Zachariah and Rastegar 2012), the diverse nature of MeCP2 target genes and the opposing effects on the studied genes (activate or repress) pose questions on the exact function of MeCP2 as a transcriptional regulator. Hence, recent studies define MeCP2 as a genome-wide epigenetic modulator rather than a transcriptional regulator (Della Ragione et al. 2012). However, to date, it remains elusive how a single protein can modulate a variety of opposing functions.
MeCP2 in Chromatin Structure
As a major DNA binding protein, MeCP2 is also involved in controlling chromatin structure (Liyanage et al. 2012; Zlatanova 2005; Chadwick and Wade 2007). MeCP2 can modulate chromatin architecture by condensing DNA through regulating the long-range interactions (Horike et al. 2005), formation of higher-order chromatin structures (Georgel et al. 2003; Agarwal et al. 2011), and formation of chromatin loops and DNA bridges (Georgel et al. 2003; Nikitina et al. 2007b; Yasui et al. 2007). The localization of MeCP2 to DAPI-counterstained heterochromatin regions of the nuclei (Zachariah et al. 2012; Craig et al. 2003), which are usually referred to as chromocenters, colocalization of MeCP2 with the heterochromatin-binding protein (HP1) at the chromocenters, and chromocenter-clustering upon MeCP2 overexpression (Brero et al. 2005), further add evidence for the role of MeCP2 as a chromatin architectural protein. Moreover, significant differences have been observed in the sizes and numbers of chromocenters in Mecp2-deficient and Mecp2-WT neurons, further supporting role of MeCP2 in chromatin organization (Singleton et al. 2011). More importantly, RTT-causing MECP2 mutations have been shown to disrupt the formation of higher-order chromatin structures (Nikitina et al. 2007a; Agarwal et al. 2011; Kumar et al. 2008).
MeCP2 in RNA Splicing
As described previously, even though several MeCP2 target genes have been identified, gene expression studies in Mecp2-deficient mice did not reveal global changes in gene expression. The broad spectrum of RTT phenotypes could not be explained by the altered expression of few specific genes (Tudor et al. 2002). However, changes in alternative splicing of genes such as Dlx5, Fgf2–5, Fut8 and Nf1 could be observed in a mouse model of RTT (Mecp2 308/Y), implying the potential role of MeCP2 in RNA splicing (Young et al. 2005). Supporting this notion, interactions of MeCP2 with an RNA binding protein YB-1 (Young et al. 2005) and spliceosome-associated protein PRPF3 (Long et al. 2011) have been reported. Moreover, RTT-causing truncating mutations are shown to disrupt the interaction between MeCP2 and spliceosome complex containing PRPF3 (Long et al. 2011). Recently, it has also been shown that the DNA methylation-dependent binding of MeCP2 to exonic sequences modulates alternative splicing (Maunakea et al. 2013). Altered RNA splicing of synaptic genes have been reported in autism as well as Rett syndrome (Smith and Sadee 2011). However, whether MeCP2 plays a role in RNA splicing in autism is currently unknown. MeCP2 is also known to regulate the alternative splicing of NMDA receptor subunit NR1 (Young et al. 2005), splice variants of which are frequently altered in mouse models of fetal alcohol spectrum disorders (FASD) (Brady et al. 2013).
MeCP2 in microRNA Regulation
Apart from the role of MeCP2 in regulating the coding genes, the most recently described function of MeCP2 is the regulation of the expression of microRNAs (miRNAs). MicroRNA-137 (miR-137) is one such miRNA which is involved in regulating neural stem cells (NSCs) proliferation and differentiation. The expression of miR-137 is epigenetically regulated by MeCP2 (activated by Mecp2-deficiency) in adult NSCs that undergo neuronal differentiation. The authors also reported the activation of miR-187 and miR-193a and the repression of miR-199a, miR-197, miR-221 and miR-222 in the absence of MeCP2 (Szulwach et al. 2010). In embryonic cortical neurons, binding of MeCP2 to the promoter of miR-184 represses its expression in an activity-dependent manner (Nomura et al. 2008). Interestingly, the role of MeCP2 in regulating miRNA and its contribution to RTT pathology have been demonstrated in mouse models of RTT (which lack MeCP2 expression), by the altered expression of a spectrum of miRNAs [miR-146a, miR-146b, miR-130, miR-122a, miR-342 and miR-409 (downregulated) and miR-29b, miR329, miR-199b, miR-382, miR-296, miR-221 and miR-92 (upregulated)] (Urdinguio et al. 2010). Loss of MeCP2 in the cerebellum of a RTT mouse model also led to altered expression of miRNAs (Wu et al. 2010). The role of MeCP2 in RTT pathology through regulating the expression of miRNAs was further strengthened by the observation of altered transcriptome of long noncoding RNAs (lncRNA) and specific upregulation of AK087060 and AK081227 in the brains of Mecp2-null RTT mouse model (Petazzi et al. 2013).
Other Biological Functions of MeCP2
As a multifunctional epigenetic modulator, MeCP2 participates in numerous biological functions both in vitro and in vivo. Within neurons, MeCP2 plays critical roles in neuronal maturation (Kishi and Macklis 2004; Fukuda et al. 2005; Kishi and Macklis 2010), terminal neuronal differentiation (Matarazzo et al. 2004; Tsujimura et al. 2009), modulating the neuronal morphology (Ballas et al. 2009; Belichenko et al. 2009b; Wang et al. 2013a; Rastegar et al. 2009; Belichenko et al. 2009a; Larimore et al. 2009) and synaptic plasticity (Chao et al. 2007; Qiu et al. 2012; Na et al. 2012, 2013; Zhong et al. 2012). Most recent studies demonstrate the role of MeCP2 in regulating protein synthesis, and it is suggested that the reduced protein synthesis in MeCP2-deficient cells is contributing to the RTT phenotypes detected in these cells (Li et al. 2013; Ricciardi et al. 2011). Further confirming the role of MeCP2 in Rett syndrome pathogenesis, the aforesaid functions are impaired in RTT patients (Kim et al. 2011; Chapleau et al. 2009) and also in RTT mouse models (Asaka et al. 2006; Belichenko et al. 2009).
Mouse Models to Study Rett Syndrome
In order to investigate the molecular mechanisms of RTT pathogenesis and design therapeutic strategies, rodent models, specifically mouse models, are extensively employed. Several different strategies are used to alter/abolish the Mecp2 expression and/or function in mice that include Mecp2 knockout, Mecp2 mutant and MECP2 knock-in strategies (Calfa et al. 2011; Ricceri et al. 2008, 2013). Most frequently used Mecp2-RTT mouse models are summarized in Table 1.
The Mecp2-deficient/Mecp2-knockout mouse models which are also referred as “null MeCP2” models have been generated through deletion of exon 3 or 4 or 3 and 4 to abolish the expression of whole MeCP2 protein. The examples of such null MeCP2 mouse models include Mecp2 tm1.1Bird, Mecp2 Jae and Mecp2 neoTam which demonstrate many RTT phenotypes (Guy et al. 2001; Chen et al. 2001; Pelka et al. 2006). Moreover, a mouse model lacking MeCP2E1 expression recapitulated RTT-like phenotypes (Yasui et al. 2014), in comparison with Mecp2e2-knockout model which lacked neurological phenotypes (Itoh et al. 2012). Similarly, the mouse models harboring known RTT-causing MECP2 mutations have been helpful in determining the role of these mutations in RTT pathology. The examples include, the mutations which result in truncated MeCP2 protein (MeCP2308 and R168X) (Shahbazian et al. 2002a; Schaevitz et al. 2013; Lawson-Yuen et al. 2007) as well as mutations which cause loss of function or loss of protein/DNA interactions (MECP2 T308A, MECP2 R306C and MECP2 T158A) (Lyst et al. 2013; Goffin et al. 2011; Ebert et al. 2013). The consequences of these mutations, phenotypes and life spans of each mouse model are summarized in Table 1.
Another RTT rodent model was generated with reduced Mecp2 expression in the rat brain, which showed no significant RTT phenotypes but exhibited transient abnormality in neurobehavior and reduced Bdnf expression in brain (Jin et al. 2008). Apart from these rodent models, other animal models are also being used to study the etiology of RTT and MeCP2 functions. A zebrafish model of Rett syndrome has been generated by introducing a C187T transition mutation leading to MeCP2 protein truncation at the position 63 (Mecp2Q63*). Unlike most RTT mouse models, this zebrafish RTT model showed milder RTT phenotypes such as motor deficits, however, was viable and fertile (Pietri et al. 2013). Moreover, a Drosophila model for RTT was generated using MECP2 overexpression and known RTT mutations (R106W, R294X, and Δ166), which resulted in abnormalities in locomotor functions and disrupted eye structure (Cukier et al. 2008).
MECP2 Mutations and Rett Syndrome
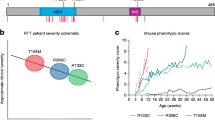
Rett syndrome is caused by de novo mutations in the MECP2 gene (Christodoulou et al. 2003), which can disrupt molecular functions of MeCP2. Approximately 600 mutations have been detected so far within the MECP2 gene [(Christodoulou et al. 2003) and (RettBASE: http://mecp2.chw.edu.au)]. These mutations include missense, nonsense and silent mutations, as well as 5′ UTR and 3′ UTR variations, intronic variations, insertions (frameshift, in-frame) and deletions (exonic deletions, frameshift, in-frame) (RettBASE: http://mecp2.chw.edu.au). Recent studies have deciphered the impact of MECP2 mutations in MeCP2 function contributing to RTT pathogenesis. Figure 2 illustrates distribution of RTT mutation hot spots and the abolished functions/interactions of MeCP2 due to these mutations.

Distribution of known MeCP2 mutations and the interactions/functions abolished due to mutations. The well-known mutations found in Rett Syndrome patients and the loss of interactions and functions of MeCP2 due these mutations are illustrated. MBD methyl-CpG-binding domain, ID inter-domain, TRD transcriptional repression domain, CTD C-terminal domain, NTD N-terminal domain, AT AT-hook domain, 5mC 5-methylcytosine, 5hmC 5-hydroxymethylcytosine. The amino acid numbers are according to the location on MeCP2E2 isoform
MeCP2 Chromatin Binding
As stated earlier, MeCP2 is a chromatin architectural protein that localizes to the chromocenters, and it is involved in chromocenter-clustering (Liyanage et al. 2012). Supporting the chromatin architectural role of MeCP2, several MECP2 mutants fail to localize into heterochromatin regions of the nucleus and to cluster heterochromatin (Agarwal et al. 2011). This is not surprising since the majority of MECP2 mutations are found within the MBD domain, which is required for methylation-dependent chromatin binding (Kudo et al. 2003). Disruption of MBD domain not only results in diffused nuclear staining due to the inability of the MeCP2 protein localization to heterochromatin, but also causes retention of the protein in the cytoplasm (Stuss et al. 2013). Moreover, T158 amino acid within the MBD domain has been implicated in stabilization of MeCP2 and DNA binding (Ho et al. 2008). A knock-in mouse model harboring T158A mutation demonstrated RTT phenotypes, decreased MeCP2 stability, and reduced DNA binding (Goffin et al. 2011). Recently, it was shown that MeCP2 can bind to both methyl marks: 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) with similar affinities, and it is the major protein that binds to 5hmC in brain (Mellen et al. 2012). Interestingly, MECP2 missense mutation R133C within the MBD hindered the MeCP2 binding to 5hmC, while D121G mutation inhibited MeCP2 binding to 5mC. The MBD domain recognizes and binds to methylated DNA, whereas the TRD domain mediates binding to AT-rich DNA through an AT-hook domain. AT-hooks are a type of DNA binding motifs, which allow binding to DNA at adenine–thymine (AT)-rich regions through a characteristic RGR domain (Reeves and Nissen 1990). In the MeCP2 protein, the disruption of RGR domain (3 amino acids between 257 and 272) within the AT-hook causes failure of MeCP2 to bind to chromatin. For this reason, the mutations R270X and G273X within the TRD-AT-hook fail to bind to chromatin and was able to disrupt the function of TRD domain and its repressor activity (Baker et al. 2013). MeCP2 recruits ATRX to heterochromatin in association with the Cohesin and CTCF, which are known to regulate certain imprinting genes such as H19 (Kernohan et al. 2010; Nan et al. 2007). The MeCP2 mutation R270X in the AT-hook domain as well as mutations in MBD domain can disrupt localization of ATRX to heterochromatin (Baker et al. 2013; Nan et al. 2007), implicating the potential of this mutation in disrupting chromatin remodeling as well as transcription regulation of MeCP2 target genes. These observations further support the role of MeCP2 mutations in abolishing DNA binding properties of MeCP2, which are required for MeCP2 function as a transcriptional regulator and chromatin architectural protein.
Even though MeCP2 is mostly known as a methyl binding protein, several studies have shown its binding to unmethylated DNA as well (Baubec et al. 2013; Hansen et al. 2010). In the absence of DNA methylation, MeCP2 is localized to genomic regions with open/accessible chromatin structure, independent of MeCP2 protein interacting partners (Baubec et al. 2013). Interestingly, the MBD domain, which is known to mediate the methylated DNA binding with high affinity also has lower affinity to unmethylated DNA (Ghosh et al. 2010a, b). Other than MBD, other domains of MeCP2 including ID, TRD and CTD have been shown to mediate unmethylated DNA binding (Ghosh et al. 2010b). Moreover, MeCP2 has been shown to cause compaction of chromatin independent of DNA methylation or through binding to unmethylated DNA (Georgel et al. 2003).
MeCP2 Binding to Repressor Complexes
Formation of multi-protein complexes containing repressor proteins such as HDAC, NCoR and SIN3 is required for transcriptional repression of MeCP2 target genes. A recent study demonstrated that neuronal activity-dependent phosphorylation of threonine residue at 308 position (T308) prevents the binding of MeCP2 to NCoR and thus prevents transcriptional repression. Interestingly, R306C, a missense MECP2 mutation, prevented the phosphorylation at T308, whereas T308A mutation prevented T308 phosphorylation and led to altered expression of MeCP2 target genes such as Npas4 and Bdnf (Ebert et al. 2013). Further confirming the contribution of RTT mutations in disrupting MeCP2 functions and thereby contributing to the RTT pathology, another group discovered a novel cluster of mutations (P302R, K304E, K305R, and R306C) within the TRD domain. These mutations disrupted the interaction of MeCP2 with the repressor complex proteins NCoR and SMRT (Lyst et al. 2013). Together, these studies suggest that the common MECP2 mutations abolish the bridge between MeCP2, DNA and MeCP2-interacting proteins.
MECP2 Mutations in Non-Rett Syndrome Cases
Although MECP2 mutations are considered to be the most prevalent cause of Rett syndrome and there is a strong correlation between MECP2 mutations and RTT phenotypes, several cases of RTT patients are found without MECP2 mutations (Temudo et al. 2011; Hoffbuhr et al. 2001; Huppke et al. 2005). Similarly, known RTT-causing MECP2 mutations are found in patients who do not show classical RTT phenotypes (Suter et al. 2013). Other than RTT, MECP2 mutations have been found in association with neurological/neuropsychiatric disorders such as, schizophrenia (Cohen et al. 2002), FASD (Zoll et al. 2004), PPM-X syndrome (Klauck et al. 2002), autism (Beyer et al. 2002), Prader-Willi syndrome (Tejada et al. 2006) and Angelman syndrome (Watson et al. 2001). These reports emphasize that the diagnosis of RTT as well as other MeCP2-related neurological disorders should not be limited to the detection of MECP2 mutations, but rather should be carried out in conjunction with clinical features. They also highlight the importance of understanding the function of MECP2 mutations in RTT as well as other MeCP2-related disorders.
MeCP2 Posttranslational Modifications
Posttranslational modifications such as phosphorylation, acetylation, methylation and ubiquitination are required for proper function and stability of proteins (Liyanage et al. 2012). Similarly, several PTMs have been detected in MeCP2 protein and shown to be important for its function and stability. These PTMs include phosphorylation, ubiquitination, SUMOylation and acetylation, which are described in detail below.
Phosphorylation
Among the MeCP2 posttranslational modifications, phosphorylation is the mostly studied PTM and has shown implications in neuronal activity-dependent transcriptional regulation and modulating MeCP2 protein–protein interactions. Several serine residues of MeCP2 are phosphorylated, including S13, S80, S149, S229, S274, S401 and S421 (amino acid numbers are based on the MeCP2E2 isoform, unless specifically mentioned). Moreover, phosphorylation at threonine residues is also implicated in MeCP2 activity-dependent functions (Ebert et al. 2013). Examples of such phosphorylation sites, the interactions assigned for some of these phosphorylated serine residues and known RTT mutations corresponding to these sites are summarized in Table 2. A complete list of phosphorylation sites reported so far can be found in the phosphosite Plus/http://www.phosphosite.org. These phosphorylated sites on MeCP2 protein mediate formation of repressor complexes containing Sin3A, HP1, SMC3 and NCoR and allow the subsequent binding to target promoters. Known RTT mutations including S86C, S229L, R306C and S401 N abolish MeCP2 interaction with the repressor proteins. Therefore, it is likely that disruption of these phosphorylation sites would contribute to RTT disease development.
Acetylation
Acetylation of MeCP2 can modulate its binding to the target gene promoters. The human MeCP2 is acetylated at lysine 461 by p300 (Choudhary et al. 2009). It is also reported that MeCP2E1 isoform is acetylated at lysine 464 in HEK-293 cells and primary mouse cortical neurons by p300 (Zocchi and Sassone-Corsi 2012). The same group showed the interaction of SIRT1 with MeCP2E1 isoform and thereby mediating deacetylation of K464 residue. Interestingly, reduced activity of SIRT1 preventing the deacetylation of MeCP2 induced the binding of MeCP2 to BDNF promoter, implying that AcK464 is required for MeCP2 binding to BDNF promoter. A recent study discovered two more sites that are acetylated within the TRD (AcK305/307) and CTD (AcK321) domains in human MeCP2E2 in SH-SY5Y cells (Gonzales et al. 2012). Implicating the potential importance of MeCP2 acetylation in RTT pathogenesis, a previously reported RTT mutation (K305E/R) corresponded to AcK305/307 site.
SUMOylation
SUMOylation is a modification which usually regulates nuclear transportation of proteins. However, SUMOylation of MeCP2 protein was reported in both cytoplasm and the nucleus of Neuro2a cells (Miyake and Nagai 2007), questioning whether this modification actually plays a role in nuclear transportation of MeCP2. The authors speculated that the SUMOylation might be involved in determining MeCP2 protein stability and its interaction with other proteins such as HDAC and DNMT. Confirming the role of SUMOylation in MeCP2 function, SUMOylation at the K223 was shown to be required for the recruitment of HDAC1/2 complex and transcriptional repression in mouse primary cortical neurons (Cheng et al. 2013).
Ubiquitination
Protein ubiquitination functions in controlling intracellular signaling events and proteolytic degradation of proteins. So far, approximately 10 ubiquitination sites at the lysine residues of MeCP2 are reported at positions 12, 82, 119, 130, 135, 233, 256, 271, 249, and 321. The functional roles of MeCP2 ubiquitination at different residues are currently unknown, but they might be involved in determining MeCP2 protein stability and degradation. Also, it is also possible that MeCP2 ubiquitination might be contributing to RTT pathogenesis, because known RTT-causing MECP2 mutations K12N, K82R and K135E correspond to three of the ubiquitination sites (Gonzales et al. 2012). Moreover, other ubiquitination sites were predicted within two putative PEST domains (enriched for proline, glutamate, serine and threonine). The two PEST domains found in the N- and C-terminal domains of MeCP2 harbored several putative and previously reported phosphorylation, and SUMOylation sites. The authors suggest that the two PEST domains might be important in proteolytic degradation of MeCP2 and thereby modulating the turnover rate of MeCP2 (Thambirajah et al. 2009).
MeCP2 Isoforms
As described in a previous section, MeCP2 is a single protein functioning in opposing ways. For instance, MeCP2 functions both as a transcriptional activator and repressor; it binds to methylated and non-methylated DNA; it binds to both 5mC and 5hmC; and either overexpression or its deficiency causes similar neurological problems (Zachariah and Rastegar 2012; Ezeonwuka and Rastegar 2014). One possibility of multifunctional properties of a single protein is to have isoforms with different properties. Alternative splicing of both human and mouse MECP2/Mecp2 gene generates two isoforms MeCP2E1 (previously described as MeCP2B/α) and MeCP2E2 (previously described as MeCP2A/β) (Fig. 1c–d) (Mnatzakanian et al. 2004; Kriaucionis and Bird 2004). MeCP2E1 isoform uses the translation start site (ATG) in the exon 1 and is comprised of exons 1, 3 and 4. In comparison, MeCP2E2 isoform uses a translation start site in the exon 2 and it is comprised of exons 2, 3 and 4. The two isoforms share the MBD, TRD and CTD domains, while they differ only at their N-termini (Fig. 1c–d).
Differential Distribution of MeCP2 Isoforms
MeCP2E1 isoform is the major isoform found in brain (Kriaucionis and Bird 2004; Zachariah et al. 2012). A previous study has demonstrated distribution of the two isoforms at the transcript levels in three stages of mouse brain development, namely new born (P1), juvenile (P21) and adult (P60) (Dragich et al. 2007). Mecp2e2 distribution was relatively similar to that of total Mecp2 at P1, whereas between P21 and P60, Mecp2e2 localization was restricted to dorsal thalamus and cortical layer V. The difference between the total Mecp2 detection throughout the adult mouse brain and the restricted detection of Mecp2e2 in thalamus and cortex layers suggested that Mecp2e1 might be the predominant Mecp2 isoform in the adult mouse brain at the transcript levels. A relatively higher expression of MECP2E2 isoform at the transcript levels is reported in the skeletal muscles, placenta, liver, and prostate gland. The authors also showed that in the whole human brain, MECP2E2 transcript levels are approximately 12-fold lower than the levels of MECP2E1 transcripts. In the human cerebellum, MECP2E2 transcript levels are approximately ninefold lower than that of MECP2E1 (Mnatzakanian et al. 2004).
Until 2012, determination of the distribution and functions of the two MeCP2 isoforms at the protein levels in brain or neural cell types had been hindered by the lack of MeCP2 isoforms-specific antibodies. In 2012, for the first time, we reported the generation of a MeCP2E1-specific antibody and showed that neuronal expression of MeCP2E1 protein is approximately 5 times higher than that of astrocytes. Moreover, distribution of MeCP2E1 isoform in the adult murine brain regions was relatively similar to that of the total MeCP2, which suggested that MeCP2E1 is the major isoform in the brain (Zachariah et al. 2012). Moreover, using a newly generated MeCP2E2-specific antibody, our very recent studies show that similar to total MeCP2 and MeCP2E1, MeCP2E2 protein is localized to the heterochromatin regions of the nucleus (Olson et al. 2014). Within the adult murine brain hippocampus, neurons, astrocytes and oligodendrocytes show the expression of both MeCP2E1 and MeCP2E2 expression within the nucleus. Further analysis of the nuclear expression levels of MeCP2 isoforms in adult brain regions (olfactory bulb, striatum, cortex, hippocampus, thalamus, brain stem and cerebellum), showed relatively uniform expression levels of MeCP2E1 as compared to the differential expression levels of MeCP2E2. During mouse brain development both isoforms showed similar expression profiles, but MeCP2E2 showed a later onset of expression as compared to the MeCP2E1 (Olson et al. 2014).
Presence of Mutations and Relevance to RTT
Approximately 80–85 % of the MECP2 mutations are localized within the MBD, TRD and CTD domains which are shared by both isoforms. Previously, exon 1 was thought to be a noncoding exon and thus was excluded from the sequencing and mutation analysis studies on Rett syndrome. Since the discovery of MeCP2E1 isoform, scientists have started to re-evaluate the mutation analysis of exon 1. To date, several RTT-causing mutations are reported within exon 1 encoding for MeCP2E1 (Gianakopoulos et al. 2012; Saunders et al. 2009; Chunshu et al. 2006; Quenard et al. 2006; Bartholdi et al. 2006; Saxena et al. 2006; Ravn et al. 2005). Studies conducted in 2005–2006 suggested that mutations in exon 1 are rare in RTT, because they could only detect exon 1 mutations in 0.03–1 % of the tested samples from RTT patients (Amir et al. 2005; Quenard et al. 2006; Evans et al. 2005b). Regardless, another study showed that the RTT patients with mutations in exon 1 had more severe phenotypes than the patients without exon 1 mutations (Bartholdi et al. 2006). Another study in 2009 showed that mutations found within exon 1 affect the translation of Mecp2e1, but not the transcription or translation of Mecp2e2 (Fichou et al. 2009). These observations also suggest that MeCP2E2 expression alone is unable to compensate for the loss of MeCP2E1 in RTT patients. So far, no mutations are reported in exon 2 encoding for MeCP2E2 isoform. The absence of exon 2 mutations has led to the belief that MeCP2E2 is not important for RTT pathology. Moreover, the Mecp2e2 isoform was suggested to be dispensable for RTT pathology as loss of Mecp2e2 in mice did not result in neurological phenotypes that mimic RTT phenotypes (Itoh et al. 2012). However, maternally transferred Mecp2e2 null allele reduced the embryonic viability implicating that loss of Mecp2e2 expression and/or function during embryonic development might not lead to a live birth. This might also explain the absence of exon 2-specific mutations in RTT. However, several studies on RTT patients picked up the same 11 bp deletion in the MECP2 exon 1 (c.47_57del11nt) (Saxena et al. 2006; Ravn et al. 2005; Amir et al. 2005), which was shown to affect the translation of not only MECP2E1 but also MECP2E2, because the deletion disrupts the 5′-UTR of the MECP2E2 which has been suggested to be essential for its translation (Saxena et al. 2006). A recent study demonstrated that in a girl with typical RTT, a novel mutation activates a splice-donor site upstream of exon 1, leading to premature splicing of MECP2. Even though the splice-acceptor site in intron 2-exon 3 was unaffected, the mutation caused a 16 amino acid deletion and a truncated MeCP2E1 protein. This deletion was predicted to cause a shift in the open reading frame for MeCP2E2, which might affect the MeCP2E2 translation efficiency. In the same patient, decreased MECP2E1 transcript levels and increased MECP2E2 transcript levels were observed (Sheikh et al. 2013). Therefore, the possibility of the contribution of MECP2E2 in RTT etiology should not be excluded. Providing more evidence to the altered MECP2 splice variants in RTT, in a cohort of Israeli patients, mutations in a splice-donor site found within the intron 1 were correlated with significant reduction in both MECP2E1 and MECP2E2 in patient’s blood samples. Interestingly, not only the splice-donor site-specific mutations, but also different mutations found throughout the MECP2 gene differentially affected the expression of MECP2E1 and MECP2E2. Moreover, in patients with atypical RTT phenotypes and no MECP2 mutations, expression of both MECP2 isoforms was affected (Petel-Galil et al. 2006). The concept on the importance of MeCP2E2 isoform in RTT is strengthened by the ability of Mecpe2e2 expression alone to rescue RTT phenotypes in mice (Luikenhuis et al. 2004; Jugloff et al. 2008), which also indicates that Mecp2e2 is required and able to maintain normal brain functions. However, the efficiency of the rescue of RTT phenotypes differs between the two Mecp2 isoforms, where Mecp2e1 is more efficient in rescuing RTT phenotypes than Mecp2e2 (Kerr et al. 2012). Together, these reports highlight that even though, the exon 1 mutations are found in rare cases of RTT and no mutations are found in exon 2, both MECP2 isoforms seem to be affected in Rett syndrome patients. Hence, the expression, regulation and the contribution of both MECP2 isoforms in RTT pathology warrants further investigations.
MeCP2 Isoform-Specific Functions and Interacting Protein Partners
The two MeCP2 isoforms share the majority of the functional domains of MeCP2, and hence, it was assumed that the two isoforms are functionally redundant. However, few recent studies point out that these isoforms might also have non-redundant functions. MeCP2E1 is considered as the major isoform in the brain and is critical for basic brain function. Our previous studies show that MeCP2E1 shows a distribution similar to that of the total MeCP2 in adult murine brain, neurons and astrocytes (Zachariah et al. 2012), and therefore, it is likely that its main functions might be similar to that of total MeCP2. Furthermore, we demonstrated the role of MeCP2E1 isoform in dendrite branching of neurons and the ability of MeCP2E1 in rescuing the aberrant neuronal morphology in RTT neurons (Rastegar et al. 2009). Similarly, the role of MeCP2E2 in early events of neurite formation was demonstrated in PC12 cells (Cusack et al. 2004). As the major protein isoform in brain, MeCP2E1 is suggested to be the relevant isoform for RTT. Even though a direct relationship of MeCP2E2 with RTT has not been shown so far, its function in placental development has been established (Itoh et al. 2012). The authors also showed the misregulation of Peg-1 in extra-embryonic tissue carrying Mecpe2e2 null allele, suggesting that Peg-1 might be a Mecp2e2-target gene. Moreover, indicating the proper expression of Mecp2e2 in neuronal cells, the overexpression of Mecp2e2, but not Mecp2e1 has been shown to cause cerebellar neuronal death (Dastidar et al. 2012). Further supporting the concept of MeCP2 isoform-specific functions, MeCP2E2 has been shown to interact with FOXG1, mutations of which are associated with RTT (Philippe et al. 2010). In comparison, MeCP2E1 isoform interacts with YB-1, Matrin 3 and SFPQ proteins in human neuronal cell-line SH-SY5Y (Yasui et al. 2014). Moreover, a MeCP2E1-specific phosphorylation site (S10) has been reported (Trinidad et al. 2008) and RettBASE mutation database shows that MECP2 frameshift/deletion mutations (Table 2) can cause alterations in this phosphorylation site (p.S10RfsX45 and p.S10fs). Although a functional role of phosphorylation at the S10 is currently unknown, future studies might lead to discovery of MeCP2E1-specific functions.
Rescue of RTT Phenotypes
Numerous therapeutic strategies are currently under development to treat Rett syndrome. However, alternate therapeutic approaches do not rescue the full spectrum of RTT phenotypes. Independent research groups have shown that introduction of MeCP2 into RTT mouse models can rescue the phenotypes. Interestingly, both MeCP2 isoforms can rescue the RTT phenotypes to different extents, where MeCP2E1 application is more successful as expression of Mecp2e1 alone can rescue RTT phenotypes (Kerr et al. 2012). However, other studies show that expression of Mecp2e2 alone can prevent the disease progression in Mecp2 null mouse models (Luikenhuis et al. 2004; Giacometti et al. 2007; Jugloff et al. 2008). Our previous studies also show that gene therapy delivery of MECP2E1 to neurons lacking exogenous MeCP2 expression can rescue the aberrant neuronal morphology (Rastegar et al. 2009).
Sensitivity to Environmental Insults/Drugs
MeCP2 is known to modulate sensitivity to different external environmental insults such as exposure to ethanol (Repunte-Canonigo et al. 2013), drugs such as Cocaine (Im et al. 2010) and Methamphetamine (Lewis et al. 2013). On the other hand, MeCP2 expression is also sensitive to these aforementioned stimuli. Implicating the potential role of MeCP2 isoforms in modulating the effects of such environmental insults, a recent study showed differential sensitivity of Mecp2 isoforms to Bisphenol A, which altered Mecp2e1 expression at low concentrations (20 μM) and Mecp2e2 expression at high concentrations (200 μM) (Warita et al. 2013). A recent study from our lab demonstrated that the DNA demethylating agent Decitabine can upregulate Mecp2e1/MeCP2E1 expression, but not Mecp2e2 expression in differentiating neural stem cells (Liyanage et al. 2013), an in vitro model we have established to mimic neural development (Liyanage et al. 2013; Barber et al. 2013; Rastegar et al. 2009). This observation provides insights on inducing a specific Mecp2/MeCP2 isoform by treatment with Decitabine (Liyanage et al. 2013). In contrast, withdrawal of Decitabine resulted in downregulation of both Mecp2 isoforms, where the effect on Mecp2e1 was greater than that of Mecp2e2. Another study showed that the expression of both Mecp2 isoforms was affected by spinal nerve injury in a study of persistent pain states. The two isoforms responded to the injection of Complete Freund’s Adjuvant (CFA) in different ways (Tochiki et al. 2012). Taken together, these studies indicate that the two MeCP2 isoforms respond to drugs at different doses, and thus, future drug therapies will have to be designed with caution not to exert detrimental effects on the two isoforms.
Regulation of Mecp2/MECP2 Isoforms
The studies on the regulation of Mecp2/MECP2 were limited to Mecp2/MECP2 in total (without differentiating between the two isoforms). Recently, we reported for the first time the potential role of DNA methylation at the regulatory elements within the Mecp2 promoter and intron 1 in regulating Mecp2 isoforms during in vitro neural differentiation (Liyanage et al. 2013). In response to Decitabine-mediated DNA demethylation at the Mecp2 promoter and intron 1 elements, Mecp2e1/MeCP2E1, but not Mecp2e2 expression was elevated significantly (Fig. 3). The nature of the correlation (positive or negative) between Mecp2 isoform-specific expression and DNA methylation at regulatory elements was dependent on specific stages of NSC differentiation, suggesting that DNA methylation might have differential impact on the Mecp2 isoform-specific expression depending on the type of cells and stages during neural development (Liyanage et al. 2013). Furthermore, the differential expression of transcript Mecp2 isoforms in the adult mouse brain regions (olfactory bulb, striatum, cortex, hippocampus, thalamus, brain stem and cerebellum) was associated with significantly different changes in DNA methylation patterns at previously reported the regulatory elements found within the Mecp2 promoter and intron 1 (Olson et al. 2014). Therefore, our studies show the potential impact of DNA methylation at Mecp2 regulatory elements in isoform-specific Mecp2 expression in both in vitro and in vivo.

Summary of DNA methylation-mediated regulation of Mecp2 isoforms in response to Decitabine exposure during neural stem cell differentiation. DNA methylation at three regulatory elements found within the Mecp2 promoter (R1-R3) and Mecp2 intron 1 (R4-R6) was analyzed by bisulfite pyrosequencing, in untreated control and Decitabine-treated neural stem cells. DNA Methylation at specific Mecp2 regulatory elements (R2 in the Mecp2 promoter and R4-R6 in the intron 1) were reduced (DNA demethylation), in response to Decitabine exposure. These changes in DNA methylation are correlated with upregulation (↑) of Mecp2/MeCP2 (total), Mecp2e1/MeCP2E1 and unchanged levels (↕) of Mecp2e2. The effects of Decitabine on MeCP2E2 protein levels are unknown (?). The information was extracted from (Liyanage et al. 2013). R region, C cytosine, 5mC 5-methylcytosine
Regulatory Mechanisms of MECP2 in Brain
Previous research studies on MeCP2 have shown that both deficiency (Nagarajan et al. 2006; Squillaro et al. 2012; Smrt et al. 2007) and overexpression (Bodda et al. 2013; Na et al. 2012; Collins et al. 2004) of MeCP2 lead to severe neurological complications. However, expression and function of MeCP2 in neurodevelopmental disorders are the focus of most MeCP2-related research work. Even though therapeutic strategies to restore the expression of MeCP2 are advancing, any therapeutic application should be administered with caution. In order to maintain proper expression levels of MeCP2, a thorough knowledge on MECP2/Mecp2/MeCP2 regulation is required. However, studying MECP2/Mecp2/MeCP2 regulatory mechanisms is perhaps the least studied topic by the MeCP2-RTT research community, yet is an essential aspect to be understood. The MECP2/Mecp2 gene is regulated at transcript level as well as posttranscriptionally by different mechanisms (Fig. 4).

Known regulatory mechanisms of MECP2/Mecp2 gene. The MECP2/Mecp2 gene is regulated transcriptionally and posttranscriptionally by multiple mechanisms. (1) The MECP2 promoter contains positive and negative regulatory elements (+RE and -RE) which are known to regulate MECP2 expression. Both MECP2/Mecp2 genes harbor CpG islands and several CpG dinucleotides regulated by DNA methylation. (2) Cis-regulatory elements are found in intragenic regions of MECP2 gene as well as intragenic regions. There are two silencer elements (S1–S2) and four enhancer elements (E1–E4). P promoter. (3) MECP2/Mecp2 3′UTR contains polyadenylation sites and Cis-acting elements (yellow boxes) which harbor binding sites for trans-acting factors involved in polyadenylation. There transcript variants are generated by polyadenylation (1.8, 7.5 and 10.2 kb). (4) Binding of miRNAs to miRNA response elements (MRE) at the 3′UTR involves in posttranscriptional regulation of MECP2/Mecp2. (5) Changes in histone modifications in response to binding of regulatory proteins regulate Mecp2/MECP2 expression. For example, binging of HMGN1 induces modifications in histones and thereby changes the chromatin structure. There is a differential enrichment of histone posttranslational modifications (PTMs) across the MECP2/Mecp2 gene. As an example, the distribution of active histone marks H3K9Ac and H3K27Ac across the MECP2 gene in three cell types (HeLa, HepG2 and HUVEC) is shown (data extracted from http://genome.ucsc.edu/; February 2009 assembly)
Promoter Elements
The human MECP2 core promoter lies between −179 and −309 (relative to transcription start site) and harbors several transcription factor binding sites such as SP1 and STAT (Liu and Francke 2006). The authors also reported the presence of two negative regulatory elements [(−309 to −370) and (−553 to −681)] and one positive regulatory element (−847 to −1,071) in the promoter region upstream of the MECP2 exon 1 (−1,071 to +9), activity of which are cell-type specific (Liu and Francke 2006). Later on, another study on the encyclopedia of DNA elements (ENCODE) [http://genome.ucsc.edu/] for MECP2/Mecp2 gene showed that the same promoter region and upstream of that region harbors binding sites for transcription factors such as SP1, SP3, TAP1, RNA Pol II, C/EBP, E2F1 and CTCF, binding of which were confirmed by chromatin immunoprecipitation experiments in different cell lines (Singh et al. 2008). Adachi et al., in 2005 reported that only a segment of Mecp2 promoter (−677/−56) is essential to drive the Mecp2 expression in neuronal cell lines and cortical neurons (Adachi et al. 2005). However, the same region does not drive the expression of Mecp2 in non-neuronal cells such as astrocytes. The extracts obtained from mouse brain cortex, striatum, cerebellum and brain stem showed binding of transcription factors such as SP1 within this promoter region. The Mecp2 promoter is a TATA-less promoter, and it does not have any canonical TATA-box and thus resembles the nature of a house keeping gene (Adachi et al. 2005). Reporting the first preclinical MECP2 isoform-specific gene therapy vectors, we previously showed that this internal mouse Mecp2 promoter (MeP) is able to drive the MECP2 expression only in neurons and glia, but not in neural stem cells (Rastegar et al. 2009).
Cis-Regulatory Elements
Human MECP2 gene is regulated at transcript level by six cis-regulatory elements: four enhancers and two silencers (Liu and Francke 2006). The two silencers are found within intron 1 (Silencer 1: S1) and MECP2 3′UTR (S2). The S2 harbors binding sites for regulatory proteins such as REST, Brn2 and BCL6. The S1 silencer element within intron 1 was proposed to be important for tissue-specific MECP2 expression or its alternative splicing. However, binding proteins for S1 were not identified in this study. Providing evidence for the potential role of this silencer element in alternative splicing, we recently showed that the expression of Mecp2 isoforms during NSC differentiation is dynamically correlated with DNA methylation at this silencer element. Furthermore, DNA demethylation of this silencer element was associated with upregulation of Mecp2e1, but not Mecp2e2 in differentiating NSCs (Liyanage et al. 2013). Moreover, our recently published studies show preferential correlation of Mecp2 isoforms with the DNA methylation at the regulatory elements found within this silencer element. For example, Mecp2e2 expression negatively correlates with the DNA methylation within this silencer in striatum, brain stem and hippocampus, in contrast to the positive correlation between Mecp2e1 and DNA methylation at the silencer element only in the olfactory bulb (Olson et al. 2014).
The four enhancer elements (E1–E4) are located within the MECP2 3′UTR (E1), downstream intergenic region between IRAK1 and CXorf12 genes (E2 and E3) and within HCFC1 intron 1 (E4). These enhancers harbor binding sites for Brn3-4, STAT, SP1, OLF1 and MYT1 (Liu and Francke 2006). The presence of cis-regulatory G-quadruplexes at the MECP2 5′UTR was described by a recent bioinformatics-based study. These conserved G-quadruplexes have been proposed to be important in regulating many features of MECP2 including translation, polyadenylation and modulating cis/trans interactions (Bagga and D’Antonio 2013).
Polyadenylation
There are several 3′polyadenylation sites in the MECP2/Mecp2 gene, which generates transcripts with different lengths (1.9, 7.5 and 10 kb) and regulate its expression in different tissues (Coy et al. 1999; Reichwald et al. 2000). The distribution of these transcript variants is tissue-specific. For instance, the short product (~8.5 kb) is the most abundant transcript, which is found in tissues such as lymphoid system and muscles, while the longer product (~10 kb) is predominantly found in the brain (Coy et al. 1999; Pelka et al. 2005). Studies in control and autistic patients suggest that there are numerous variations in 3′UTR between them, and these transcripts have different translational efficiencies. Some of these transcript variants are more unstable and undergo transcriptional degradation (Coutinho et al. 2007). Newnham et al., reported the presence of cis-acting elements which can regulate the polyadenylation of human MECP2 gene and mutations of which can reduce the polyadenylation efficiency. The putative Cis-acting regulatory elements include, CstF binding site, G-rich element and upstream sequence elements. Moreover, binding of the trans-acting factor hnRNP F to these cis-regulatory elements (G-rich element) can further regulate the polyadenylation (Newnham et al. 2010).
Epigenetic mechanisms such as promoter DNA methylation, activity of miRNAs and histone PTMs are also involved in regulating Mecp2/MECP2 expression.
Promoter DNA Methylation
As stated earlier, MECP2/Mecp2 gene undergoes changes according to X-chromosome inactivation (D’Esposito et al. 1996). Analysis of MECP2 promoter regions spanning between −531 and −243 bp showed increased overall promoter methylation (2–2.5 %) in the cerebral cortex of male autistic patients, relative to control brains. This significantly increased promoter methylation negatively correlated with reduced MECP2 expression, implying the potential involvement of promoter methylation in MECP2 downregulation. Within the 15 CpG sites analyzed in this study, two CpG sites (−496 and −445) showed significantly higher methylation over other CpG sites. Moreover, the same promoter region shows binding sites for RNA polymerase II, as well as transcription factors SP1 and SP3 (Nagarajan et al. 2006). The same research group later demonstrated that X-chromosome inactivation by DNA methylation influences MECP2 expression in males through modulating DNA methylation over a boundary between MECP2 promoter and heterochromatin regions (−700 to −509 bp). The binding of CTCF to this methylation boundary (−595 to −389) prevents MECP2 silencing in human neuronal cells (Nagarajan et al. 2008). A comparison of MECP2 promoter methylation in RTT monozygotic twins had been conducted before, which indicated no differences in the MECP2 promoter methylation between the twins (Miyake et al. 2013). However, to conclude that promoter methylation does not play a role in RTT, a direct comparison of RTT patients to age- and gender- matched healthy individuals is necessary.
The potential role of Mecp2 promoter methylation in regulating Mecp2 gene transcription was demonstrated by the association of increased promoter methylation at individual CpG sites (2–5 %) over a 164 bp region (1 to −164 bp) with reduced Mecp2 expression in different samples collected from pups, which underwent stress due to maternal separation. These samples were sperms and brain cortex in three generations (F1 to F3). DNA methylation changes at the studied promoter region were observed in these generations in association with reduced Mecp2 expression, indicating that the epigenetic memory of early stress is transmitted through generations and results in altered expression of Mecp2 (Franklin et al. 2010).
Further strengthening the role of DNA methylation in regulating Mecp2 expression, we recently showed a dynamic relationship between Mecp2 expression and DNA methylation at three regulatory elements found within the Mecp2 promoter, and three regulatory elements found within the Mecp2 intron 1 in in vitro differentiating neural stem cells (Liyanage et al. 2013). The reported intron 1 regions overlap with a previously described silencer element (Liu and Francke 2006). Treatment with DNA demethylating agent Decitabine upregulated Mecp2 expression in an isoform-specific manner, in association with demethylation of specific elements within the promoter and intron 1 (Fig. 3). In contrast, Decitabine withdrawal caused downregulation of both Mecp2 isoforms without significant changes in promoter and intron 1 DNA methylation. We observed a negative correlation between Mecp2 isoform-specific expression and Mecp2 promoter methylation. The correlation between the expression of Mecp2 isoforms and intron 1 DNA methylation was dynamic (negative or positive correlation) depending on the stage of NSC differentiation, and our results suggested that DNA methylation may play dynamic roles at different stages of neural development (Liyanage et al. 2013). Similar positive/negative correlations between Mecp2 isoform-specific expression and DNA methylation at the Mecp2 regulatory elements were also observed in vivo in the adult mouse brain regions, confirming our finding in in vitro studies (Olson et al. 2014). Taken together, these results suggest the involvement of DNA methylation at these studied Mecp2 regulatory elements in controlling Mecp2 expression in a brain region-specific manner.
In addition to these reports, treatment with DNA demethylating agents has been shown to downregulate Mecp2 expression both in vivo and in vitro. Delivery of intrathecal 5-azacytidine to rats following chronic constriction injury in the spinal cord resulted in downregulation of Mecp2/MeCP2 in association with reduced global DNA methylation within the spinal cord cells (Wang et al. 2011). Treatment of lens epithelial myofibroblasts with Zebularine, another DNA demethylating agent also resulted in reduced levels of MeCP2 (Zhou et al. 2011). However, it is unknown whether these Zebularine- or 5-azacytidine-mediated changes in global DNA methylation led to any changes of gene-specific DNA methylation at the Mecp2 promoter causing the reduced levels of MeCP2.
Regulation by MicroRNAs
Activity of several miRNAs has been shown to influence the expression of MeCP2 in different systems. The 3′UTRs of MECP2/Mecp2 gene have been predicted to provide different effects on the translation efficiency (Reichwald et al. 2000). The long 3′UTR part of the transcripts harbors binding sites for miRNAs (miRNA response elements/MRE), which can regulate expression of MECP2/Mecp2 expression posttranscriptionally (Klein et al. 2007).
The MREs found within the long MECP2 3′UTR in fetal human brains provide binding sites for miR-483-5p and suppress the expression of human MeCP2 during human brain development as well as in cells obtained from Beckwith–Wiedemann syndrome patients (Han et al. 2013). In schizophrenic brain neocortex, miR130b has been shown to target 3′UTR of MECP2 (Burmistrova et al. 2007). Both miR-155 and miR-802 can downregulate MeCP2 expression in Downs Syndrome brains and thereby cause altered expression of MeCP2 target genes Creb1 and Mef2c (Kuhn et al. 2010). Under ischemic conditions, MeCP2 protein, but not transcript expression, is regulated by miR-132, in the mouse cortex (Lusardi et al. 2010). The same miRNA (miR-132) is involved in homeostatic feedback loop regulation of MeCP2 in rat neurons through binding to the Mecp2 3′ UTR miRNA recognition element (Klein et al. 2007). Suggesting the potential involvement of miRNAs in regulating MeCP2 expression in fetal alcohol syndrome, MeCP2 was detected as a common target for miR-152, miR199a-3p and miR-685 in cortical neurons exposed to ethanol (Guo et al. 2012). Even though irrelevant for MeCP2 expression in brain and neurodevelopmental disorders, miR-212, miR-224, miR-452, miR-181c and miR-340 have been shown to impact MeCP2 expression in gastric cancer conditions (Wada et al. 2010; Hashimoto et al. 2013). Furthermore, MeCP2 is a direct target of miR-30a-3p in human umbilical vein endothelial cells, which can significantly reduce MeCP2 protein levels (Volkmann et al. 2013).
Histone Posttranslational Modifications
The binding of high mobility group N1 protein (HMGN1) to MECP2/Mecp2 promoter in human and mouse brain extracts acts as a negative regulator for MECP2/Mecp2, downregulating its expression upon HMGN1 binding. The binding of HMGN1 induces chromatin structural changes at the binding sites within the promoter. HMGN1 binding reduced the enrichment of active histone mark H3K9Ac, while increasing the enrichment of H3K9me2, an inactive histone mark (Abuhatzira et al. 2011). Chromatin immunoprecipitation results shown in ENCODE demonstrate unique distribution of histone PTMs throughout the MECP2/Mecp2 gene. For example, active histone marks H3K4me2 and H3K4me3 are clustered around the transcription start sites of MECP2/Mecp2 [(Singh et al. 2008) and (http://genome.ucsc.edu/)]. Further showing the role of histone acetylation in regulating MECP2 expression in glioma cells, inhibition of HDAC activity by valproic acid induced MECP2 transcript expression in a time-dependent manner (Kim et al. 2008). However, during synapse maturation in hippocampal neurons, treatment with Trichostatin A (TSA) resulted in downregulation of Mecp2 transcript and MeCP2 protein levels (Akhtar et al. 2009). These reports also suggest that MECP2/Mecp2 regulatory mechanisms might be cell-type specific and thus should be taken into consideration when regulatory mechanisms are used in therapeutic interventions.
Even though majority of these regulatory mechanisms are not directly linked to RTT physiology, it is important to investigate whether any of these mechanisms contribute in reduced expression and/or function of MECP2 in RTT patients. This is significantly important especially in the absence of known RTT-causing MECP2 mutations.
Advances in Therapeutic Approaches for Rett Syndrome
Currently, Rett syndrome has no cure. However, giving more hope for the patients suffering from RTT, intensive efforts from multiple research groups have proven that certain RTT phenotypes can be rescued in mouse models of RTT (Ricceri et al. 2013), and thus, there might be possible effective treatments for RTT. The proposed therapeutic strategies for RTT include restoring the expression and function of MeCP2 and/or targeting downstream targets of MeCP2. Moreover, for translating the preclinical mice model research into human, there are multiple clinical trials taking place in human RTT patients. Examples of some of the therapeutic strategies occupied so far in RTT mouse models as well as in human RTT patients are described in Table 3.
MeCP2 and Other Human Diseases
Autism Spectrum Disorders
Autism spectrum disorders (ASD) are a spectrum of pervasive neurodevelopmental disorders, which are characterized by impaired social interactions and repetitive stereotyped behaviors (Rapin and Tuchman 2008). Altered expression of MECP2 is found in patients with autism (Nagarajan et al. 2006; Samaco et al. 2004). Reduced MECP2 expression in autistic patients has been associated with increased MECP2 promoter DNA methylation (Nagarajan et al. 2006, 2008). Moreover, MECP2 mutations within the coding regions (Carney et al. 2003; Beyer et al. 2002; Lam et al. 2000; Loat et al. 2008) and sequence variants within the MECP2 3′UTR (Shibayama et al. 2004; Coutinho et al. 2007) are also found in autistic patients, indicating the potential involvement of these sequence variations in altered expression of MECP2 in autism. Not only reduced MECP2 expression, but also overexpression of MECP2 is found in ASD patients (Kuwano et al. 2011). Recent studies also demonstrate that the patients with MECP2 duplication (MECP2 duplication syndrome) share similar features to ASD (Peters et al. 2013; Xu et al. 2012b).
Fetal Alcohol Spectrum Disorders
Prenatal exposure to ethanol leads to a spectrum of neurodevelopmental disorders referred to as FASD. Demonstrating a potential link between MeCP2, RTT and FASD, a mutation (R270X) within the TRD-AT-hook domain was found in a FASD patient with combined phenotypes of both RTT and FASD (Zoll et al. 2004). With the accumulating evidence on the role of epigenetic mechanisms in FASD pathogenesis, several recent studies have investigated the expression patterns of MeCP2 in rodent FASD models. These studies show the aberrant expression of MeCP2 in rodent brains exposed to ethanol, where the effect of ethanol on MeCP2 expression seems to be dependent on multiple factors such as amount of ethanol, time of exposure as well as duration of ethanol exposure (Chen et al. 2013; Kim et al. 2013; Romano-Lopez et al. 2012; Repunte-Canonigo et al. 2013; Tunc-Ozcan et al. 2013). Moreover, the role of MeCP2 in sensitivity to alcohol and addiction to alcohol have also been demonstrated using a RTT mouse model [MeCP2308/Y] (Repunte-Canonigo et al. 2013). These reports implicate the potential role of MeCP2 in other neurodevelopmental disorders and hence highlight the critical role of MeCP2 in nervous system function.
Cancer
MeCP2 is mainly discussed in association with neurological disorders such as RTT and autism. Interestingly, recent reports also show the epigenetic role of MeCP2 and other MBD proteins in different cancers (Parry and Clarke 2011). This is not surprising, because many cancer-related genes are silenced by epigenetic mechanisms, specifically promoter hypermethylation (Fukushige and Horii 2013). The relation between MeCP2 and breast cancer is well established, where MeCP2 is involved in epigenetic regulation of breast cancer-related genes (Mirza et al. 2013; Ray et al. 2013; Sapkota et al. 2012; Muller et al. 2003). Moreover, the expression and potential importance of MeCP2 in prostate cancer were shown previously (Shu et al. 2011; Yaqinuddin et al. 2008; Bernard et al. 2006). MeCP2 regulates cell proliferation, cell growth and apoptosis in prostate cancer cells (Bernard et al. 2006; Yaqinuddin et al. 2008). Apart from these reports, MeCP2 has also been linked to cervical cancer (Wang et al. 2013) and ductal carcinomas (Xu et al. 2012a). Altered MeCP2 expression in gastric cancer has been shown to be mediated by the action of miR-212 (Wada et al. 2010). Also, showing a link between brain tumors and Rett syndrome, one study reported the presence of brain-stem tumor in a small girl with RTT phenotypes (Vanhala et al. 1998).
Unanswered Questions and Closing Remarks
Despite the tremendous progress that has been made with regard to understanding the molecular mechanisms by which MECP2 mutations and MeCP2 expression and/or functional abnormalities cause RTT and to design potential therapeutic strategies, there are numerous unanswered questions yet to be disclosed. To design an effective therapeutic strategy for Rett syndrome, comprehensive understanding of the mechanisms by which Mecp2/MeCP2 expression (total and isoform-specific) is regulated in brain, and in specific cell types of the brain is critical. Moreover, it is still unclear how a single protein can perform opposing functions such as transcriptional activation and repression and how a single protein can bind to unmethylated and methylated DNA (5mC and 5hmC). Further, it is also important to investigate whether this multifunctional property can be explained by the nature of the two MeCP2 isoforms, the MeCP2 interacting protein partners or conformational changes of the protein. Most of the proposed therapeutic approaches are designed to be used in developing animal models. It is important to know whether any of these therapies can be studied in adult RTT patients. The next challenge would be to translate the knowledge obtained from animal models to human. Even though the mouse and human Mecp2/MECP2 are highly conserved, differences between mouse and human brain structure, immune system and response to environmental factors would challenge the knowledge translation.
Abbreviations
- 5hmC:
-
5-Hydroxymethylcytosine
- 5mC:
-
5-Methylcytosine
- ALC:
-
Acetyl-l-carnitine
- ASD:
-
Autism spectrum disorders
- BDNF:
-
Brain-derived neurotrophic factor
- CREB:
-
cAMP response element-binding protein
- CTD:
-
C-terminal domain
- DNMT:
-
DNA methyltransferases
- E:
-
Enhancer
- EE:
-
Environmental enrichment
- ENCODE:
-
Encyclopaedia of DNA elements
- FASD:
-
Fetal alcohol spectrum disorders
- GA:
-
Glatiramer acetate
- HDAC:
-
Histone deacetylases
- HMGN1:
-
High mobility group N1 protein
- HP1:
-
Heterochromatin binding protein 1
- ID:
-
Inter-domain
- IGF1:
-
Insulin-like growth factor 1
- iPSC:
-
Induced pluripotent stem cells
- IRAK1:
-
Interleukin-1 receptor associated kinase gene
- lncRNA:
-
Long noncoding RNAs
- MBD:
-
Methyl binding domain
- MeCP2:
-
Methyl CpG binding protein 2
- MeP:
-
Mecp2 promoter
- miRNAs:
-
MicroRNAs
- mnt:
-
Months
- MRE:
-
miRNA response elements
- NG:
-
Nodose cranial sensory ganglial cells
- NSCs:
-
Neural stem cells
- NTD:
-
N-terminal domain
- PTMs:
-
Posttranslational modifications
- RCP:
-
Red opsin gene
- RTT:
-
Rett syndrome
- S:
-
Silencer
- TM:
-
Tamoxifen
- TRD:
-
Transcription repression domain
- TSA:
-
Trichostatin A
- wks:
-
Weeks
- yr:
-
Years
- ω-3 PUFAs:
-
ω-3 polyunsaturated fatty acids
References
Abuhatzira, L., Makedonski, K., Kaufman, Y., Razin, A., & Shemer, R. (2007). MeCP2 deficiency in the brain decreases BDNF levels by REST/CoREST-mediated repression and increases TRKB production. Epigenetics, 2(4), 214–222.
Abuhatzira, L., Shamir, A., Schones, D. E., Schaffer, A. A., & Bustin, M. (2011). The chromatin-binding protein HMGN1 regulates the expression of methyl CpG-binding protein 2 (MECP2) and affects the behavior of mice. Journal of Biological Chemistry, 286(49), 42051–42062. doi:10.1074/jbc.M111.300541.
Acampa, M., & Guideri, F. (2006). Cardiac disease and Rett syndrome. Archives of Disease in Childhood, 91(5), 440–443. doi:10.1136/adc.2005.090290.
Adachi, M., Autry, A. E., Covington, H. E, 3rd, & Monteggia, L. M. (2009). MeCP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of Rett syndrome. Journal of Neuroscience, 29(13), 4218–4227. doi:10.1523/JNEUROSCI.4225-08.2009.
Adachi, M., Keefer, E. W., & Jones, F. S. (2005). A segment of the Mecp2 promoter is sufficient to drive expression in neurons. Human Molecular Genetics, 14(23), 3709–3722. doi:10.1093/hmg/ddi402.
Adams, V. H., McBryant, S. J., Wade, P. A., Woodcock, C. L., & Hansen, J. C. (2007). Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. Journal of Biological Chemistry, 282(20), 15057–15064. doi:10.1074/jbc.M700855200.
Agarwal, N., Becker, A., Jost, K. L., Haase, S., Thakur, B. K., Brero, A., et al. (2011). MeCP2 Rett mutations affect large scale chromatin organization. Human Molecular Genetics, 20(21), 4187–4195. doi:10.1093/hmg/ddr346.
Akhtar, M. W., Raingo, J., Nelson, E. D., Montgomery, R. L., Olson, E. N., Kavalali, E. T., et al. (2009). Histone deacetylases 1 and 2 form a developmental switch that controls excitatory synapse maturation and function. Journal of Neuroscience, 29(25), 8288–8297. doi:10.1523/JNEUROSCI.0097-09.2009.
Amir, R. E., Fang, P., Yu, Z., Glaze, D. G., Percy, A. K., Zoghbi, H. Y., et al. (2005). Mutations in exon 1 of MECP2 are a rare cause of Rett syndrome. Journal of Medical Genetics, 42(2), e15. doi:10.1136/jmg.2004.026161.
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., & Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics, 23(2), 185–188. doi:10.1038/13810.
Amir, R. E., & Zoghbi, H. Y. (2000). Rett syndrome: Methyl-CpG-binding protein 2 mutations and phenotype–genotype correlations. American Journal of Medical Genetics, 97(2), 147–152.
Asaka, Y., Jugloff, D. G., Zhang, L., Eubanks, J. H., & Fitzsimonds, R. M. (2006). Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiology of Diseases, 21(1), 217–227. doi:10.1016/j.nbd.2005.07.005.
Bagga, J. S., & D’Antonio, L. A. (2013). Role of conserved cis-regulatory elements in the post-transcriptional regulation of the human MECP2 gene involved in autism. Human Genomics, 7(1), 19. doi:10.1186/1479-7364-7-19.
Baker, S. A., Chen, L., Wilkins, A. D., Yu, P., Lichtarge, O., & Zoghbi, H. Y. (2013). An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell, 152(5), 984–996. doi:10.1016/j.cell.2013.01.038.
Ballas, N., Lioy, D. T., Grunseich, C., & Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nature Neuroscience, 12(3), 311–317. doi:10.1038/nn.2275.
Barber, B. A., Liyanage, V. R., Zachariah, R. M., Olson, C. O., Bailey, M. A., & Rastegar, M. (2013). Dynamic expression of MEIS1 homeoprotein in E14.5 forebrain and differentiated forebrain-derived neural stem cells. Annals of Anatomy, 195(5), 431–440. doi:10.1016/j.aanat.2013.04.005.
Barber, B. A., & Rastegar, M. (2010). Epigenetic control of Hox genes during neurogenesis, development, and disease. Annals of Anatomy, 192(5), 261–274. doi:10.1016/j.aanat.2010.07.009.
Bartholdi, D., Klein, A., Weissert, M., Koenig, N., Baumer, A., Boltshauser, E., et al. (2006). Clinical profiles of four patients with Rett syndrome carrying a novel exon 1 mutation or genomic rearrangement in the MECP2 gene. Clinical Genetics, 69(4), 319–326. doi:10.1111/j.1399-0004.2006.00604.x.
Baubec, T., Ivanek, R., Lienert, F., & Schubeler, D. (2013). Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell, 153(2), 480–492. doi:10.1016/j.cell.2013.03.011.
Belichenko, N. P., Belichenko, P. V., & Mobley, W. C. (2009a). Evidence for both neuronal cell autonomous and nonautonomous effects of methyl-CpG-binding protein 2 in the cerebral cortex of female mice with Mecp2 mutation. Neurobiology of Diseases, 34(1), 71–77. doi:10.1016/j.nbd.2008.12.016.
Belichenko, P. V., Wright, E. E., Belichenko, N. P., Masliah, E., Li, H. H., Mobley, W. C., et al. (2009b). Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: Evidence for disruption of neuronal networks. Journal of Comparative Neurology, 514(3), 240–258. doi:10.1002/cne.22009.
Ben-Zeev, B., Aharoni, R., Nissenkorn, A., & Arnon, R. (2011). Glatiramer acetate (GA, Copolymer-1) an hypothetical treatment option for Rett syndrome. Medical Hypotheses, 76(2), 190–193. doi:10.1016/j.mehy.2010.09.015.
Bernard, D., Gil, J., Dumont, P., Rizzo, S., Monte, D., Quatannens, B., et al. (2006). The methyl-CpG-binding protein MECP2 is required for prostate cancer cell growth. Oncogene, 25(9), 1358–1366. doi:10.1038/sj.onc.1209179.
Beyer, K. S., Blasi, F., Bacchelli, E., Klauck, S. M., Maestrini, E., & Poustka, A. (2002). Mutation analysis of the coding sequence of the MECP2 gene in infantile autism. Human Genetics, 111(4–5), 305–309. doi:10.1007/s00439-002-0786-3.
Bienvenu, T., Carrie, A., de Roux, N., Vinet, M. C., Jonveaux, P., Couvert, P., et al. (2000). MECP2 mutations account for most cases of typical forms of Rett syndrome. Human Molecular Genetics, 9(9), 1377–1384.
Bodda, C., Tantra, M., Mollajew, R., Arunachalam, J. P., Laccone, F. A., Can, K., et al. (2013). Mild overexpression of Mecp2 in mice causes a higher susceptibility toward seizures. American Journal of Pathology, 183(1), 195–210. doi:10.1016/j.ajpath.2013.03.019.
Brady, M. L., Diaz, M. R., Iuso, A., Everett, J. C., Valenzuela, C. F., & Caldwell, K. K. (2013). Moderate prenatal alcohol exposure reduces plasticity and alters NMDA receptor subunit composition in the dentate gyrus. Journal of Neuroscience, 33(3), 1062–1067. doi:10.1523/JNEUROSCI.1217-12.2013.
Brendel, C., Belakhov, V., Werner, H., Wegener, E., Gartner, J., Nudelman, I., et al. (2011). Readthrough of nonsense mutations in Rett syndrome: Evaluation of novel aminoglycosides and generation of a new mouse model. Journal of Molecular Medicine (Berlin), 89(4), 389–398. doi:10.1007/s00109-010-0704-4.
Brendel, C., Klahold, E., Gartner, J., & Huppke, P. (2009). Suppression of nonsense mutations in Rett syndrome by aminoglycoside antibiotics. Pediatric Research, 65(5), 520–523. doi:10.1203/PDR.0b013e31819d9ebc.
Brero, A., Easwaran, H. P., Nowak, D., Grunewald, I., Cremer, T., Leonhardt, H., et al. (2005). Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. Journal of Cell Biology, 169(5), 733–743. doi:10.1083/jcb.200502062.
Buchthal, B., Lau, D., Weiss, U., Weislogel, J. M., & Bading, H. (2012). Nuclear calcium signaling controls methyl-CpG-binding protein 2 (MeCP2) phosphorylation on serine 421 following synaptic activity. Journal of Biological Chemistry, 287(37), 30967–30974. doi:10.1074/jbc.M112.382507.
Budden, S. S., & Gunness, M. E. (2003). Possible mechanisms of osteopenia in Rett syndrome: Bone histomorphometric studies. Journal of Child Neurology, 18(10), 698–702.
Burmistrova, O. A., Goltsov, A. Y., Abramova, L. I., Kaleda, V. G., Orlova, V. A., & Rogaev, E. I. (2007). MicroRNA in schizophrenia: Genetic and expression analysis of miR-130b (22q11). Biochemistry (Moscow), 72(5), 578–582.
Byard, R. W. (2006). Forensic issues and possible mechanisms of sudden death in Rett syndrome. Journal of Clinical Forensic Medicine, 13(2), 96–99. doi:10.1016/j.jcfm.2005.08.013.
Calfa, G., Percy, A. K., & Pozzo-Miller, L. (2011). Experimental models of Rett syndrome based on Mecp2 dysfunction. Experimental Biology and Medicine (Maywood), 236(1), 3–19. doi:10.1258/ebm.2010.010261.
Carney, R. M., Wolpert, C. M., Ravan, S. A., Shahbazian, M., Ashley-Koch, A., Cuccaro, M. L., et al. (2003). Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatric Neurology, 28(3), 205–211.
Carter, P., Downs, J., Bebbington, A., Williams, S., Jacoby, P., Kaufmann, W. E., et al. (2010). Stereotypical hand movements in 144 subjects with Rett syndrome from the population-based Australian database. Movement Disorders, 25(3), 282–288. doi:10.1002/mds.22851.
Cartron, P. F., Nadaradjane, A., Lepape, F., Lalier, L., Gardie, B., & Vallette, F. M. (2013). Identification of TET1 partners that control its DNA-demethylating function. Genes Cancer, 4(5–6), 235–241. doi:10.1177/1947601913489020.
Chadwick, L. H., & Wade, P. A. (2007). MeCP2 in Rett syndrome: Transcriptional repressor or chromatin architectural protein? Current Opinion in Genetics & Development, 17(2), 121–125. doi:10.1016/j.gde.2007.02.003.
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science, 320(5880), 1224–1229. doi:10.1126/science.1153252.
Chahrour, M., & Zoghbi, H. Y. (2007). The story of Rett syndrome: From clinic to neurobiology. Neuron, 56(3), 422–437. doi:10.1016/j.neuron.2007.10.001.
Chang, Q., Khare, G., Dani, V., Nelson, S., & Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron, 49(3), 341–348. doi:10.1016/j.neuron.2005.12.027.
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature, 468(7321), 263–269. doi:10.1038/nature09582.
Chao, H. T., Zoghbi, H. Y., & Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron, 56(1), 58–65. doi:10.1016/j.neuron.2007.08.018.
Chapleau, C. A., Calfa, G. D., Lane, M. C., Albertson, A. J., Larimore, J. L., Kudo, S., et al. (2009). Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiology of Diseases, 35(2), 219–233. doi:10.1016/j.nbd.2009.05.001.
Chen, R. Z., Akbarian, S., Tudor, M., & Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nature Genetics, 27(3), 327–331. doi:10.1038/85906.
Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A. E., Griffith, E. C., et al. (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science, 302(5646), 885–889. doi:10.1126/science.1086446.
Chen, Y., Ozturk, N. C., & Zhou, F. C. (2013). DNA methylation program in developing hippocampus and its alteration by alcohol. PLoS ONE, 8(3), e60503. doi:10.1371/journal.pone.0060503.
Cheng, J., Huang, M., Zhu, Y., Xin, Y. J., Zhao, Y. K., Huang, J., et al. (2013). SUMOylation of MeCP2 is essential for transcriptional repression and hippocampal synapse development. Journal of Neurochemistry,. doi:10.1111/jnc.12523.
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science, 325(5942), 834–840. doi:10.1126/science.1175371.
Christodoulou, J., Grimm, A., Maher, T., & Bennetts, B. (2003). RettBASE: The IRSA MECP2 variation database—A new mutation database in evolution. Human Mutation, 21(5), 466–472. doi:10.1002/humu.10194.
Chunshu, Y., Endoh, K., Soutome, M., Kawamura, R., & Kubota, T. (2006). A patient with classic Rett syndrome with a novel mutation in MECP2 exon 1. Clinical Genetics, 70(6), 530–531. doi:10.1111/j.1399-0004.2006.00712.x.
Cirignotta, F., Lugaresi, E., & Montagna, P. (1986). Breathing impairment in Rett syndrome. American Journal of Medical Genetics, Supplement 1, 167–173.
Cohen, S., Gabel, H. W., Hemberg, M., Hutchinson, A. N., Sadacca, L. A., Ebert, D. H., et al. (2011). Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron, 72(1), 72–85. doi:10.1016/j.neuron.2011.08.022.
Cohen, D., Lazar, G., Couvert, P., Desportes, V., Lippe, D., Mazet, P., et al. (2002). MECP2 mutation in a boy with language disorder and schizophrenia. American Journal of Psychiatry, 159(1), 148–149.
Collins, A. L., Levenson, J. M., Vilaythong, A. P., Richman, R., Armstrong, D. L., Noebels, J. L., et al. (2004). Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Human Molecular Genetics, 13(21), 2679–2689. doi:10.1093/hmg/ddh282.
Coutinho, A. M., Oliveira, G., Katz, C., Feng, J., Yan, J., Yang, C., et al. (2007). MECP2 coding sequence and 3′UTR variation in 172 unrelated autistic patients. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 144B(4), 475–483. doi:10.1002/ajmg.b.30490.
Coy, J. F., Sedlacek, Z., Bachner, D., Delius, H., & Poustka, A. (1999). A complex pattern of evolutionary conservation and alternative polyadenylation within the long 3″-untranslated region of the methyl-CpG-binding protein 2 gene (MeCP2) suggests a regulatory role in gene expression. Human Molecular Genetics, 8(7), 1253–1262.
Craig, J. M., Earle, E., Canham, P., Wong, L. H., Anderson, M., & Choo, K. H. (2003). Analysis of mammalian proteins involved in chromatin modification reveals new metaphase centromeric proteins and distinct chromosomal distribution patterns. Human Molecular Genetics, 12(23), 3109–3121. doi:10.1093/hmg/ddg330.
Cukier, H. N., Perez, A. M., Collins, A. L., Zhou, Z., Zoghbi, H. Y., & Botas, J. (2008). Genetic modifiers of MeCP2 function in Drosophila. PLoS Genetics, 4(9), e1000179. doi:10.1371/journal.pgen.1000179.
Cusack, S. M., Rohn, T. T., Medeck, R. J., Irwin, K. M., Brown, R. J., Mercer, L. M., et al. (2004). Suppression of MeCP2β expression inhibits neurite extension in PC12 cells. Experimental Cell Research, 299(2), 442–453. doi:10.1016/j.yexcr.2004.05.035.
Dastidar, S. G., Bardai, F. H., Ma, C., Price, V., Rawat, V., Verma, P., et al. (2012). Isoform-specific toxicity of Mecp2 in postmitotic neurons: Suppression of neurotoxicity by FoxG1. Journal of Neuroscience, 32(8), 2846–2855. doi:10.1523/JNEUROSCI.5841-11.2012.
De Felice, C., Signorini, C., Durand, T., Ciccoli, L., Leoncini, S., D’Esposito, M., et al. (2012). Partial rescue of Rett syndrome by omega-3 polyunsaturated fatty acids (PUFAs) oil. Genes & Nutrition, 7(3), 447–458. doi:10.1007/s12263-012-0285-7.
Delcuve, G. P., Rastegar, M., & Davie, J. R. (2009). Epigenetic control. Journal of Cellular Physiology, 219(2), 243–250. doi:10.1002/jcp.21678.
Della Ragione, F., Filosa, S., Scalabri, F., & D’Esposito, M. (2012). MeCP2 as a genome-wide modulator: The renewal of an old story. Frontiers in Genetics, 3, 181. doi:10.3389/fgene.2012.00181.
Deogracias, R., Yazdani, M., Dekkers, M. P., Guy, J., Ionescu, M. C., Vogt, K. E., et al. (2012). Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proceedings of the National Academy of Sciences USA, 109(35), 14230–14235. doi:10.1073/pnas.1206093109.
Dephoure, N., Zhou, C., Villen, J., Beausoleil, S. A., Bakalarski, C. E., Elledge, S. J., et al. (2008). A quantitative atlas of mitotic phosphorylation. Proceedings of the National Academy of Sciences USA, 105(31), 10762–10767. doi:10.1073/pnas.0805139105.
Derecki, N. C., Cronk, J. C., Lu, Z., Xu, E., Abbott, S. B., Guyenet, P. G., et al. (2012). Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature, 484(7392), 105–109. doi:10.1038/nature10907.
D’Esposito, M., Quaderi, N. A., Ciccodicola, A., Bruni, P., Esposito, T., D’Urso, M., et al. (1996). Isolation, physical mapping, and northern analysis of the X-linked human gene encoding methyl CpG-binding protein, MECP2. Mammalian Genome, 7(7), 533–535.
Dragich, J. M., Kim, Y. H., Arnold, A. P., & Schanen, N. C. (2007). Differential distribution of the MeCP2 splice variants in the postnatal mouse brain. Journal of Comparative Neurology, 501(4), 526–542. doi:10.1002/cne.21264.
Ebert, D. H., Gabel, H. W., Robinson, N. D., Kastan, N. R., Hu, L. S., Cohen, S., et al. (2013). Activity-dependent phosphorylation of MECP2 threonine 308 regulates interaction with NcoR. Nature,. doi:10.1038/nature12348.
Evans, J. C., Archer, H. L., Colley, J. P., Ravn, K., Nielsen, J. B., Kerr, A., et al. (2005a). Early onset seizures and Rett-like features associated with mutations in CDKL5. European Journal of Human Genetics, 13(10), 1113–1120. doi:10.1038/sj.ejhg.5201451.
Evans, J. C., Archer, H. L., Whatley, S. D., Kerr, A., Clarke, A., & Butler, R. (2005b). Variation in exon 1 coding region and promoter of MECP2 in Rett syndrome and controls. European Journal of Human Genetics, 13(1), 124–126. doi:10.1038/sj.ejhg.5201270.
Ezeonwuka, C., & Rastegar, M. (2014). MeCP2-related diseases and animal models. Diseases, 2(1), 45–70.
Fichou, Y., Nectoux, J., Bahi-Buisson, N., Rosas-Vargas, H., Girard, B., Chelly, J., et al. (2009). The first missense mutation causing Rett syndrome specifically affecting the MeCP2_e1 isoform. Neurogenetics, 10(2), 127–133. doi:10.1007/s10048-008-0161-1.
Franklin, T. B., Russig, H., Weiss, I. C., Graff, J., Linder, N., Michalon, A., et al. (2010). Epigenetic transmission of the impact of early stress across generations. Biological Psychiatry, 68(5), 408–415. doi:10.1016/j.biopsych.2010.05.036.
Fukuda, T., Itoh, M., Ichikawa, T., Washiyama, K., & Goto, Y. (2005). Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. Journal of Neuropathology and Experimental Neurology, 64(6), 537–544.
Fukushige, S., & Horii, A. (2013). DNA methylation in cancer: A gene silencing mechanism and the clinical potential of its biomarkers. Tohoku Journal of Experimental Medicine, 229(3), 173–185.
Fyffe, S. L., Neul, J. L., Samaco, R. C., Chao, H. T., Ben-Shachar, S., Moretti, P., et al. (2008). Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron, 59(6), 947–958. doi:10.1016/j.neuron.2008.07.030.
Garg, S. K., Lioy, D. T., Cheval, H., McGann, J. C., Bissonnette, J. M., Murtha, M. J., et al. (2013). Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. Journal of Neuroscience, 33(34), 13612–13620. doi:10.1523/JNEUROSCI.1854-13.2013.
Gemelli, T., Berton, O., Nelson, E. D., Perrotti, L. I., Jaenisch, R., & Monteggia, L. M. (2006). Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biological Psychiatry, 59(5), 468–476. doi:10.1016/j.biopsych.2005.07.025.
Georgel, P. T., Horowitz-Scherer, R. A., Adkins, N., Woodcock, C. L., Wade, P. A., & Hansen, J. C. (2003). Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation. Journal of Biological Chemistry, 278(34), 32181–32188. doi:10.1074/jbc.M305308200.
Ghosh, R. P., Horowitz-Scherer, R. A., Nikitina, T., Shlyakhtenko, L. S., & Woodcock, C. L. (2010a). MeCP2 binds cooperatively to its substrate and competes with histone H1 for chromatin binding sites. Molecular and Cellular Biology, 30(19), 4656–4670. doi:10.1128/MCB.00379-10.
Ghosh, R. P., Nikitina, T., Horowitz-Scherer, R. A., Gierasch, L. M., Uversky, V. N., Hite, K., et al. (2010b). Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry, 49(20), 4395–4410. doi:10.1021/bi9019753.
Giacometti, E., Luikenhuis, S., Beard, C., & Jaenisch, R. (2007). Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proceedings of the National Academy of Sciences USA, 104(6), 1931–1936. doi:10.1073/pnas.0610593104.
Gianakopoulos, P. J., Zhang, Y., Pencea, N., Orlic-Milacic, M., Mittal, K., Windpassinger, C., et al. (2012). Mutations in MECP2 exon 1 in classical Rett patients disrupt MECP2_e1 transcription, but not transcription of MECP2_e2. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 159B(2), 210–216. doi:10.1002/ajmg.b.32015.
Gibson, J. H., Williamson, S. L., Arbuckle, S., & Christodoulou, J. (2005). X chromosome inactivation patterns in brain in Rett syndrome: Implications for the disease phenotype. Brain Development, 27(4), 266–270. doi:10.1016/j.braindev.2004.07.002.
Gillberg, C. (1986). Autism and Rett syndrome: Some notes on differential diagnosis. American Journal of Medical Genetics, Supplement 1, 127–131.
Goffin, D., Allen, M., Zhang, L., Amorim, M., Wang, I. T .J., Reyes, A. -R. S., et al. (2011). Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nature Neuroscience, 15(2), 274–283. http://www.nature.com/neuro/journal/v15/n2/abs/nn.2997.html#supplementary-information.
Gonzales, M. L., Adams, S., Dunaway, K. W., & LaSalle, J. M. (2012). Phosphorylation of distinct sites in MeCP2 modifies cofactor associations and the dynamics of transcriptional regulation. Molecular and Cellular Biology, 32(14), 2894–2903. doi:10.1128/MCB.06728-11.
Guideri, F., & Acampa, M. (2005). Sudden death and cardiac arrhythmias in Rett syndrome. Pediatric Cardiology, 26(1), 111. doi:10.1007/s00246-004-0701-x.
Guideri, F., Acampa, M., Blardi, P., de Lalla, A., Zappella, M., & Hayek, Y. (2004). Cardiac dysautonomia and serotonin plasma levels in Rett syndrome. Neuropediatrics, 35(1), 36–38. doi:10.1055/s-2004-815789.
Guo, Y., Chen, Y., Carreon, S., & Qiang, M. (2012). Chronic intermittent ethanol exposure and its removal induce a different miRNA expression pattern in primary cortical neuronal cultures. Alcoholism, Clinical and Experimental Research, 36(6), 1058–1066. doi:10.1111/j.1530-0277.2011.01689.x.
Guy, J., Cheval, H., Selfridge, J., & Bird, A. (2011). The role of MeCP2 in the brain. Annual Review of Cell and Developmental Biology, 27, 631–652. doi:10.1146/annurev-cellbio-092910-154121.
Guy, J., Gan, J., Selfridge, J., Cobb, S., & Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science, 315(5815), 1143–1147. doi:10.1126/science.1138389.
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., & Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nature Genetics, 27(3), 322–326. doi:10.1038/85899.
Hagberg, B. (2002). Clinical manifestations and stages of Rett syndrome. Mental Retardation and Developmental Disabilities, 8(2), 61–65. doi:10.1002/mrdd.10020.
Hagberg, B., Aicardi, J., Dias, K., & Ramos, O. (1983). A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Annals of Neurology, 14(4), 471–479. doi:10.1002/ana.410140412.
Hagebeuk, E. E., Koelman, J. H., Duran, M., Abeling, N. G., Vyth, A., & Poll-The, B. T. (2011). Clinical and electroencephalographic effects of folinic acid treatment in Rett syndrome patients. Journal of Child Neurology, 26(6), 718–723. doi:10.1177/0883073810390037.
Han, K., Gennarino, V. A., Lee, Y., Pang, K., Hashimoto-Torii, K., Choufani, S., et al. (2013). Human-specific regulation of MeCP2 levels in fetal brains by microRNA miR-483-5p. Genes & Development, 27(5), 485–490. doi:10.1101/gad.207456.112.
Hansen, J. C., Ghosh, R. P., & Woodcock, C. L. (2010). Binding of the Rett syndrome protein, MeCP2, to methylated and unmethylated DNA and chromatin. IUBMB Life, 62(10), 732–738. doi:10.1002/iub.386.
Hashimoto, Y., Akiyama, Y., & Yuasa, Y. (2013). Multiple-to-multiple relationships between microRNAs and target genes in gastric cancer. PLoS ONE, 8(5), e62589. doi:10.1371/journal.pone.0062589.
Heilstedt, H. A., Shahbazian, M. D., & Lee, B. (2002). Infantile hypotonia as a presentation of Rett syndrome. American Journal of Medical Genetics, 111(3), 238–242. doi:10.1002/ajmg.10633.
Hite, K. C., Kalashnikova, A. A., & Hansen, J. C. (2012). Coil-to-helix transitions in intrinsically disordered methyl CpG binding protein 2 and its isolated domains. Protein Science, 21(4), 531–538. doi:10.1002/pro.2037.
Ho, K. L., McNae, I. W., Schmiedeberg, L., Klose, R. J., Bird, A. P., & Walkinshaw, M. D. (2008). MeCP2 binding to DNA depends upon hydration at methyl-CpG. Molecular Cell, 29(4), 525–531. doi:10.1016/j.molcel.2007.12.028.
Hoffbuhr, K., Devaney, J. M., LaFleur, B., Sirianni, N., Scacheri, C., Giron, J., et al. (2001). MeCP2 mutations in children with and without the phenotype of Rett syndrome. Neurology, 56(11), 1486–1495.
Hoffbuhr, K. C., Moses, L. M., Jerdonek, M. A., Naidu, S., & Hoffman, E. P. (2002). Associations between MeCP2 mutations, X-chromosome inactivation, and phenotype. Mental Retardation and Developmental Disabilities Research Reviews, 8(2), 99–105. doi:10.1002/mrdd.10026.
Holm, V. A., & King, H. A. (1990). Scoliosis in the Rett syndrome. Brain Development, 12(1), 151–153.
Horike, S., Cai, S., Miyano, M., Cheng, J. F., & Kohwi-Shigematsu, T. (2005). Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nature Genetics, 37(1), 31–40. doi:10.1038/ng1491.
Hung, M. S., & Shen, C. K. (2003). Eukaryotic methyl-CpG-binding domain proteins and chromatin modification. Eukaryotic Cell, 2(5), 841–846.
Huppke, P., Ohlenbusch, A., Brendel, C., Laccone, F., & Gartner, J. (2005). Mutation analysis of the HDAC 1, 2, 8 and CDKL5 genes in Rett syndrome patients without mutations in MECP2. American Journal of Medical Genetics Part A, 137(2), 136–138. doi:10.1002/ajmg.a.30764.
Hutchinson, A. N., Deng, J. V., Cohen, S., & West, A. E. (2012). Phosphorylation of MeCP2 at Ser421 contributes to chronic antidepressant action. Journal of Neuroscience, 32(41), 14355–14363. doi:10.1523/JNEUROSCI.2156-12.2012.
Im, H. I., Hollander, J. A., Bali, P., & Kenny, P. J. (2010). MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nature Neuroscience, 13(9), 1120–1127. doi:10.1038/nn.2615.
Isaacs, J. S., Murdock, M., Lane, J., & Percy, A. K. (2003). Eating difficulties in girls with Rett syndrome compared with other developmental disabilities. Journal of the American Dietetic Association, 103(2), 224–230. doi:10.1053/jada.2003.50026.
Ishii, T., Makita, Y., Ogawa, A., Amamiya, S., Yamamoto, M., Miyamoto, A., et al. (2001). The role of different X-inactivation pattern on the variable clinical phenotype with Rett syndrome. Brain Development, 23(Suppl 1), S161–S164.
Itoh, M., Tahimic, C. G., Ide, S., Otsuki, A., Sasaoka, T., Noguchi, S., et al. (2012). Methyl CpG-binding protein isoform MeCP2_e2 is dispensable for Rett syndrome phenotypes but essential for embryo viability and placenta development. Journal of Biological Chemistry, 287(17), 13859–13867. doi:10.1074/jbc.M111.309864.
Jan, M. M., Dooley, J. M., & Gordon, K. E. (1999). Male Rett syndrome variant: Application of diagnostic criteria. Pediatric Neurology, 20(3), 238–240.
Jedele, K. B. (2007). The overlapping spectrum of Rett and Angelman syndromes: A clinical review. Seminars in Pediatric Neurology, 14(3), 108–117. doi:10.1016/j.spen.2007.07.002.
Jentarra, G. M., Olfers, S. L., Rice, S. G., Srivastava, N., Homanics, G. E., Blue, M., et al. (2010). Abnormalities of cell packing density and dendritic complexity in the MeCP2 A140V mouse model of Rett syndrome/X-linked mental retardation. BMC Neuroscience, 11, 19. doi:10.1186/1471-2202-11-19.
Jin, J., Bao, X., Wang, H., Pan, H., Zhang, Y., & Wu, X. (2008). RNAi-induced down-regulation of Mecp2 expression in the rat brain. International Journal of Developmental Neuroscience, 26(5), 457–465. doi:10.1016/j.ijdevneu.2008.02.009.
Jugloff, D. G., Vandamme, K., Logan, R., Visanji, N. P., Brotchie, J. M., & Eubanks, J. H. (2008). Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. Human Molecular Genetics, 17(10), 1386–1396. doi:10.1093/hmg/ddn026.
Julu, P. O., Kerr, A. M., Hansen, S., Apartopoulos, F., & Jamal, G. A. (1997). Immaturity of medullary cardiorespiratory neurones leading to inappropriate autonomic reactions as a likely cause of sudden death in Rett’s syndrome. Archives of Disease in Childhood, 77(5), 464–465.
Katz, D. M., Dutschmann, M., Ramirez, J. M., & Hilaire, G. (2009). Breathing disorders in Rett syndrome: Progressive neurochemical dysfunction in the respiratory network after birth. Respiratory Physiology & Neurobiology, 168(1–2), 101–108. doi:10.1016/j.resp.2009.04.017.
Kernohan, K. D., Jiang, Y., Tremblay, D. C., Bonvissuto, A. C., Eubanks, J. H., Mann, M. R., et al. (2010). ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Developmental Cell, 18(2), 191–202. doi:10.1016/j.devcel.2009.12.017.
Kerr, B., Silva, P. A., Walz, K., & Young, J. I. (2010). Unconventional transcriptional response to environmental enrichment in a mouse model of Rett syndrome. PLoS ONE, 5(7), e11534. doi:10.1371/journal.pone.0011534.
Kerr, B., Soto, C. J., Saez, M., Abrams, A., Walz, K., & Young, J. I. (2012). Transgenic complementation of MeCP2 deficiency: Phenotypic rescue of Mecp2-null mice by isoform-specific transgenes. European Journal of Human Genetics, 20(1), 69–76. doi:10.1038/ejhg.2011.145.
Kim, K. Y., Hysolli, E., & Park, I. H. (2011). Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proceedings of the National Academy of Sciences USA, 108(34), 14169–14174. doi:10.1073/pnas.1018979108.
Kim, P., Park, J. H., Choi, C. S., Choi, I., Joo, S. H., Kim, M. K., et al. (2013). Effects of ethanol exposure during early pregnancy in hyperactive, inattentive and impulsive behaviors and MeCP2 expression in rodent offspring. Neurochemical Research, 38(3), 620–631. doi:10.1007/s11064-012-0960-5.
Kim, B., Rincon Castro, L. M., Jawed, S., & Niles, L. P. (2008). Clinically relevant concentrations of valproic acid modulate melatonin MT(1) receptor, HDAC and MeCP2 mRNA expression in C6 glioma cells. European Journal of Pharmacology, 589(1–3), 45–48. doi:10.1016/j.ejphar.2008.04.058.
Kishi, N., & Macklis, J. D. (2004). MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Molecular and Cellular Neuroscience, 27(3), 306–321. doi:10.1016/j.mcn.2004.07.006.
Kishi, N., & Macklis, J. D. (2010). MeCP2 functions largely cell-autonomously, but also non-cell-autonomously, in neuronal maturation and dendritic arborization of cortical pyramidal neurons. Experimental Neurology, 222(1), 51–58. doi:10.1016/j.expneurol.2009.12.007.
Klauck, S. M., Lindsay, S., Beyer, K. S., Splitt, M., Burn, J., & Poustka, A. (2002). A mutation hot spot for nonspecific X-linked mental retardation in the MECP2 gene causes the PPM-X syndrome. American Journal of Human Genetics, 70(4), 1034–1037. doi:10.1086/339553.
Klein, M. E., Lioy, D. T., Ma, L., Impey, S., Mandel, G., & Goodman, R. H. (2007). Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nature Neuroscience, 10(12), 1513–1514. doi:10.1038/nn2010.
Kline, D. D., Ogier, M., Kunze, D. L., & Katz, D. M. (2010). Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. Journal of Neuroscience, 30(15), 5303–5310. doi:10.1523/JNEUROSCI.5503-09.2010.
Kondo, M., Gray, L. J., Pelka, G. J., Christodoulou, J., Tam, P. P., & Hannan, A. J. (2008). Environmental enrichment ameliorates a motor coordination deficit in a mouse model of Rett syndrome–Mecp2 gene dosage effects and BDNF expression. European Journal of Neuroscience, 27(12), 3342–3350. doi:10.1111/j.1460-9568.2008.06305.x.
Kriaucionis, S., & Bird, A. (2004). The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Research, 32(5), 1818–1823. doi:10.1093/nar/gkh349.
Kudo, S., Nomura, Y., Segawa, M., Fujita, N., Nakao, M., Hammer, S., et al. (2002). Functional characterisation of MeCP2 mutations found in male patients with X linked mental retardation. Journal of Medical Genetics, 39(2), 132–136.
Kudo, S., Nomura, Y., Segawa, M., Fujita, N., Nakao, M., Schanen, C., et al. (2003). Heterogeneity in residual function of MeCP2 carrying missense mutations in the methyl CpG binding domain. Journal of Medical Genetics, 40(7), 487–493.
Kuhn, D. E., Nuovo, G. J., Terry, A. V, Jr, Martin, M. M., Malana, G. E., Sansom, S. E., et al. (2010). Chromosome 21-derived microRNAs provide an etiological basis for aberrant protein expression in human Down syndrome brains. Journal of Biological Chemistry, 285(2), 1529–1543. doi:10.1074/jbc.M109.033407.
Kumar, A., Kamboj, S., Malone, B. M., Kudo, S., Twiss, J. L., Czymmek, K. J., et al. (2008). Analysis of protein domains and Rett syndrome mutations indicate that multiple regions influence chromatin-binding dynamics of the chromatin-associated protein MECP2 in vivo. Journal of Cell Science, 121(Pt 7), 1128–1137. doi:10.1242/jcs.016865.
Kuwano, Y., Kamio, Y., Kawai, T., Katsuura, S., Inada, N., Takaki, A., et al. (2011). Autism-associated gene expression in peripheral leucocytes commonly observed between subjects with autism and healthy women having autistic children. PLoS ONE, 6(9), e24723. doi:10.1371/journal.pone.0024723.
Lam, C. W., Yeung, W. L., Ko, C. H., Poon, P. M., Tong, S. F., Chan, K. Y., et al. (2000). Spectrum of mutations in the MECP2 gene in patients with infantile autism and Rett syndrome. Journal of Medical Genetics, 37(12), E41.
Larimore, J. L., Chapleau, C. A., Kudo, S., Theibert, A., Percy, A. K., & Pozzo-Miller, L. (2009). Bdnf overexpression in hippocampal neurons prevents dendritic atrophy caused by Rett-associated MECP2 mutations. Neurobiology of Diseases, 34(2), 199–211. doi:10.1016/j.nbd.2008.12.011.
Lawson-Yuen, A., Liu, D., Han, L., Jiang, Z. I., Tsai, G. E., Basu, A. C., et al. (2007). Ube3a mRNA and protein expression are not decreased in Mecp2R168X mutant mice. Brain Research, 1180, 1–6. doi:10.1016/j.brainres.2007.08.039.
Lee, J., Leonard, H., Piek, J., & Downs, J. (2013). Early development and regression in Rett syndrome. Clinical Genetics,. doi:10.1111/cge.12110.
Lewis, C. R., Staudinger, K., Scheck, L., & Olive, M. F. (2013). The effects of maternal separation on adult methamphetamine self-administration, extinction, reinstatement, and MeCP2 immunoreactivity in the nucleus accumbens. Frontiers in Psychiatry, 4, 55. doi:10.3389/fpsyt.2013.00055.
Li, Y., Wang, H., Muffat, J., Cheng, A. W., Orlando, D. A., Loven, J., et al. (2013). Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell, 13(4), 446–458. doi:10.1016/j.stem.2013.09.001.
Li, H., Zhong, X., Chau, K. F., Williams, E. C., & Chang, Q. (2011). Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nature Neuroscience, 14(8), 1001–1008. doi:10.1038/nn.2866.
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., et al. (2011). A role for glia in the progression of Rett’s syndrome. Nature, 475(7357), 497–500. doi:10.1038/nature10214.
Liu, J., & Francke, U. (2006). Identification of cis-regulatory elements for MECP2 expression. Human Molecular Genetics, 15(11), 1769–1782. doi:10.1093/hmg/ddl099.
Liyanage, V. R. B., Zachariah, R. M., Delcuve, G. P., Davie, J. R., & Rastegar, M. (2012). New developments in chromatin research: An epigenetic perspective. In N. M. Simpson & V. J. Stewart (Eds.), New developments in chromatin research (pp. 29–58). New York: Nova Science.
Liyanage, V., Zachariah, R., & Rastegar, M. (2013). Decitabine alters the expression of Mecp2 isoforms via dynamic DNA methylation at the Mecp2 regulatory elements in neural stem cells. Molecular Autism, 4(1), 46. doi:10.1186/2040-2392-4-46.
Loat, C. S., Curran, S., Lewis, C. M., Duvall, J., Geschwind, D., Bolton, P., et al. (2008). Methyl-CpG-binding protein 2 polymorphisms and vulnerability to autism. Genes, Brain and Behavior, 7(7), 754–760. doi:10.1111/j.1601-183X.2008.00414.x.
Lonetti, G., Angelucci, A., Morando, L., Boggio, E. M., Giustetto, M., & Pizzorusso, T. (2010). Early environmental enrichment moderates the behavioral and synaptic phenotype of MeCP2 null mice. Biological Psychiatry, 67(7), 657–665. doi:10.1016/j.biopsych.2009.12.022.
Long, S. W., Ooi, J. Y., Yau, P. M., & Jones, P. L. (2011). A brain-derived MeCP2 complex supports a role for MeCP2 in RNA processing. Bioscience Reports, 31(5), 333–343. doi:10.1042/BSR20100124.
Luikenhuis, S., Giacometti, E., Beard, C. F., & Jaenisch, R. (2004). Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proceedings of the National Academy of Sciences USA, 101(16), 6033–6038. doi:10.1073/pnas.0401626101.
Lusardi, T. A., Farr, C. D., Faulkner, C. L., Pignataro, G., Yang, T., Lan, J., et al. (2010). Ischemic preconditioning regulates expression of microRNAs and a predicted target, MeCP2, in mouse cortex. Journal of Cerebral Blood Flow and Metabolism, 30(4), 744–756. doi:10.1038/jcbfm.2009.253.
Lyst, M. J., Ekiert, R., Ebert, D. H., Merusi, C., Nowak, J., Selfridge, J., et al. (2013). Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nature Neuroscience, 16(7), 898–902. doi:10.1038/nn.3434.
Madan, N., Levine, M., Pourmoghadam, K., & Sokoloski, M. (2004). Severe sinus bradycardia in a patient with Rett syndrome: A new cause for a pause? Pediatric Cardiology, 25(1), 53–55. doi:10.1007/s00246-003-0341-6.
Maezawa, I., & Jin, L. W. (2010). Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. Journal of Neuroscience, 30(15), 5346–5356. doi:10.1523/JNEUROSCI.5966-09.2010.
Maezawa, I., Swanberg, S., Harvey, D., LaSalle, J. M., & Jin, L. W. (2009). Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. Journal of Neuroscience, 29(16), 5051–5061. doi:10.1523/JNEUROSCI.0324-09.2009.
Mao, L. M., Horton, E., Guo, M. L., Xue, B., Jin, D. Z., Fibuch, E. E., et al. (2011). Cocaine increases phosphorylation of MeCP2 in the rat striatum in vivo: A differential role of NMDA receptors. Neurochemistry International, 59(5), 610–617. doi:10.1016/j.neuint.2011.04.013.
Marchetto, M. C., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y., et al. (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell, 143(4), 527–539. doi:10.1016/j.cell.2010.10.016.
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., et al. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science, 302(5646), 890–893. doi:10.1126/science.1090842.
Matarazzo, V., Cohen, D., Palmer, A. M., Simpson, P. J., Khokhar, B., Pan, S. J., et al. (2004). The transcriptional repressor Mecp2 regulates terminal neuronal differentiation. Molecular and Cellular Neuroscience, 27(1), 44–58. doi:10.1016/j.mcn.2004.05.005.
Maunakea, A. K., Chepelev, I., Cui, K., & Zhao, K. (2013). Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Research,. doi:10.1038/cr.2013.110.
McCauley, M. D., Wang, T., Mike, E., Herrera, J., Beavers, D. L., Huang, T. W., et al. (2011). Pathogenesis of lethal cardiac arrhythmias in Mecp2 mutant mice: Implication for therapy in Rett syndrome. Science Translational Medicine, 3(113), 113ra125. doi:10.1126/scitranslmed.3002982.
McGraw, C. M., Samaco, R. C., & Zoghbi, H. Y. (2011). Adult neural function requires MeCP2. Science, 333(6039), 186. doi:10.1126/science.1206593.
Mellen, M., Ayata, P., Dewell, S., Kriaucionis, S., & Heintz, N. (2012). MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell, 151(7), 1417–1430. doi:10.1016/j.cell.2012.11.022.
Mirza, S., Sharma, G., Parshad, R., Gupta, S. D., Pandya, P., & Ralhan, R. (2013). Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. Journal of Breast Cancer, 16(1), 23–31. doi:10.4048/jbc.2013.16.1.23.
Miyake, K., & Nagai, K. (2007). Phosphorylation of methyl-CpG binding protein 2 (MeCP2) regulates the intracellular localization during neuronal cell differentiation. Neurochemistry International, 50(1), 264–270. doi:10.1016/j.neuint.2006.08.018.
Miyake, K., Yang, C., Minakuchi, Y., Ohori, K., Soutome, M., Hirasawa, T., et al. (2013). Comparison of genomic and epigenomic expression in monozygotic twins discordant for Rett syndrome. PLoS ONE, 8(6), e66729. doi:10.1371/journal.pone.0066729.
Mnatzakanian, G. N., Lohi, H., Munteanu, I., Alfred, S. E., Yamada, T., MacLeod, P. J., et al. (2004). A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nature Genetics, 36(4), 339–341. doi:10.1038/ng1327.
Moog, U., Smeets, E. E., van Roozendaal, K. E., Schoenmakers, S., Herbergs, J., Schoonbrood-Lenssen, A. M., et al. (2003). Neurodevelopmental disorders in males related to the gene causing Rett syndrome in females (MECP2). European Journal of Paediatric Neurology, 7(1), 5–12.
Morton, R. E., Bonas, R., Minford, J., Kerr, A., & Ellis, R. E. (1997). Feeding ability in Rett syndrome. Developmental Medicine and Child Neurology, 39(5), 331–335.
Motil, K. J., Caeg, E., Barrish, J. O., Geerts, S., Lane, J. B., Percy, A. K., et al. (2012). Gastrointestinal and nutritional problems occur frequently throughout life in girls and women with Rett syndrome. Journal of Pediatric Gastroenterology and Nutrition, 55(3), 292–298. doi:10.1097/MPG.0b013e31824b6159.
Muller, H. M., Fiegl, H., Goebel, G., Hubalek, M. M., Widschwendter, A., Muller-Holzner, E., et al. (2003). MeCP2 and MBD2 expression in human neoplastic and non-neoplastic breast tissue and its association with oestrogen receptor status. British Journal of Cancer, 89(10), 1934–1939. doi:10.1038/sj.bjc.6601392.
Murakami, J. W., Courchesne, E., Haas, R. H., Press, G. A., & Yeung-Courchesne, R. (1992). Cerebellar and cerebral abnormalities in Rett syndrome: A quantitative MR analysis. AJR. American Journal of Roentgenology, 159(1), 177–183. doi:10.2214/ajr.159.1.1609693.
Na, E. S., Nelson, E. D., Adachi, M., Autry, A. E., Mahgoub, M. A., Kavalali, E. T., et al. (2012). A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. Journal of Neuroscience, 32(9), 3109–3117. doi:10.1523/JNEUROSCI.6000-11.2012.
Na, E. S., Nelson, E. D., Kavalali, E. T., & Monteggia, L. M. (2013). The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology, 38(1), 212–219. doi:10.1038/npp.2012.116.
Nag, N., Mellott, T. J., & Berger-Sweeney, J. E. (2008). Effects of postnatal dietary choline supplementation on motor regional brain volume and growth factor expression in a mouse model of Rett syndrome. Brain Research, 1237, 101–109. doi:10.1016/j.brainres.2008.08.042.
Nagarajan, R. P., Hogart, A. R., Gwye, Y., Martin, M. R., & LaSalle, J. M. (2006). Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics, 1(4), e1–e11.
Nagarajan, R. P., Patzel, K. A., Martin, M., Yasui, D. H., Swanberg, S. E., Hertz-Picciotto, I., et al. (2008). MECP2 promoter methylation and X chromosome inactivation in autism. Autism Research, 1(3), 169–178. doi:10.1002/aur.24.
Nan, X., Hou, J., Maclean, A., Nasir, J., Lafuente, M. J., Shu, X., et al. (2007). Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proceedings of the National Academy of Sciences USA, 104(8), 2709–2714. doi:10.1073/pnas.0608056104.
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N., et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature, 393(6683), 386–389. doi:10.1038/30764.
Neul, J. L., & Zoghbi, H. Y. (2004). Rett syndrome: A prototypical neurodevelopmental disorder. Neuroscientist, 10(2), 118–128. doi:10.1177/1073858403260995.
Newnham, C. M., Hall-Pogar, T., Liang, S., Wu, J., Tian, B., Hu, J., et al. (2010). Alternative polyadenylation of MeCP2: Influence of cis-acting elements and trans-acting factors. RNA Biology, 7(3), 361–372.
Nguyen, M. V., Du, F., Felice, C. A., Shan, X., Nigam, A., Mandel, G., et al. (2012). MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. Journal of Neuroscience, 32(29), 10021–10034. doi:10.1523/JNEUROSCI.1316-12.2012.
Nguyen, M. V., Felice, C. A., Du, F., Covey, M. V., Robinson, J. K., Mandel, G., et al. (2013). Oligodendrocyte lineage cells contribute unique features to Rett syndrome neuropathology. Journal of Neuroscience, 33(48), 18764–18774. doi:10.1523/JNEUROSCI.2657-13.2013.
Nikitina, T., Ghosh, R. P., Horowitz-Scherer, R. A., Hansen, J. C., Grigoryev, S. A., & Woodcock, C. L. (2007a). MeCP2-chromatin interactions include the formation of chromatosome-like structures and are altered in mutations causing Rett syndrome. Journal of Biological Chemistry, 282(38), 28237–28245. doi:10.1074/jbc.M704304200.
Nikitina, T., Shi, X., Ghosh, R. P., Horowitz-Scherer, R. A., Hansen, J. C., & Woodcock, C. L. (2007b). Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Molecular and Cellular Biology, 27(3), 864–877. doi:10.1128/MCB.01593-06.
Nomura, T., Kimura, M., Horii, T., Morita, S., Soejima, H., Kudo, S., et al. (2008). MeCP2-dependent repression of an imprinted miR-184 released by depolarization. Human Molecular Genetics, 17(8), 1192–1199. doi:10.1093/hmg/ddn011.
Nomura, Y., & Segawa, M. (1992). Motor symptoms of the Rett syndrome: Abnormal muscle tone, posture, locomotion and stereotyped movement. Brain & Development, 14(Suppl), S21–S28.
Ogier, M., & Katz, D. M. (2008). Breathing dysfunction in Rett syndrome: Understanding epigenetic regulation of the respiratory network. Respiratory Physiology & Neurobiology, 164(1–2), 55–63. doi:10.1016/j.resp.2008.04.005.
Ogier, M., Wang, H., Hong, E., Wang, Q., Greenberg, M. E., & Katz, D. M. (2007). Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. Journal of Neuroscience, 27(40), 10912–10917. doi:10.1523/JNEUROSCI.1869-07.2007.
Okabe, Y., Takahashi, T., Mitsumasu, C., Kosai, K., Tanaka, E., & Matsuishi, T. (2012). Alterations of gene expression and glutamate clearance in astrocytes derived from an MeCP2-null mouse model of Rett syndrome. PLoS ONE, 7(4), e35354. doi:10.1371/journal.pone.0035354.
Olson, C. O., Zachariah, R. M., Ezeonwuka, C. D., Liyanage, V. R. B., & Rastegar, M. (2014). Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS ONE, 9(3), e90645. doi:10.1371/journal.pone.0090645.
Olsson, B., & Rett, A. (1987). Autism and Rett syndrome: Behavioural investigations and differential diagnosis. Developmental Medicine and Child Neurology, 29(4), 429–441.
Olynik, B. M., & Rastegar, M. (2012). The genetic and epigenetic journey of embryonic stem cells into mature neural cells. Frontiers in Genetics, 3, 81. doi:10.3389/fgene.2012.00081.
Panighini, A., Duranti, E., Santini, F., Maffei, M., Pizzorusso, T., Funel, N., et al. (2013). Vascular dysfunction in a mouse model of rett syndrome and effects of curcumin treatment. PLoS ONE, 8(5), e64863. doi:10.1371/journal.pone.0064863.
Parry, L., & Clarke, A. R. (2011). The roles of the Methyl-CpG binding proteins in cancer. Genes & Cancer, 2(6), 618–630. doi:10.1177/1947601911418499.
Pelka, G. J., Watson, C. M., Christodoulou, J., & Tam, P. P. (2005). Distinct expression profiles of Mecp2 transcripts with different lengths of 3′UTR in the brain and visceral organs during mouse development. Genomics, 85(4), 441–452. doi:10.1016/j.ygeno.2004.12.002.
Pelka, G. J., Watson, C. M., Radziewic, T., Hayward, M., Lahooti, H., Christodoulou, J., et al. (2006). Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain, 129(Pt 4), 887–898. doi:10.1093/brain/awl022.
Petazzi, P., Sandoval, J., Szczesna, K., Jorge, O. C., Roa, L., Sayols, S., et al. (2013). Dysregulation of the long non-coding RNA transcriptome in a Rett syndrome mouse model. RNA Biology, 10(7), 1197–1203. doi:10.4161/rna.24286.
Petel-Galil, Y., Benteer, B., Galil, Y. P., Zeev, B. B., Greenbaum, I., Vecsler, M., et al. (2006). Comprehensive diagnosis of Rett’s syndrome relying on genetic, epigenetic and expression evidence of deficiency of the methyl-CpG-binding protein 2 gene: Study of a cohort of Israeli patients. Journal of Medical Genetics, 43(12), e56. doi:10.1136/jmg.2006.041285.
Peters, S. U., Hundley, R. J., Wilson, A. K., Warren, Z., Vehorn, A., Carvalho, C. M., et al. (2013). The behavioral phenotype in MECP2 duplication syndrome: A comparison with idiopathic autism. Autism Research, 6(1), 42–50. doi:10.1002/aur.1262.
Philippe, C., Amsallem, D., Francannet, C., Lambert, L., Saunier, A., Verneau, F., et al. (2010). Phenotypic variability in Rett syndrome associated with FOXG1 mutations in females. Journal of Medical Genetics, 47(1), 59–65. doi:10.1136/jmg.2009.067355.
Pietri, T., Roman, A. C., Guyon, N., Romano, S. A., Washbourne, P., Moens, C. B., et al. (2013). The first mecp2-null zebrafish model shows altered motor behaviors. Frontiers in Neural Circuits, 7, 118. doi:10.3389/fncir.2013.00118.
Pini, G., Scusa, M. F., Congiu, L., Benincasa, A., Morescalchi, P., Bottiglioni, I., et al. (2012). IGF1 as a potential treatment for Rett syndrome: Safety assessment in six Rett patients. Autism Research and Treatment, 2012, 679801. doi:10.1155/2012/679801.
Popescu, A. C., Sidorova, E., Zhang, G., & Eubanks, J. H. (2010). Aminoglycoside-mediated partial suppression of MECP2 nonsense mutations responsible for Rett syndrome in vitro. Journal of Neuroscience Research, 88(11), 2316–2324. doi:10.1002/jnr.22409.
Qiu, Z., Sylwestrak, E. L., Lieberman, D. N., Zhang, Y., Liu, X. Y., & Ghosh, A. (2012). The Rett syndrome protein MeCP2 regulates synaptic scaling. Journal of Neuroscience, 32(3), 989–994. doi:10.1523/JNEUROSCI.0175-11.2012.
Quenard, A., Yilmaz, S., Fontaine, H., Bienvenu, T., Moncla, A., des Portes, V., et al. (2006). Deleterious mutations in exon 1 of MECP2 in Rett syndrome. European Journal of Medical Genetics, 49(4), 313–322. doi:10.1016/j.ejmg.2005.11.002.
Ramirez, J. M., Ward, C. S., & Neul, J. L. (2013). Breathing challenges in Rett Syndrome: Lessons learned from humans and animal models. Respiratory Physiology & Neurobiology,. doi:10.1016/j.resp.2013.06.022.
Rapin, I., & Tuchman, R. F. (2008). Autism: Definition, neurobiology, screening, diagnosis. Pediatric Clinics of North America, 55(5), 1129–1146. doi:10.1016/j.pcl.2008.07.005.
Rastegar, M., Hotta, A., Pasceri, P., Makarem, M., Cheung, A. Y., Elliott, S., et al. (2009). MECP2 isoform-specific vectors with regulated expression for Rett syndrome gene therapy. PLoS ONE, 4(8), e6810. doi:10.1371/journal.pone.0006810.
Ravn, K., Nielsen, J. B., & Schwartz, M. (2005). Mutations found within exon 1 of MECP2 in Danish patients with Rett syndrome. Clinical Genetics, 67(6), 532–533. doi:10.1111/j.1399-0004.2005.00444.x.
Ray, B. K., Dhar, S., Henry, C., Rich, A., & Ray, A. (2013). Epigenetic regulation by Z-DNA silencer function controls cancer-associated ADAM-12 expression in breast cancer: Cross-talk between MeCP2 and NF1 transcription factor family. Cancer Research, 73(2), 736–744. doi:10.1158/0008-5472.CAN-12-2601.
Reeves, R., & Nissen, M. S. (1990). The AT-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. Journal of Biological Chemistry, 265(15), 8573–8582.
Reichwald, K., Thiesen, J., Wiehe, T., Weitzel, J., Poustka, W. A., Rosenthal, A., et al. (2000). Comparative sequence analysis of the MECP2-locus in human and mouse reveals new transcribed regions. Mammalian Genome, 11(3), 182–190.
Reiss, A. L., Faruque, F., Naidu, S., Abrams, M., Beaty, T., Bryan, R. N., et al. (1993). Neuroanatomy of Rett syndrome: A volumetric imaging study. Annals of Neurology, 34(2), 227–234. doi:10.1002/ana.410340220.
Renieri, A., Meloni, I., Longo, I., Ariani, F., Mari, F., Pescucci, C., et al. (2003). Rett syndrome: The complex nature of a monogenic disease. Journal of Molecular Medicine (Berlin), 81(6), 346–354. doi:10.1007/s00109-003-0444-9.
Repunte-Canonigo, V., Chen, J., Lefebvre, C., Kawamura, T., Kreifeldt, M., Basson, O., et al. (2013). MeCP2 regulates ethanol sensitivity and intake. Addiction Biology,. doi:10.1111/adb.12047.
Rett, A. (1966). On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wiener Medizinische Wochenschrift, 116(37), 723–726.
Ricceri, L., De Filippis, B., & Laviola, G. (2008). Mouse models of Rett syndrome: From behavioural phenotyping to preclinical evaluation of new therapeutic approaches. Behavioural Pharmacology, 19(5–6), 501–517. doi:10.1097/FBP.0b013e32830c3645.
Ricceri, L., De Filippis, B., & Laviola, G. (2013). Rett syndrome treatment in mouse models: Searching for effective targets and strategies. Neuropharmacology, 68, 106–115. doi:10.1016/j.neuropharm.2012.08.010.
Ricciardi, S., Boggio, E. M., Grosso, S., Lonetti, G., Forlani, G., Stefanelli, G., et al. (2011). Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Human Molecular Genetics, 20(6), 1182–1196. doi:10.1093/hmg/ddq563.
Romano-Lopez, A., Mendez-Diaz, M., Ruiz-Contreras, A. E., Carrisoza, R., & Prospero-Garcia, O. (2012). Maternal separation and proclivity for ethanol intake: A potential role of the endocannabinoid system in rats. Neuroscience, 223, 296–304. doi:10.1016/j.neuroscience.2012.07.071.
Roux, J. C., Dura, E., Moncla, A., Mancini, J., & Villard, L. (2007). Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. European Journal of Neuroscience, 25(7), 1915–1922. doi:10.1111/j.1460-9568.2007.05466.x.
Roze, E., Cochen, V., Sangla, S., Bienvenu, T., Roubergue, A., Leu-Semenescu, S., et al. (2007). Rett syndrome: An overlooked diagnosis in women with stereotypic hand movements, psychomotor retardation, parkinsonism, and dystonia? Movement Disorders, 22(3), 387–389. doi:10.1002/mds.21276.
Samaco, R. C., Nagarajan, R. P., Braunschweig, D., & LaSalle, J. M. (2004). Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Human Molecular Genetics, 13(6), 629–639. doi:10.1093/hmg/ddh063.
Sapkota, Y., Robson, P., Lai, R., Cass, C. E., Mackey, J. R., & Damaraju, S. (2012). A two-stage association study identifies methyl-CpG-binding domain protein 2 gene polymorphisms as candidates for breast cancer susceptibility. European Journal of Human Genetics, 20(6), 682–689. doi:10.1038/ejhg.2011.273.
Saunders, C. J., Minassian, B. E., Chow, E. W., Zhao, W., & Vincent, J. B. (2009). Novel exon 1 mutations in MECP2 implicate isoform MeCP2_e1 in classical Rett syndrome. American Journal of Medical Genetics: Part A, 149A(5), 1019–1023. doi:10.1002/ajmg.a.32776.
Saxena, A., de Lagarde, D., Leonard, H., Williamson, S. L., Vasudevan, V., Christodoulou, J., et al. (2006). Lost in translation: Translational interference from a recurrent mutation in exon 1 of MECP2. Journal of Medical Genetics, 43(6), 470–477. doi:10.1136/jmg.2005.036244.
Schaevitz, L. R., Gómez, N. B., Zhen, D. P., & Berger-Sweeney, J. E. (2013). MeCP2 R168X male and female mutant mice exhibit Rett-like behavioral deficits. Genes, Brain and Behavior,. doi:10.1111/gbb.12070.
Schaevitz, L. R., Nicolai, R., Lopez, C. M., D’Iddio, S., Iannoni, E., & Berger-Sweeney, J. E. (2012). Acetyl-L-carnitine improves behavior and dendritic morphology in a mouse model of Rett syndrome. PLoS ONE, 7(12), e51586. doi:10.1371/journal.pone.0051586.
Shahbazian, M. D., Antalffy, B., Armstrong, D. L., & Zoghbi, H. Y. (2002a). Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Human Molecular Genetics, 11(2), 115–124.
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., et al. (2002b). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron, 35(2), 243–254.
Shahbazian, M. D., & Zoghbi, H. Y. (2002). Rett syndrome and MeCP2: Linking epigenetics and neuronal function. American Journal of Human Genetics, 71(6), 1259–1272. doi:10.1086/345360.
Sheikh, T. I., Mittal, K., Willis, M. J., & Vincent, J. B. (2013). A synonymous change, p.Gly16Gly in MECP2 Exon 1, causes a cryptic splice event in a Rett syndrome patient. Orphanet Journal of Rare Diseases, 8, 108. doi:10.1186/1750-1172-8-108.
Shibayama, A., Cook, E. H, Jr, Feng, J., Glanzmann, C., Yan, J., Craddock, N., et al. (2004). MECP2 structural and 3′-UTR variants in schizophrenia, autism and other psychiatric diseases: A possible association with autism. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 128B(1), 50–53. doi:10.1002/ajmg.b.30016.
Shu, L., Khor, T. O., Lee, J. H., Boyanapalli, S. S., Huang, Y., Wu, T. Y., et al. (2011). Epigenetic CpG demethylation of the promoter and reactivation of the expression of Neurog1 by curcumin in prostate LNCaP cells. AAPS Journal, 13(4), 606–614. doi:10.1208/s12248-011-9300-y.
Singh, J., Saxena, A., Christodoulou, J., & Ravine, D. (2008). MECP2 genomic structure and function: Insights from ENCODE. Nucleic Acids Research, 36(19), 6035–6047. doi:10.1093/nar/gkn591.
Singleton, M. K., Gonzales, M. L., Leung, K. N., Yasui, D. H., Schroeder, D. I., Dunaway, K., et al. (2011). MeCP2 is required for global heterochromatic and nucleolar changes during activity-dependent neuronal maturation. Neurobiology of Diseases, 43(1), 190–200. doi:10.1016/j.nbd.2011.03.011.
Skene, P. J., Illingworth, R. S., Webb, S., Kerr, A. R., James, K. D., Turner, D. J., et al. (2010). Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Molecular Cell, 37(4), 457–468. doi:10.1016/j.molcel.2010.01.030.
Smith, R. M., & Sadee, W. (2011). Synaptic signaling and aberrant RNA splicing in autism spectrum disorders. Frontiers in Synaptic Neuroscience, 3, 1. doi:10.3389/fnsyn.2011.00001.
Smrt, R. D., Eaves-Egenes, J., Barkho, B. Z., Santistevan, N. J., Zhao, C., Aimone, J. B., et al. (2007). Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiology of Diseases, 27(1), 77–89. doi:10.1016/j.nbd.2007.04.005.
Squillaro, T., Alessio, N., Cipollaro, M., Melone, M. A., Hayek, G., Renieri, A., et al. (2012). Reduced expression of MECP2 affects cell commitment and maintenance in neurons by triggering senescence: New perspective for Rett syndrome. Molecular Biology of the Cell, 23(8), 1435–1445. doi:10.1091/mbc.E11-09-0784.
Stettner, G. M., Huppke, P., Gartner, J., Richter, D. W., & Dutschmann, M. (2008). Disturbances of breathing in Rett syndrome: Results from patients and animal models. Advances in Experimental Medicine and Biology, 605, 503–507. doi:10.1007/978-0-387-73693-8_88.
Stuss, D. P., Cheema, M., Ng, M. K., de Paz, A. M., Williamson, B., Missiaen, K., et al. (2013). Impaired in vivo binding of MeCP2 to chromatin in the absence of its DNA methyl-binding domain. Nucleic Acids Research, 41(9), 4888–4900. doi:10.1093/nar/gkt213.
Subramaniam, B., Naidu, S., & Reiss, A. L. (1997). Neuroanatomy in Rett syndrome: Cerebral cortex and posterior fossa. Neurology, 48(2), 399–407.
Suter, B., Treadwell-Deering, D., Zoghbi, H. Y., Glaze, D. G., & Neul, J. L. (2013). Brief report: MECP2 mutations in people without Rett syndrome. Journal of Autism and Developmental Disorders,. doi:10.1007/s10803-013-1902-z.
Szulwach, K. E., Li, X., Smrt, R. D., Li, Y., Luo, Y., Lin, L., et al. (2010). Cross talk between microRNA and epigenetic regulation in adult neurogenesis. Journal of Cell Biology, 189(1), 127–141. doi:10.1083/jcb.200908151.
Tao, J., Hu, K., Chang, Q., Wu, H., Sherman, N. E., Martinowich, K., et al. (2009). Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proceedings of the National Academy of Sciences USA, 106(12), 4882–4887. doi:10.1073/pnas.0811648106.
Tejada, M. I., Penagarikano, O., Rodriguez-Revenga, L., Martinez-Bouzas, C., Garcia, B., Badenas, C., et al. (2006). Screening for MECP2 mutations in Spanish patients with an unexplained mental retardation. Clinical Genetics, 70(2), 140–144. doi:10.1111/j.1399-0004.2006.00647.x.
Temudo, T., Santos, M., Ramos, E., Dias, K., Vieira, J. P., Moreira, A., et al. (2011). Rett syndrome with and without detected MECP2 mutations: An attempt to redefine phenotypes. Brain Development, 33(1), 69–76. doi:10.1016/j.braindev.2010.01.004.
Thambirajah, A. A., Eubanks, J. H., & Ausio, J. (2009). MeCP2 post-translational regulation through PEST domains: Two novel hypotheses: potential relevance and implications for Rett syndrome. BioEssays, 31(5), 561–569. doi:10.1002/bies.200800220.
Thatcher, K. N., & LaSalle, J. M. (2006). Dynamic changes in Histone H3 lysine 9 acetylation localization patterns during neuronal maturation require MeCP2. Epigenetics, 1(1), 24–31.
Tochiki, K. K., Cunningham, J., Hunt, S. P., & Geranton, S. M. (2012). The expression of spinal methyl-CpG-binding protein 2, DNA methyltransferases and histone deacetylases is modulated in persistent pain states. Molecular Pain, 8, 14. doi:10.1186/1744-8069-8-14.
Trinidad, J. C., Thalhammer, A., Specht, C. G., Lynn, A. J., Baker, P. R., Schoepfer, R., et al. (2008). Quantitative analysis of synaptic phosphorylation and protein expression. Molecular and Cellular Proteomics, 7(4), 684–696. doi:10.1074/mcp.M700170-MCP200.
Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., McCurry, C., Fu, D. D., et al. (2009). Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proceedings of the National Academy of Sciences USA, 106(6), 2029–2034. doi:10.1073/pnas.0812394106.
Tsujimura, K., Abematsu, M., Kohyama, J., Namihira, M., & Nakashima, K. (2009). Neuronal differentiation of neural precursor cells is promoted by the methyl-CpG-binding protein MeCP2. Experimental Neurology, 219(1), 104–111. doi:10.1016/j.expneurol.2009.05.001.
Tudor, M., Akbarian, S., Chen, R. Z., & Jaenisch, R. (2002). Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proceedings of the National Academy of Sciences USA, 99(24), 15536–15541. doi:10.1073/pnas.242566899.
Tunc-Ozcan, E., Ullmann, T. M., Shukla, P. K., & Redei, E. E. (2013). Low-dose thyroxine attenuates autism-associated adverse effects of fetal alcohol in male offspring’s social behavior and hippocampal gene expression. Alcoholism, Clinical and Experimental Research,. doi:10.1111/acer.12183.
Urdinguio, R. G., Fernandez, A. F., Lopez-Nieva, P., Rossi, S., Huertas, D., Kulis, M., et al. (2010). Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics, 5(7), 656–663. doi:10.4161/epi.5.7.13055.
Vanhala, R., Gaily, E., Paetau, A., & Riikonen, R. (1998). Pons tumour behind a phenotypic Rett syndrome presentation. Developmental Medicine and Child Neurology, 40(12), 836–839.
Vecsler, M., Ben Zeev, B., Nudelman, I., Anikster, Y., Simon, A. J., Amariglio, N., et al. (2011). Ex vivo treatment with a novel synthetic aminoglycoside NB54 in primary fibroblasts from Rett syndrome patients suppresses MECP2 nonsense mutations. PLoS ONE, 6(6), e20733. doi:10.1371/journal.pone.0020733.
Volkmann, I., Kumarswamy, R., Pfaff, N., Fiedler, J., Dangwal, S., Holzmann, A., et al. (2013). MicroRNA-mediated epigenetic silencing of Sirtuin1 contributes to impaired angiogenic responses. Circulation Research,. doi:10.1161/CIRCRESAHA.113.301702.
Vora, P., Mina, R., Namaka, M., & Frost, E. E. (2010). A novel transcriptional regulator of myelin gene expression: Implications for neurodevelopmental disorders. NeuroReport, 21(14), 917–921. doi:10.1097/WNR.0b013e32833da500.
Wada, R., Akiyama, Y., Hashimoto, Y., Fukamachi, H., & Yuasa, Y. (2010). miR-212 is downregulated and suppresses methyl-CpG-binding protein MeCP2 in human gastric cancer. International Journal of Cancer, 127(5), 1106–1114. doi:10.1002/ijc.25126.
Wang, Y., Liu, C., Guo, Q. L., Yan, J. Q., Zhu, X. Y., Huang, C. S., et al. (2011). Intrathecal 5-azacytidine inhibits global DNA methylation and methyl-CpG-binding protein 2 expression and alleviates neuropathic pain in rats following chronic constriction injury. Brain Research, 1418, 64–69. doi:10.1016/j.brainres.2011.08.040.
Wang, I. T., Reyes, A. R., & Zhou, Z. (2013a). Neuronal morphology in MeCP2 mouse models is intrinsically variable and depends on age, cell type, and Mecp2 mutation. Neurobiology of Diseases, 58C, 3–12. doi:10.1016/j.nbd.2013.04.020.
Wang, J. T., Wu, T. T., Bai, L., Ding, L., Hao, M., & Wang, Y. (2013b). Effect of folate in modulating the expression of DNA methyltransferase 1 and methyl-CpG-bingding protein 2 in cervical cancer cell lines. Zhonghua Liu Xing Bing Xue Za Zhi, 34(2), 173–177.
Warita, K., Mitsuhashi, T., Ohta, K., Suzuki, S., Hoshi, N., Miki, T., et al. (2013). Gene expression of epigenetic regulatory factors related to primary silencing mechanism is less susceptible to lower doses of bisphenol A in embryonic hypothalamic cells. Journal of Toxicological Sciences, 38(2), 285–289.
Watson, P., Black, G., Ramsden, S., Barrow, M., Super, M., Kerr, B., et al. (2001). Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. Journal of Medical Genetics, 38(4), 224–228.
Weaving, L. S., Christodoulou, J., Williamson, S. L., Friend, K. L., McKenzie, O. L., Archer, H., et al. (2004). Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. American Journal of Human Genetics, 75(6), 1079–1093. doi:10.1086/426462.
Weaving, L. S., Williamson, S. L., Bennetts, B., Davis, M., Ellaway, C. J., Leonard, H., et al. (2003). Effects of MECP2 mutation type, location and X-inactivation in modulating Rett syndrome phenotype. American Journal of Medical Genetics: Part A, 118A(2), 103–114. doi:10.1002/ajmg.a.10053.
Wither, R. G., Lang, M., Zhang, L., & Eubanks, J. H. (2013). Regional MeCP2 expression levels in the female MeCP2-deficient mouse brain correlate with specific behavioral impairments. Experimental Neurology, 239, 49–59. doi:10.1016/j.expneurol.2012.09.005.
Woodyatt, G. C., & Ozanne, A. E. (1993). A longitudinal study of cognitive skills and communication behaviours in children with Rett syndrome. Journal of Intellectual Disability Research, 37(Pt 4), 419–435.
Wu, S. H., & Camarena, V. (2009). MeCP2 function in the basolateral amygdala in Rett syndrome. Journal of Neuroscience, 29(32), 9941–9942. doi:10.1523/JNEUROSCI.2540-09.2009.
Wu, W., Gu, W., Xu, X., Shang, S., & Zhao, Z. (2012). Downregulation of CNPase in a MeCP2 deficient mouse model of Rett syndrome. Neurological Research, 34(2), 107–113. doi:10.1179/016164111X13214359296301.
Wu, H., Tao, J., Chen, P. J., Shahab, A., Ge, W., Hart, R. P., et al. (2010). Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proceedings of the National Academy of Sciences USA, 107(42), 18161–18166. doi:10.1073/pnas.1005595107.
Xinhua, B., Shengling, J., Fuying, S., Hong, P., Meirong, L., & Wu, X. R. (2008). X chromosome inactivation in Rett Syndrome and its correlations with MECP2 mutations and phenotype. Journal of Child Neurology, 23(1), 22–25. doi:10.1177/0883073807307077.
Xu, X., Jin, H., Liu, Y., Liu, L., Wu, Q., Guo, Y., et al. (2012a). The expression patterns and correlations of claudin-6, methy-CpG binding protein 2, DNA methyltransferase 1, histone deacetylase 1, acetyl-histone H3 and acetyl-histone H4 and their clinicopathological significance in breast invasive ductal carcinomas. Diagnostic Pathology, 7, 33. doi:10.1186/1746-1596-7-33.
Xu, X., Xu, Q., Zhang, Y., Zhang, X., Cheng, T., Wu, B., et al. (2012b). A case report of Chinese brothers with inherited MECP2-containing duplication: Autism and intellectual disability, but not seizures or respiratory infections. BMC Medical Genetics, 13, 75. doi:10.1186/1471-2350-13-75.
Yaqinuddin, A., Abbas, F., Naqvi, S. Z., Bashir, M. U., Qazi, R., & Qureshi, S. A. (2008). Silencing of MBD1 and MeCP2 in prostate-cancer-derived PC3 cells produces differential gene expression profiles and cellular phenotypes. Bioscience Reports, 28(6), 319–326. doi:10.1042/BSR20080032.
Yasui, D. H., Gonzales, M. L., Aflatooni, J. O., Crary, F. K., Hu, D. J., Gavino, B. J., et al. (2014). Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Human Molecular Genetics,. doi:10.1093/hmg/ddt640.
Yasui, D. H., Peddada, S., Bieda, M. C., Vallero, R. O., Hogart, A., Nagarajan, R. P., et al. (2007). Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proceedings of the National Academy of Sciences USA, 104(49), 19416–19421. doi:10.1073/pnas.0707442104.
Yasui, D. H., Xu, H., Dunaway, K. W., Lasalle, J. M., Jin, L. W., & Maezawa, I. (2013). MeCP2 modulates gene expression pathways in astrocytes. Molecular Autism, 4(1), 3. doi:10.1186/2040-2392-4-3.
Young, D. J., Bebbington, A., Anderson, A., Ravine, D., Ellaway, C., Kulkarni, A., et al. (2008). The diagnosis of autism in a female: Could it be Rett syndrome? European Journal of Pediatrics, 167(6), 661–669. doi:10.1007/s00431-007-0569-x.
Young, J. I., Hong, E. P., Castle, J. C., Crespo-Barreto, J., Bowman, A. B., Rose, M. F., et al. (2005). Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proceedings of the National Academy of Sciences USA, 102(49), 17551–17558. doi:10.1073/pnas.0507856102.
Yu, D., Sakurai, F., & Corey, D. R. (2011). Clonal Rett Syndrome cell lines to test compounds for activation of wild-type MeCP2 expression. Bioorganic & Medicinal Chemistry Letters, 21(18), 5202–5205. doi:10.1016/j.bmcl.2011.07.053.
Zachariah, R. M., Olson, C. O., Ezeonwuka, C., & Rastegar, M. (2012). Novel MeCP2 isoform-specific antibody reveals the endogenous MeCP2E1 expression in murine brain, primary neurons and astrocytes. PLoS ONE, 7(11), e49763. doi:10.1371/journal.pone.0049763.
Zachariah, R. M., & Rastegar, M. (2012). Linking epigenetics to human disease and Rett syndrome: The emerging novel and challenging concepts in MeCP2 research. Neural Plasticity, 2012, 415825. doi:10.1155/2012/415825.
Zanella, S., Mebarek, S., Lajard, A. M., Picard, N., Dutschmann, M., & Hilaire, G. (2008). Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respiratory Physiology & Neurobiology, 160(1), 116–121. doi:10.1016/j.resp.2007.08.009.
Zhong, X., Li, H., & Chang, Q. (2012). MeCP2 phosphorylation is required for modulating synaptic scaling through mGluR5. Journal of Neuroscience, 32(37), 12841–12847. doi:10.1523/JNEUROSCI.2784-12.2012.
Zhou, Z., Hong, E. J., Cohen, S., Zhao, W. N., Ho, H. Y., Schmidt, L., et al. (2006). Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron, 52(2), 255–269. doi:10.1016/j.neuron.2006.09.037.
Zhou, P., Lu, Y., & Sun, X. H. (2011). Zebularine suppresses TGF-beta-induced lens epithelial cell-myofibroblast transdifferentiation by inhibiting MeCP2. Molecular Vision, 17, 2717–2723.
Zlatanova, J. (2005). MeCP2: The chromatin connection and beyond. Biochemistry and Cell Biology, 83(3), 251–262. doi:10.1139/o05-048.
Zocchi, L., & Sassone-Corsi, P. (2012). SIRT1-mediated deacetylation of MeCP2 contributes to BDNF expression. Epigenetics, 7(7), 695–700. doi:10.4161/epi.20733.
Zoll, B., Huppke, P., Wessel, A., Bartels, I., & Laccone, F. (2004). Fetal alcohol syndrome in association with Rett syndrome. Genetic Counseling, 15(2), 207–212.
Acknowledgments
We apologize that due to space limitations, many excellent papers could not be included in this review. We thank all members of the Rastegar laboratory for helpful discussions. The research in the Rastegar Laboratory is supported by funds from Natural Sciences and Engineering Research Council of Canada (NSERC Discovery Grant 372405-2009), Canadian Institute of Health Research (CIHR Team Grant TEC-128094, and CIHR Catalyst Grant CEN-132383), The Health Sciences Centre Foundation (HSCF), Scottish Rite Charitable Foundation of Canada (SRCFC, Grant 10110), and Graduate Enhancement of Tri-council Stipend (GETS). VRBL is a recipient of MHRC/UMGF studentship award.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liyanage, V.R.B., Rastegar, M. Rett Syndrome and MeCP2. Neuromol Med 16, 231–264 (2014). https://doi.org/10.1007/s12017-014-8295-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-014-8295-9