
Abstract
Posttranslational modification of proteins by the small ubiquitin-like modifier (SUMO) is a potent regulator of various cellular events. Hundreds of substrates have been identified, many of them involved in vital processes like transcriptional regulation, signal transduction, protein degradation, cell cycle regulation, DNA repair, chromatin organization, and nuclear transport. In recent years, protein sumoylation increasingly attracted attention, as it could be linked to heart failure, cancer, and neurodegeneration. However, underlying mechanisms involving how modification by SUMO contributes to disease development are still scarce thus necessitating further research. This review aims to critically discuss currently available concepts of the SUMO pathway, thereby highlighting regulation in the healthy versus diseased organism, focusing on neurologic aspects. Better understanding of differential regulation in health and disease may finally allow to uncover pathogenic mechanisms and contribute to the development of disease-specific therapies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Posttranslational modifications are efficient tools within a cellular system to quickly and reversibly modulate protein functions without the need of de novo protein synthesis. Such a fine-tuning mechanism usually results in regulating the choice of inter- and intra-molecular binding surfaces that can have diverse consequences, like on protein stability, conformation, activity, and intracellular localization to adapt a target protein to the changing needs of a cell. Small chemical modifiers, such as phosphorylation, acetylation, and methylation, are well established as cellular regulators, as they are heavily investigated since a long time. Modification by ubiquitin or the small ubiquitin-like modifier (SUMO) stands apart in that the modifier itself is a small polypeptide. Ubiquitination is best known for its role in protein degradation (reviewed in Pickart 2001), but has also several non-proteolytic regulatory functions (reviewed in Komander and Rape 2012). SUMO is structurally related to ubiquitin and is also conjugated to target lysines via a hierarchical enzymatic cascade (Bayer et al. 1998; Kim et al. 2002; Pickart 2001). In the past two decades, hundreds of substrates have been identified, with numbers steadily rising. Many of these substrates were found to play crucial roles in vital processes like transcriptional regulation, signal transduction, protein degradation, cell cycle regulation, DNA repair, chromatin organization or nuclear transport (reviewed in, e.g., Cubenas-Potts and Matunis 2013; Garcia-Dominguez and Reyes 2009; Geiss-Friedlander and Melchior 2007; Geoffroy and Hay 2009; Jackson and Durocher 2013; Flotho and Melchior 2013). Thus, it is not surprising that defects in the sumoylation pathway are associated with severe diseases, such as neurodegeneration, heart failure, and cancer (Bettermann et al. 2012; Krumova and Weishaupt 2013; J. Wang 2011; Wilkinson and Henley 2010).
The consequences of protein sumoylation can be manifold, and to further complicate matters, investigation is usually hampered by the highly transient nature of this modification and the crosstalk with other modifications. The important role of SUMO in neurologic functions and diseases becomes more apparent (compare Fig. 1), but we are only at the beginning to understand the underlying mechanistic concepts. In this review, we provide an overview on the prevailing concepts of regulation in the SUMO system. Direct disease-associated sumoylation will be discussed separately in more specialized chapters of this issue.

SUMO is a regulator of neurological functions and diseases. A selected overview of neurological functions and diseases that are associated with the SUMO pathway
The SUMO Pathway
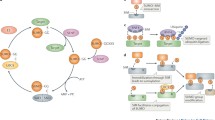
SUMO is a small protein (about 11 kDa in size) that belongs to the ubiquitin-like family of modifiers (Ubls). Protein sumoylation is a highly dynamic process depending on the balance of conjugation and deconjugation (see Fig. 2). Similar to the ubiquitin system, SUMO is expressed as a precursor protein. Maturation is achieved by specific SUMO proteases, which expose the C-terminal di-glycine that is critical for conjugation (e.g., Hickey et al. 2012). The matured SUMO is subsequently conjugated to substrates via a hierarchical enzymatic cascade (Fig. 2). In an ATP-dependent step, SUMO is attached to an internal cysteine of the heterodimeric E1 activating enzyme Aos1/Uba2, forming an energy-rich thioester bond (Desterro et al. 1999; Johnson et al. 1997). SUMO is then transferred to the catalytic cysteine of the E2 conjugating enzyme Ubc9, again resulting in a thioester bond (Desterro et al. 1997; Gong et al. 1997; Johnson and Blobel 1997). In contrast to ubiquitination, the SUMO E2 enzyme Ubc9 is able to directly recognize and conjugate SUMO to its substrates by forming an isopeptide bond between the C-terminal glycine of SUMO and the ε-amino group of the target lysine (Okuma et al. 1999; Mahajan et al. 1998). Usually, this reaction is very weak, and SUMO E3 ligases (see below for detailed discussion) greatly enhance the efficiency of SUMO conjugation (Johnson and Gupta 2001; Sachdev et al. 2001; Pichler et al. 2002). SUMO E3 ligases are thought to ensure substrate specificity, but although hundreds of proteins have been identified as SUMO targets, only a handful E3 ligases are described (see below and reviewed in Gareau and Lima 2010; Geiss-Friedlander and Melchior 2007; Flotho and Melchior 2013). Targets can be modified with a single SUMO, with multiple SUMOs, or with SUMO chains. The consequences of sumoylation result in changed binding interfaces that can have implications on diverse protein functions like intracellular localization, activity, stability, and conformational changes.

The SUMO pathway. Prior to entering the cycle of reversible SUMO conjugation, the expressed SUMO precursor protein has to be matured. Proteolytic cleavage by SUMO proteases exposes a di-glycine motif (GG) that is critical for subsequent activation/conjugation. The mature SUMO is activated in an ATP-dependent step by the E1 activating enzyme Aos1/Uba2, establishing a thioester linkage between the C-terminal glycine of SUMO and the catalytic cysteine residue of the E1 enzyme. Subsequently, SUMO is transferred to the E2 conjugating enzyme Ubc9, again forming a thioester bond. Ubc9 can then catalyze target modification either directly or in conjunction with a SUMO E3 ligase, by forming an isopeptide bond between the ε-amino group of the target lysine and the C-terminal glycine of SUMO. Next to the maturation of SUMO precursors, SUMO proteases are able to deconjugate substrates, thus feeding SUMO back in the conjugation cycle
SUMO conjugation is a reversible process, and deconjugation is mediated by specific SUMO proteases (see below and reviewed in Hickey et al. 2012; Kim and Baek 2009).
The maintenance of balanced SUMO conjugation and deconjugation in the cell is critical for survival. Knockout experiments in mice showed embryonic lethality not only when abolishing SUMO conjugation by targeting the SUMO E2-enzyme Ubc9 (Nacerddine et al. 2005), but also when preventing deconjugation of substrates by targeting the SUMO proteases SENP1 and SENP2 (Cheng et al. 2007; Chiu et al. 2008). Interestingly, a SUMO1 knockout is viable and all essential functions can be carried out by its paralogs under unchallenged conditions, suggesting that SUMO2/3 species at least partly compensate for the loss of SUMO1 (Evdokimov et al. 2008; Zhang et al. 2008). It will be interesting to see, if SUMO1 could also compensate for loss of SUMO2/3 species.
In the following, we will discuss various issues we think are important to understand the complexity and the power of the SUMO system.
SUMO Paralogs
Less complex eukaryotic organisms express a single SUMO protein, whereas plants and vertebrates encode different SUMO variants. The human genome encodes four SUMO paralogs: SUMO1, the nearly identical SUMO2 and SUMO3 and SUMO4. In contrast to the ubiquitously expressed SUMO1-3, SUMO4 is limited to immune cells, pancreatic islands, and the kidney (Bohren et al. 2004; Guo et al. 2004; Wang and She 2008). All SUMO paralogs are synthesized as precursors with C-terminal extensions of different length. For conjugation, these extensions need to be processed to expose a di-glycine motif. However, for SUMO4, it is presently unclear how it gets processed, as the residue Pro90 appears to prevent cleavage by known SUMO proteases (Owerbach et al. 2005), although it gets cleaved upon stress induction (Wei et al. 2008).
In contrast to ubiquitin, all SUMO variants share an unstructured and flexible N-terminus. This domain represents the major acceptor site for SUMO chain assembly, assuming these chains are rather extended and highly flexible. SUMO2/3 and also the single SUMO in yeast SMT3 are equipped with one or even three SUMO consensus sites, respectively. Not surprisingly, these sites represent indeed the major linkage sites for SUMO chain formation. Mass spectrometry analysis of both in vitro and in vivo samples indicated that SUMO2 forms chains via Lys 11 (Tatham et al. 2001; Matic et al. 2008b) and SMT3 involves all three consensus site lysines, Lys 11, 15, and 19 for chain formation (Bencsath et al. 2002; Klug et al. 2013; Matic et al. 2008b). SUMO1 has no SUMO consensus site, but can at least in vitro heavily form SUMO chains in the presence of a short E3 ligase fragment (Pichler et al. 2002). In vitro assembled SUMO1 chains are linked via the N-terminal lysines Lys 7, 16, and 17 (Pedrioli et al. 2006). Further evidence for in vivo SUMO chain formation via non-consensus site lysines comes from different mass spectrometry studies identifying the non-consensus linkages via Lys 7, 17, and 25 for SUMO1, Lys 5, 7, 32, and 34 for SUMO2 and Lys 5, 33, and 35 for SUMO3 (Blomster et al. 2010; Bruderer et al. 2011; Matic et al. 2010; Tatham et al. 2001). However, it is not clear to which extent such non-consensus SUMO chains are formed in comparison with consensus SUMO chains, nor if they have indeed distinct biological functions. Alternatively, in case of SUMO1, it is even discussed that it rather functions as a chain terminator (reviewed in Praefcke et al. 2012).
Understanding of the exciting role of the different SUMO chains is emerging and insights into their synthesis and associated biological functions are eagerly awaited.
How SUMO isoforms are regulated themselves is still poorly understood, but several studies point to a regulation at transcriptional and posttranscriptional levels. As mentioned above, SUMO4 expression is restricted to specific tissues and despite a ubiquitous expression of SUMO1-3, their levels appear to differ across tissues and during development (Loriol et al. 2012; Xu and Au 2005). In cells, SUMO1 is found mostly conjugated to substrates and is less abundant in its free form. In contrast, free SUMO2/3 levels are high under normal conditions, however, get conjugated upon various stimuli including heat shock and arsenic treatment (Lallemand-Breitenbach et al. 2008; Saitoh and Hinchey 2000; Tatham et al. 2008; Weisshaar et al. 2008). Other stimuli, like DNA repair, involve diverse SUMO paralogs (Golebiowski et al. 2009). Under all these conditions, waves of specific, but also rather global changes in sumoylation can take place (Johnson and Blobel 1999; Psakhye and Jentsch 2012; Tatham et al. 2005; Yin et al. 2012).
Especially interesting for neurobiologists is the finding that SUMO1 conjugation is induced in response to hypoxia (Comerford et al. 2003; Shao et al. 2004). Furthermore, several studies identified a global increase in mainly SUMO2/3 species upon ischemic challenge, including hypothermia (Cimarosti et al. 2008; Datwyler et al. 2011; Lee et al. 2007; Loftus et al. 2009; Yang et al. 2008), which appears to protect brains against focal cerebral ischemic damage (Lee et al. 2011b). In addition, SUMO1-3 species are found to be increased in human astrocytic brain tumors and have functions in glioblastoma cell survival (Yang et al. 2013).
Another important regulator of SUMO proteins are posttranslational modifications. It had been previously demonstrated that SUMO1 is modified by both acetylation and phosphorylation (Lallemand-Breitenbach et al. 2008; Matic et al. 2008a). Only recently, an exciting function was identified for acetylation, where modification at Lys 37 in SUMO1 and Lys 33 in SUMO2 affected specific non-covalent SUMO interactions by neutralizing the required basic charge of SUMO (Ullmann et al. 2012). Interestingly, SUMO variants can also be modified with ubiquitin, e.g., Lys 20 and 32 of SUMO3 (Lamoliatte et al. 2013), raising the pressing question of whether ubiquitination of SUMO regulates the abundance of SUMO proteins or if other non-proteolytic regulatory functions are involved. As SUMO4 appears to be highly instable in most tissues and only gets stabilized and matured in response to stress (Wei et al. 2008), it is likely that ubiquitin-dependent pathways are involved. The best example of how ubiquitin-dependent degradation regulates the SUMO pathway comes from the identification of SUMO2 chains, which are assembled upon stress. These chains are recognized by so-called SUMO-targeted ubiquitin E3 ligases (StUbls), which are equipped with multiple SUMO interaction motifs (SIMs, see also below). The important role of StUbls is demonstrated from yeast to humans and is associated with functions in DNA repair and PML degradation (Erker et al. 2013; Galanty et al. 2012; Guzzo et al. 2012; Li et al. 2013b; Poulsen et al. 2013; Prudden et al. 2007; Tatham et al. 2008; Vyas et al. 2013; Weisshaar et al. 2008; Xie et al. 2007; Yin et al. 2012; Uzunova et al. 2007; Mullen and Brill 2008; Burgess et al. 2007).
SUMO-Like Proteins
A group of proteins, with main functions in DNA repair and genome stability, were identified as they share characteristics of two tandem SUMO-like domains (SLD1 and SLD2). Two different classes of SLDs are described, the RENi protein family (Rad60 in fission yeast, Esc2 in bakers yeast and NIP45 in mammals) (Novatchkova et al. 2005) and USP1/UAF1, a ubiquitin protease (Yang et al. 2011a). SLDs are approximately 100 residues in size that show little sequence homology, but characteristic structural similarity to SUMO (Prudden et al. 2009; Prudden et al. 2011; Sekiyama et al. 2010). The RENi SLD2 appears closer related to SUMO1, whereas the SLDs in USP1/UAF1 show similarity to SUMO2/3 (Sekiyama et al. 2010; Yang et al. 2011a). Accordingly, these proteins show different binding properties and also different functional consequences are reported. The USP1/UAF1 SLD2 directly recognizes a motif similar to a conventional SIM (see also below) that allows binding to its targets (Yang et al. 2011a). The RENi SLD2s cannot bind such a conventional SIM, but recognize the Ubc9 backside (which we refer to as ClassII SIM, see below) in a similar manner as it was identified for SUMO. This interaction is reported to interfere with SUMO chain elongation (Prudden et al. 2009, 2011; Sekiyama et al. 2010). As binding of free SUMO to Ubc9’s backside needs to be displaced for E1 interaction (Duda et al. 2007), it would be interesting to test consequences of the RENi SLD2 on Ubc9 charging with SUMO. However, the RENi SLD1 binds to SUMO E1 and E3 enzymes, but not to Ubc9 (Prudden et al. 2009). Currently, this binding interface is not further characterized, but may resemble USP1/UAF1 SLDs and recognize more conventional SIMs. Interestingly, SLDs possess di-glycine motives, but currently there is no evidence that they get processed and conjugated.
SUMO Consensus Motifs (SCM)
With the identification of SUMO substrates and mapping of the respective SUMO attachment sites, it quickly became clear that many sites share a common motif: Ψ K × D/E (Ψ = I, V or L), which was later shown to be directly recognized by the catalytic cleft of Ubc9 (see Fig. 3b and Bernier-Villamor et al. 2002). Mass spectrometric analysis of several substrates expands the motif to (I/V/L/M/F/C) K × (D/E) and revealed that it also functions when it is inverted (E/D) × K (V/I/L/F/P) (Matic et al. 2010). Importantly, such motifs are only functional when placed in an unstructured and exposed region, as their positioning in an alpha helix changes the orientations of the essential residues and consequently prevents recognition by Ubc9 (Pichler et al. 2005).

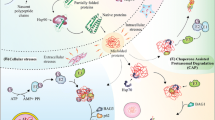
E3-independent and E3-dependent mechanisms of SUMO conjugation. a The covalent modification of substrates by SUMO is primarily directed to lysine residues located within the conserved SUMO consensus motif (SCM). Substrate modification can be facilitated either directly by the SUMO E2 enzyme Ubc9 (left panel) or in the presence of SUMO E3 ligases as well as other proteins denoted as ‘cofactors’ (right panel). Furthermore, a variety of non-covalent interactions can influence the SUMO modification event. Substrate SCMs can be directly recognized by the Ubc9~SUMO thioester moiety, resulting in limited substrate sumoylation. Certain substrates bear additional conserved SUMO interaction motifs (SIMs) that non-covalently interact with SUMO, either with SUMO charged Ubc9 (I) or with sumoylated Ubc9 (II), contributing to substrate selection, enhanced binding, and SUMO conjugation. In addition, certain substrates also have additional Ubc9 binding surfaces (III) that influence sumoylation. E3s/cofactors act as scaffold proteins that further stabilize interactions between substrate and the charged Ubc9~SUMO (IV) or enhance Ubc9–SUMO thioester discharge (V). The latter can involve secondary binding events including SIMs. The above mechanisms for SUMO modification are not mutually exclusive, as the presence of E3s/cofactors could also enhance the events depicted on the left. b SUMO consensus motifs. Sequences observed to act as SCMs are shown; Classical, inverted, hydrophobic cluster sumoylation motif (HCSM), phosphorylation-dependent SUMO motif (PSDM), and negatively charged SUMO motif (NSDM). ‘Ψ’ represents hydrophobic residues, ‘K’ is the lysine that gets modified, and ‘S P’ represents a phosphorylated serine. Also shown alongside is a ribbon cartoon representation of the interaction between Ubc9 and the SCM, as observed in RanGAP1 (PDB code 1KPS). The side chains of residues that make up the RanGAP1 SCM, as well as residues in Ubc9 involved in catalysis and SCM interaction, are labeled and shown in stick format. c SUMO interaction motifs. Conserved consensus sequences of the three SIMs are depicted: SIMa, SIMb, and SIMr. ‘Ac’ represents acidic residues. Also shown is a ribbon representation of the interaction between SUMO1 and the SIM, as observed in the Internal Repeat (IR) 1 region of RanBP2 (PDB code 1Z5S). In contrast to SCMs, SIMs form secondary structures, as observed by the packing of the RanBP2 beta strand within the hydrophobic groove, located between alpha helix-1 and beta strand-2 of SUMO1. Models were generated using Protean 3D
Several extended variations of the conventional consensus motif are reported (summarized in Fig. 3b), involving mostly negatively charged residues downstream of the core motif. This can be achieved by serine phosphorylation, as it is shown for the phosphorylation-dependent SUMO motif (PDSM): Ψ K × (D/E) × × S P (Hietakangas et al. 2006), the phosphorylation sumoylation motif (pSuM) Ψ K × S P and the extended pSuM Ψ K × S P S (S) × × S P (Picard et al. 2012). In contrast to the PDSM, the negatively charged SUMO motif Ψ K × E × × E E E E (NDSM) constitutively presents additional acidic residues downstream of the core motif (Yang et al. 2006). The hydrophobic cluster sumoylation motif (HCSM) Ψ Ψ Ψ K × D/E demonstrates another extended variation (Matic et al. 2010). Together, for several of the extended SUMO consensus motifs (PDSM, NDSM, and HCSM), it was shown that the additional residues enhance binding to Ubc9 and consequently promote conjugation (compare Fig. 3a, Mohideen et al. 2009; Yang et al. 2006; Bernier-Villamor et al. 2002). However, it is important to note that not all SUMO consensus sites are indeed sumoylated and many SUMO substrates are modified at non-consensus lysines (Lamoliatte et al. 2013; Matic et al. 2010; Pichler et al. 2005).
Non-covalent SUMO Binding
SUMO can regulate protein functions also via non-covalent binding to so-called SUMO-interaction motifs (SIMs, for an overview compare Fig. 3). Canonical SIMs are defined by a short hydrophobic core region often flanked by an acidic stretch. Currently, three types of conventional class I SIMs are described (Praefcke et al. 2012):
-
SIMa: Ψ Ψ × Ψ AcAcAcAcAc
-
SIMb: Ψ Ψ DLT
-
SIMr: AcAcAcAcAc Ψ × Ψ Ψ
-
Ψ = V, I or L, Ac = D, E or S
Such SIMs form β-strands that align with the hydrophobic groove formed between the β2-strand and the α1-helix of SUMO paralogs (compare Fig. 3c and Chang et al. 2011; Hecker et al. 2006; Reverter and Lima 2005). Electrostatic interactions between positively charged surfaces on SUMO and the negative charges in the SIM also contribute to binding. Non-covalent SUMO/SIM interactions can also be dynamically regulated by different posttranslational modifications. Such modifications can involve either the SUMO or the SIM and change the charge of the binding interface. As already mentioned above, SUMO can get acetylated which selectively modulates SIM interactions (Ullmann et al. 2012). On the other hand, the SIM itself is prone to regulation, as addition of negative charges, such as phosphorylation, stimulates binding to Lys 39 in SUMO1, Lys 35 in SUMO2, or Lys 34 in SUMO3 (Chang et al. 2011; Stehmeier and Muller 2009). These examples clearly indicate the dynamics and specificity of non-covalent interactions, although we are just beginning to understand the mechanisms regulating SIM interactions and its biological consequences. Most SIMs cannot distinguish between SUMO paralogs, though some selected SIMs do (Chang et al. 2011; Meulmeester et al. 2008; Ouyang et al. 2009), but the underlying requirements for paralog selection remain enigmatic.
In addition to these classical SIMs, there are at least two clearly distinct classes of non-covalent SUMO interactions, which we will refer to as Class II-SIMs and Class III-SIMs. Class II-SIMs bind to a very different surface on SUMO as described for canonical SIMs. So far, only two proteins are known to bind SUMO in this manner, the SUMO E2 Ubc9 via its backside (Capili and Lima 2007; Duda et al. 2007; Knipscheer et al. 2007) and the dipeptidyl peptidase-9 (DDP9) (Pilla et al. 2012). Interestingly, while Ubc9 does not show any paralog specificity, DDP9 exhibits a high preference for SUMO1. One explanation for this could be the extended SUMO binding interface observed with Ubc9 in comparison with the one mapped for DDP9, specifically involving the loop around Glu 67. Also, the corresponding surfaces are more positively charged in SUMO1 than in SUMO2, which may explain the obtained paralog specificity for a small binding interface (DDP9), but likely gets meaningless in context of a more extended interface (Ubc9) (Pilla et al. 2012; Capili and Lima 2007; Duda et al. 2007; Knipscheer et al. 2007). The binding interface with SUMO for the Class III-SIMs is not yet mapped, but it is specific in that the SIM is dependent on a zinc-coordinating motif of the ZZ type present in HERC2 (Danielsen et al. 2012). It will be interesting to gain deeper insights in the regulation and specificity of non-covalent SUMO interaction in the future.
The Heterodimeric E1 Activating Enzyme
The functional SUMO E1 activating enzyme constitutes a heterodimer of the proteins Aos1 and Uba2 (also referred to as SAE1 and SAE2). This enzyme has two important consecutive functions: First, it activates the matured SUMO di-glycine motif by adenylation, a step that consumes ATP. Subsequently, the adenylated SUMO is attacked by the conserved cysteine residue in Uba2 to form a highly reactive E1~SUMO thioester bond (see Fig. 2).
Regulation at this step of the SUMO cascade has dramatic consequences on global sumoylation as it was demonstrated for the CLEO virus protein Gam1. Gam1 functions as substrate adaptor for the ubiquitin E3 ligase complex Cul2/5-EloB/C-Roc1 and targets the E1 for proteasomal degradation (Boggio et al. 2004, 2007).
Another mechanism that is especially interesting in regard to neurodegenerative diseases is that oxidative stress can regulate global SUMO levels. Low concentrations of H2O2 were found to induce the formation of reversible disulfide bonds between the catalytic cysteines of the E1 subunit Uba2 and the E2 enzyme Ubc9. Such an impairment in the catalytic activity of the two key enzymes of SUMO conjugation results in a global loss of sumoylation by shifting the equilibrium to desumoylation of most cellular SUMO targets (Bossis and Melchior 2006). Furthermore, the activity of the E1 enzyme can be modulated by sumoylation of the catalytic subunit Uba2. This modification neither effects adenylation nor thioester formation activity, but impairs the transfer of SUMO to the E2 and accordingly global substrate modification. Upon heat shock, E1 sumoylation is reduced, which correlates with an increase in SUMO conjugation (Truong et al. 2012). Currently, it is not clear whether this increase is indeed dependent on E1 desumoylation, as it is not known how limiting the E1 enzyme is in the cell. In conclusion, regulation of the E1 enzyme results in global changes in sumoylation, and therefore, it is conceivable that we hear more about regulation of this key sumoylation enzyme in the near future.
The Single E2 Conjugating Enzyme
The broker of SUMO conjugation is the E2 enzyme Ubc9. At first, it interacts with the SUMO charged E1 and takes SUMO from the E1 to its own catalytic cysteine, again forming a thioester bond (Ubc9~SUMO). From here, SUMO is transferred to the target and this step can be performed either directly or with the help of an E3 ligase or cofactor (see Fig. 2). As already mentioned above, Ubc9 can directly recognize the core SUMO consensus motif (see Fig. 3b, Bernier-Villamor et al. 2002), but this interaction is usually too fragile for an efficient SUMO transfer. Indeed, other mechanisms have evolved to either stabilize the interaction between Ubc9~SUMO and its substrate (see Fig. 3a, model I–IV) or accelerate the SUMO transfer from Ubc9~SUMO to the substrate (compare Fig. 3a, model V).
For substrate/Ubc9~SUMO stabilization, classical concepts involve E3 ligases that bind to both Ubc9 and substrate to ensure close proximity for an efficient transfer (Fig. 3a, model IV). As outlined above, another mechanism to stabilize substrate/Ubc9~SUMO interaction is by substrates possessing an additional binding interface for Ubc9 next to a SUMO consensus motif (see Fig. 3a, model III). This was described for the acidic residues of the NSDM motif (Yang et al. 2006), for the hydrophobic cluster of the HCSM (Matic et al. 2010), and for RanGAP1 that holds in addition to a HCSM, a unique binding interface with Ubc9 outside the catalytic cleft (Bernier-Villamor et al. 2002). Continuing in this trend, selected substrates have been observed with SIM motifs that contribute to SUMO~Ubc9 recruitment and enhance conjugation (Fig. 3a, model I, Klug et al. 2013; Knipscheer et al. 2008; Meulmeester et al. 2008; Zhu et al. 2008; Chang et al. 2011).
An increase in substrate/Ubc9 binding can be further obtained by posttranslational modifications on either side. The best understood examples for substrate modification are the different phosphorylation-dependent SUMO motifs discussed above, which upon modification enhance Ubc9 binding followed by their sumoylation (Hietakangas et al. 2006; Mohideen et al. 2009; Picard et al. 2012). Alternatively, regulation of Ubc9 itself can contribute to substrate selection, as it was shown for Ubc9 sumoylation (Knipscheer et al. 2008) and recently also for its acetylation (Hsieh et al. 2013). Whereas sumoylated Ubc9 in mammals shows enhanced affinity and activity for selected SIM containing SUMO substrates (see Fig. 3a, model II, Knipscheer et al. 2008), Ubc9 acetylation impairs the affinity and activity to NSDM containing SUMO substrates (Hsieh et al. 2013). Regulation of Ubc9 sumoylation positively controls a subset of SIM containing SUMO substrates, whereas its acetylation attenuates modification of NSDM containing substrates (Hsieh et al. 2013; Knipscheer et al. 2008). To date, it is still unclear where, when, and how Ubc9 is sumoylated or acetylated and what the respective functions in vivo are. On the other hand, Ubc9 deacetylation was demonstrated to take place upon hypoxia by the deacetylase sirtuin1 (SIRT1), thus activating the sumoylation of selected NSDM substrates (Hsieh et al. 2013).
In addition, Ubc9 was shown to be a target of S-nitrosation (Qu et al. 2007) and phosphorylation (Su et al. 2012; Tomasi et al. 2012). Interestingly, Ubc9 phosphorylation by CDK1/Cyclin B enhances Ubc9–SUMO thioester formation and consequently target modification in vitro (Su et al. 2012), suggesting a general consequence on Ubc9’s catalytic activity. A similar outcome was also reported for Rhes, a small Ras-related G-protein that is enriched in the striatum (Subramaniam et al. 2009).
We are beginning to better understand the different mechanisms regulating Ubc9, and it will be important to distinguish between global regulatory and more specific events with consequences on a limited number of substrates. In that sense, Ubc9 is unique in combining both, general functions with global consequences and specific functions on single or subsets of substrates. Hence, regulating Ubc9 by different posttranslational means is an elegant mechanism to selectively regulate a group of substrates upon a certain stimulus and it will be illuminating to learn more about these mechanisms and its implication in biological processes.
Ubc9 is an essential enzyme for sumoylation and its knockout in mice is lethal in early development, whereas deletion of one allele showed no detectable phenotype (Nacerddine et al. 2005). This indicates that Ubc9 is relative abundant and not limiting in the cell. However, different pathogens have evolved mechanisms to reduce Ubc9 levels to achieve global reduction in SUMO levels, as it is demonstrated for the bacterium Listeria monocytogenes and the viral protein HPVE6 (Heaton et al. 2011; Ribet et al. 2010). It would be interesting to examine the minimal levels of Ubc9 tolerated by the cell, both under constitutive and stress conditions, as well as at which levels it becomes pathogenic. Reduced Ubc9 levels were not the only pathologic state observed for the enzyme, as it has been reported to be overexpressed in diverse types of cancer, including glioma, the most common type of primary brain tumors (Dong et al. 2013; Li et al. 2013a; Mo and Moschos 2005; Moschos et al. 2007, 2010; Ronen et al. 2009; Zhao et al. 2012; Zhu et al. 2010; Yang et al. 2013). High Ubc9 levels were reported to promote cell proliferation, cell invasion, and metastasis and correlate with poor response to chemotherapy and poor clinical prognosis (Li et al. 2013a; Zhu et al. 2010; Chen et al. 2011). In line, it was reported that Ubc9 levels are controlled by various microRNAs, which are repressed in cancer tissues (Zhao et al. 2012; F. Wu et al. 2009). Surprisingly, 21-week-old transgenic mice that overexpress high Ubc9 levels show no gross abnormal phenotypes and appear not to develop tumors. Rather the opposite is true, as high Ubc9 levels showed a beneficial effect of increased global sumoylation that protects mouse brains against focal cerebral ischemic damage (Lee et al. 2011b). Concordantly, deep hypothermia that protects organ damage induced by transient ischemia also demonstrates elevated levels in global sumoylation and triggers nuclear translocation of Ubc9 in neurons, correlating with an increase in SUMO2/3 species in the nucleus (Wang et al. 2012).
In summary, Ubc9 presents the central enzyme in the SUMO conjugation pathway, equipped with manifold mechanisms that are in place to globally or specifically influence SUMO conjugation levels. Hence, it constitutes a fascinating target to regulate substrate modification by interfering with E1 interactions, SUMO binding, posttranslational modification, altered expression levels, or by altered binding to E3 enzymes. It will still be fascinating to ascertain the multifaceted roles of Ubc9 and how it contributes to health and disease.
The E3 Ligases
Proteins that catalyze the transfer of SUMO from the charged E2 enzyme onto a substrate are described as E3 ligating enzymes. Although originally assumed to behave as molecular scaffolds by bringing substrate and charged E2 in close proximity (see Fig. 3a, model IV), increasing evidence arises that priming the E2~SUMO thioester for efficient transfer is the core function for enhanced substrate sumoylation (see Fig. 3a, model V). The catalytic mechanisms are not mutually exclusive, but can be influenced by distinct non-covalent interactions between SUMO, the E3, the E2 or the substrate as outlined in Fig. 3. Currently, only a handful of proteins have been observed to significantly enhance substrate sumoylation in vitro and in vivo. However, for many of these regulators, it has not been clearly demonstrated whether they indeed catalyze sumoylation or rather function as cofactors of sumoylation.
Yeast Siz1 and Siz2 proteins together with their mammalian homologs, the proteins inhibitor of STAT (PIAS) family, were the first SUMO E3 ligases uncovered. Several family members have been identified, so far: PIAS1, the PIASxα/PIASxβ isoforms, PIAS3, and PIASy (Kahyo et al. 2001; Sachdev et al. 2001; Sapetschnig et al. 2002; Schmidt and Muller 2002; Johnson and Gupta 2001; Kotaja et al. 2002). All PIAS and Siz proteins share a zinc finger domain that is structurally related to that of RING and U-box domain containing ubiquitin E3 ligases. Therefore, this group of SUMO E3 ligases was designated as Siz/PIAS (SP)-RING family (Hochstrasser 2001). A remote relative that also possesses such a SP-RING, but is otherwise unrelated, is the Nse2/Mms21 protein, which is a multiprotein complex component required for DNA repair (Andrews et al. 2005; Potts and Yu 2005; Zhao and Blobel 2005). In addition to the central SP-RING, the PIAS family also bears other functional domains or motifs: The N-terminal SAP (scaffold attachment factor-A/B, acinus, and PIAS) domain, a PINIT motif, a unique C-terminal domain (SP-CTD), a SIM, and a variable serine/threonine-rich C-terminal region (S/T) that flank the SP-RING domain, and together influence downstream PIAS effects not restricted to SUMO conjugation (Yunus and Lima 2009 and reviewed in Gareau and Lima 2010, Rytinki et al. 2009).
The SAP domain, the PINIT domain, and also the C-terminus are involved in protein interactions and regulate the intracellular localization and substrate modification in vivo, though these domains are dispensable for core E3 functions in vitro (Yunus and Lima 2009). This was surprising, as PIAS family E3 ligases were originally assumed to function as scaffolds to bring the charged E2 in close proximity to its substrates (compare Fig. 3). Structural and functional analysis indicated that the SP-RING, along with the SP-CTD, is sufficient to activate the charged Ubc9~SUMO for substrate conjugation. As acidic residues in the SP-CTD and basic residues on the SUMO surface are required for activity, it is proposed that the SP-RING binds to Ubc9 and the donor SUMO gets optimally positioned via the SP-CTD domain for efficient transfer (Yunus and Lima 2009). Indeed, such a mechanism for donor ubiquitin priming was recently demonstrated for a related ubiquitin RING E3 ligase (Plechanovova et al. 2012).
SP-RING E3 ligases are involved in diverse cellular functions including transcriptional regulation (e.g., reviewed in Rytinki et al. 2009; Schmidt and Muller 2003; Sharrocks 2006), DNA repair (X. L. Chen et al. 2007; Morris et al. 2009; Silver et al. 2011; Zhao and Blobel 2005 and reviewed in Branzei et al. 2006; Galanty et al. 2009; Potts 2009; Zlatanou and Stewart 2010), cell cycle (e.g., Azuma et al. 2005; Ryu and Azuma 2010), apoptosis (e.g., Zhang et al. 2010; Liu and Shuai 2001), cell migration and invasion (Castillo-Lluva et al. 2010), neuroreceptor regulation (Dutting et al. 2011), and oxidative stress response (Leitao et al. 2011). However, although different substrates for each pathway are identified, it is currently not clear whether and how substrate specificity is performed, as all SP-RING E3 ligases have broad target spectra of substrates. One could also envision that upon specific stimuli, waves of multiple substrates are sumoylated, as it is reported for cell cycle and DNA repair regulation or upon heat shock (Johnson and Blobel 1999; Psakhye and Jentsch 2012; Golebiowski et al. 2009).
Concordant with low substrate specificity of individual PIAS E3 ligases, diverse knock out studies of PIAS family members demonstrate only modest defects indicating large redundancy between the different family members (Liu et al. 2004; Roth et al. 2004; Santti et al. 2005; Wong et al. 2004). In agreement, also the yeast Siz1 and Siz2 are not essential for viability (Johnson and Gupta 2001). One idea how substrate specificity can be achieved in vivo comes from a highly regulated co-occurrence of substrate and its E3 ligase in a spatial and temporal manner that would imply transcriptional and post-transcriptional regulation. Again, one prime example comes from yeast showing that during mitosis, Siz1 gets phosphorylated and translocates from the nucleus to the bud neck, where it encounters its substrates (Johnson and Gupta 2001). Also PIASxα, PIAS1, and likely other PIAS family members were found to be regulated by phosphorylation, involving various kinases in a stimulus-dependent manner (Liu et al. 2005, 2007; Stehmeier and Muller 2009; Yang and Sharrocks 2006). In general, phosphorylation appears not to impair the catalytic activity of these E3 ligases per se, but clearly affects their ability to modulate substrate-dependent transcription in vivo (Stehmeier and Muller 2009; Yang and Sharrocks 2006). To clearly understand the underlying molecular mechanism, additional biochemical studies are required. Regulation of cellular localization is also reported for PIASy, which depends on autosumoylation for nuclear localization required for target modification (Ihara et al. 2005).
Other than phosphorylation and sumoylation, PIAS3 can be regulated by nitric oxide via S-nitrosation, which destabilizes PIAS3 by promoting its interaction with the ubiquitin E3 ligase tripartite motif-containing 32 (Trim32, Qu et al. 2007). Also the ubiquitin E3 ligase Siah2 was shown to regulate cellular PIAS levels (Depaux et al. 2007). Tight regulation of PIAS family members is important during development, as it was shown in Xenopus laevis. PIAS family members are expressed throughout early development with overlapping expression, though distinct expression patterns were observed, like particular high expression of PIASy in neural and neural crest derivatives (Burn et al. 2011). Interestingly, deregulation of PIAS by overexpression disrupts mesoderm induction and impairs body axis formation (Burn et al. 2011), further highlighting the importance of tight regulation. Concordantly, increased PIAS3 expression was reported in a variety of cancer tissues including brain, lung, breast, prostate, and colon-rectum tumors (L. Wang and Banerjee 2004), as well as increased PIAS1 in human prostate cancer (Hoefer et al. 2012). Indeed, a mechanism leading to the upregulation was discovered for PIASy upon hypoxic stress (Cai et al. 2010). Together, these findings indicate that also SP-RING E3 ligases underlie a highly complex regulatory system that leaves much room for further investigations.
Structurally very different to SP-RING SUMO E3 ligases is the Ran-binding protein 2 (RanBP2), a large nuclear pore complex component showing SUMO E3 ligase properties (Pichler et al. 2002, 2004). The minimal catalytic region of RanBP2 only requires one out of two internal repeats, which are largely unstructured bearing no resemblance to other characterized E3 ligase domains (Pichler et al. 2004; Reverter and Lima 2005; Tatham et al. 2005). As this minimal domain binds both to Ubc9 and SUMO, it is discussed that it also primes the donor SUMO for efficient transfer (see Fig. 3a, model V, Reverter and Lima 2005). However, endogenous RanBP2 forms a stable complex with sumoylated RanGAP1 and a ‘structural’ inaccessible Ubc9 species, which together occupy one of the internal repeats, leaving the second repeat remaining for E3 activity (Werner et al. 2012). Indeed, this second repeat is able to recruit an additional ‘catalytic’ Ubc9 to the complex, but the corresponding predicted SIM appears not to be required for activity, thus questioning the Ubc9~SUMO priming model in context of the larger complex (Werner et al. 2012). The identification of an unconventional second SIM is likely, as the linking region together with the second repeat entity specifically binds to SUMO1, but not to SUMO2 (Tatham et al. 2005), raising the possibility for SUMO1 specificity of the larger RanBP2 complex. Indeed, a recent structural study revealed how the second repeat increased SUMO1 binding, as well as SUMO1-specific E3 ligase activity for the RanBP2 complex (Gareau et al. 2012).
As previously observed with the minimal catalytic domain of SP-RING SUMO ligases, no substrate-binding domain could be identified in the minimal catalytic fragment of RanBP2 (Pichler et al. 2002, 2004; Reverter and Lima 2005). However, RanBP2’s catalytic entity in context of the full-length protein is flanked by binding sites for transport receptors, suggesting transport cargos as putative substrates. Spatial and temporal specific substrate sumoylation is also supposed to play a role for RanBP2. Although restricted to the nuclear envelope during interphase (Hamada et al. 2011; Walde et al. 2012; Wu et al. 1995; Yokoyama et al. 1995), it gets enriched at kinetochores and mitotic spindle upon disassembly of the nuclear envelope in mitosis (Roscioli et al. 2012; Swaminathan et al. 2004; Joseph et al. 2002), which likely involves different substrate sets for the various localizations. On a physiologic level, RanBP2 null mice are embryonic lethal (Aslanukov et al. 2006). Although viable, animals with low amounts of RanBP2 develop severe aneuploidy and are highly sensitive to tumor formation because of chromosomal segregation defects in the absence of noticeable transport defects (Dawlaty et al. 2008). In addition, RanBP2 haploinsufficiency appears to confer an increased susceptibility to neurotoxic damage in the brain (Cho et al. 2012).
Other reports of SUMO E3 ligases include the chromobox protein homolog (CBX) 4/Polycomb (Pc) 2 homolog, a member of the polycomb repressor complex 1 (Wotton and Merrill 2007), the tumor suppressor p14ARF/cyclin-dependent kinase inhibitor 2A (Xirodimas et al. 2002; Woods et al. 2004; Tago et al. 2005), the histone deacetylase (HDAC) 4 (Lee et al. 2009; Yang et al. 2011b; Zhao et al. 2005), the serine/arginine-rich protein SF2/ASF (Pelisch et al. 2010), certain members of the TRIM protein family (Chu and Yang 2011), the transcription factor Krox20 (Garcia-Gutierrez et al. 2011), and topoisomerase I interacting protein (Topors) (Weger et al. 2005), which also function as a ubiquitin E3 ligase (Rajendra et al. 2004). Interestingly, phosphorylation switches the RING dispensable SUMO E3 ligase role to that of the RING-dependent ubiquitin function (Yang et al. 2009).
Another example of dual ubiquitin/SUMO E3 ligase functions has been reported for mitochondrial ubiquitin ligase activator of NF-κB (MULAN), a mitochondrial-anchored protein ligase (MAPL) involved in mitochondrial fission/fusion dynamics (Li et al. 2008; Braschi et al. 2009). Deregulation of mitochondrial homeostasis has been observed in the pathogenesis of several neurodegenerative disorders, and an active role for SUMO in these pathways needs to be further explored.
Also the meiotic yeast Zip3 and its mammalian homolog RNF212 are reported to function as SUMO E3 ligases (Cheng et al. 2006; Reynolds et al. 2013), although these proteins possess a classical ubiquitin RING E3 motif (Perry et al. 2005) seemingly required for SUMO E3 activity (Cheng et al. 2006). However, depletion of Zip3 in yeast results in an increase in meiotic SUMO conjugation (Cheng et al. 2006; Klug et al. 2013), contrary to what one would expect for a SUMO E3 ligase.
Recently, the yeast Ubc9 was shown to become inactivated in its conventional E2 functions upon sumoylation, but gains a role as cofactor for unmodified Ubc9 and positions the donor SUMO for an efficient transfer (see Fig. 3a, model V) important for meiotic SUMO chain formation (Klug et al. 2013).
However, together all these factors enhance sumoylation to a certain extent, but for many of them it is currently not clear whether the underlying mechanism indeed resembles that of a SUMO E3 ligase. As mentioned earlier, and in Fig. 3a, the E2 enzyme is inherently capable in mediating SUMO conjugation for some of the substrates. Modest quantities of a genuine E3 enzyme, at levels much lower than target proteins, are sufficient in enhancing target sumoylation. This feature needs to be clearly established for designating a protein as a SUMO E3 ligase. Furthermore, it is crucial to examine the effects of cryptic motifs (SIMs, SCMs, and additional binding interfaces) that would perhaps allow non-covalent interactions between the substrate and SUMO/E2/‘E3’, thereby influencing sumoylation activity. Careful biochemical and functional studies are necessary to verify SUMO E3 ligase roles for some of the above proteins, as well as for other, as yet uncharacterized proteins.
SUMO Proteases
SUMO proteases carry out two important tasks in the sumoylation pathway. First, they are required for maturing the expressed SUMO precursor proteins via their endopeptidase activity, and secondly, they remove SUMO from target proteins via their isopeptidase activity, thus feeding SUMO back into the conjugation cycle (see also Fig. 2). However, in conjunction with the conjugation machinery, the isopeptidase activity determines the steady state levels of individual substrate sumoylation in the cell. The first SUMO proteases, ubiquitin-like protease (Ulp) 1 and 2, were discovered in yeast and subsequently guided the discovery of mammalian SUMO proteases (Li and Hochstrasser 1999, 2000; Gong et al. 2000; Yeh et al. 2000). A family of six sentrin-specific proteases (SENP) in humans (SENP 1–3 and 5–7) is responsible for SUMO processing, deconjugation, and depolymerization activities (reviewed in Mukhopadhyay and Dasso 2007; Hay 2007; Hickey et al. 2012). Recently, two new SUMO protease classes have been uncovered: desumoylating isopeptidases (DESI) 1 and 2, as well as ubiquitin-specific protease-like (USPL) 1 (Shin et al. 2012; Schulz et al. 2012). All of the above enzymes are cysteine isopeptidases, with SENPs (clan CE/family C48) harboring a conserved core and a catalytic triad comprised of histidine, aspartate, and cysteine residues (Gong et al. 2000). In contrast, DESIs (clan CP/family C97) bear a catalytic cysteine/histidine dyad among other structural differences (Suh et al. 2012). USPL1 (clan CA/family C98) is dissimilar to both SENPs and DESIs, rather it is distantly related to deubiquitinating enzymes (DUBs) (Schulz et al. 2012, classification data from MEROPS database Rawlings et al. 2012).
Endopeptidase-mediated processing of precursor SUMO involves specific cleavage of the C-terminal tail, after the di-glycine motif, resulting in a mature protein. In vitro studies have shown that catalytic domains of SENP2 and SENP1 are more proficient in processing SUMO2 and SUMO1 precursors, respectively, while both showing limited activity toward the SUMO3 precursor that is instead processed better by SENP5 (Reverter and Lima 2004; Xu and Au 2005; Shen et al. 2006; Di Bacco et al. 2006; Gong and Yeh 2006). Structural characterization of SENP1 and SENP2 catalytic domains revealed how residues in the precursor tails of different paralogs influence their processing (Reverter and Lima 2004; Shen et al. 2006). Moreover, the observed absence of SUMO4 processing was attributed to an atypical proline in its tail sequence (Owerbach et al. 2005). Endopeptidase activities were absent for SENP6 and SENP7, while both DESI1 and USPL1 displayed weak activity on the precursors of SUMO1 and SUMO2 (Mukhopadhyay et al. 2006; Lima and Reverter 2008; Suh et al. 2012; Schulz et al. 2012).
Isopeptidase activity or deconjugation of sumoylated proteins has been observed, to varying degrees in vitro, with nearly all of the SUMO proteases. However, in vivo isopeptidase activity is regulated by subcellular localization of the SUMO proteases, as well as by their interactions with sumoylated substrates. The importance of SUMO isopeptidase regulation through cellular localization was first understood with the yeast proteases. Ulp2 is distributed within the nucleus, while Ulp1 is restricted, via its non-catalytic amino-terminal region, to the nucleoplasmic face of the nuclear pore complex (NPC) (Li and Hochstrasser 2003; Panse et al. 2003; Mossessova and Lima 2000). A certain proportion of Ulp1 is exported to the cytoplasm, again via export sequences within the amino-terminal region, to undertake crucial isopeptidase functions required for cell division (Johnson and Blobel 1999; Takahashi et al. 1999). Deletion of Ulp1’s amino-terminal region allows its distribution throughout the cell, which can be lethal at high levels due to indiscriminate isopeptidase activity, underlining the importance of its spatial regulation.
Subcellular localizations for most of the human SENPs are predominantly in the nucleus; SENP1 and SENP2 at discrete sub-nuclear compartments and the NPC (Gong et al. 2000; Bailey and O’Hare 2002; Hang and Dasso 2002; Zhang et al. 2002), SENP3 and SENP5 within the nucleolus (Gong and Yeh 2006; Di Bacco et al. 2006), and SENP6 throughout the nucleoplasm (Mukhopadhyay et al. 2006, 2010) with sub-populations at promyelocytic leukemia protein (PML) nuclear bodies (Hattersley et al. 2011). However, the mechanisms that regulate sub-nuclear localizations of SENPs are relatively unknown. In addition, SENP1 and SENP2 bear nuclear localization signals (NLS) and nuclear export signals (NES) in their amino-terminal regions that enable shuttling between the nucleus and cytoplasm (Kim et al. 2005; Itahana et al. 2006).
Cellular SENP1 is unique in its ability to deconjugate both SUMO1 and SUMO2, while SENP2, and most of the other SENPs, showed an in vivo preference for SUMO2/3 deconjugation (Kolli et al. 2010).
All the SENPs have amino-terminal extensions of varying lengths and sequences (reviewed in Mukhopadhyay and Dasso 2007), though structural and/or functional aspects are still very limited. Analogous to the regulatory mechanisms observed for yeast Ulp1, the amino-terminus of SENP2 also appears responsible for its localization to the nucleoplasmic face of the NPC and in addition contributes to its preference in SUMO2 deconjugation (Kolli et al. 2010; Hang and Dasso 2002). Interestingly during mitosis, a fraction of nucleolar SENP5 is relocalized to the cytoplasm, where it was observed to modulate the sumoylation status of mitochondrial proteins (Zunino et al. 2007). In particular, deconjugation of SUMO1 from dynamin-related protein (DRP) 1 (a mitochondrial GTPase) promotes its mitochondrial localization, thus driving mitochondrial fragmentation and its segregation (Harder et al. 2004; Zunino et al. 2007, 2009). Cytoplasmic localization was also observed for both DESI1 and 2, with a partly nuclear profile for the former (Shin et al. 2012). Low expression profiles were observed for USPL1, exclusively at nuclear Cajal bodies (Schulz et al. 2012).
Regulation of SUMO proteases can also occur through other mechanisms including transcription (Lee et al. 2011a), phosphorylation (Baldwin et al. 2009), proteasomal degradation (Itahana et al. 2006; Kuo et al. 2008; Yan et al. 2010), various stimuli like oxidative stress, oxygen deprivation/hypoxia (Huang et al. 2009; Xu et al. 2008; Cheng et al. 2007; Xu et al. 2010), and upon heat shock (Pinto et al. 2012). In yeast, mitotic phosphorylation of Ulp2 appeared to inhibit its isopeptidase functions with concomitant stabilization of sumoylated substrates (Baldwin et al. 2009; Bachant et al. 2002). Mammalian SENP3 is observed to be phosphorylated and constitutively degraded by the ubiquitin E3 ligase C-terminus of HSP70-interaction protein (CHIP) (Kuo et al. 2008; Yan et al. 2010). However, under mild oxidative stress, SENP3 is stabilized by the chaperone heat shock protein 90 (HSP90) relaxing its restrictive nucleolar localization, thus permitting deconjugation of a different set of substrates involved in transcriptional regulation (Huang et al. 2009). Increased expression of SENP2 stimulates desumoylation of transcription activators inducing transcription (Girdwood et al. 2003; Ross et al. 2002; Yang et al. 2003). Under genotoxic stress, SENP2 gets activated leading to its own transcriptional down-regulation in a negative feedback loop (Lee et al. 2011a).
Interestingly, low levels of reactive oxygen species (ROS) can reversibly inactivate enzymes of the SUMO conjugation machinery, thus contributing to significantly reduced overall sumoylation levels (Bossis and Melchior 2006). In the case of SENP1 and Ulp1, a reversible inhibition was observed at low levels of ROS with subsequent irreversible inactivation at higher levels, together suggesting the proteases could possibly function as redox sensors in cells (Xu et al. 2008). In contrast, global increases in SUMO2/3 conjugates have been observed in cellular systems upon heat shock and induced ischemia (W. Yang et al. 2012; Golebiowski et al. 2009). At least SUMO2/3, as well as yeast SMT3, is capable of forming SUMO polymers (Bencsath et al. 2002; Klug et al. 2013; Matic et al. 2008b), though it is not clear to which extent polymers of SUMO2/3 are involved in the above increase in SUMO conjugates. That SUMO chains are important in selected biological processes is underlined by SUMO proteases specialized in depolymerization of SUMO chains like Ulp2, SENP6, and SENP7 (Li and Hochstrasser 2000; Mukhopadhyay et al. 2006; Shen et al. 2009; Lima and Reverter 2008). Concurrently, the catalytic domains of a majority of the SENPs, but not SENP6, were observed to be thermo-sensitive, clarifying to some extent the cellular increase in SUMO2/3 conjugates upon heat shock (Pinto et al. 2012). A recent study also uncovered novel roles for SENP3 at the mitochondria and in the ischemic stress response pathway (Guo et al. 2013). Oxygen/glucose deprivation induced the lysosomal degradation of SENP3 and led to concomitant increase in SUMO2/3 conjugates, including DRP1 sumoylation. Following reoxygenation, SENP3 levels recover leading to deconjugation of the SUMO-modified DRP1 and its subsequent stabilization on mitochondria. DRP1 then induces mitochondrial fission, cytochrome c release, and finally caspase-induced cell death. Mitochondrial fusion/fission cycles are essential for cells to maintain healthy mitochondria. Earlier reports have uncovered the importance of phosphorylation and ubiquitination events in mitochondrial physiology (reviewed in Chan 2012; Youle and van der Bliek 2012). The emerging role for SUMO at the mitochondria highlights the dynamic cross talk between different posttranslational modifications in regulating essential cellular processes.
Roles for SUMO proteases in other crucial cellular processes like DNA repair, transcriptional regulation, ribosome biogenesis, among others, are an area of continued interest and have been recently reviewed (Hickey et al. 2012). While the above studies have revealed several functional and regulatory features for SENPs, aspects such as functional profiles of full-length SENPs and their substrate specificity will have to be examined further and will reveal novel and exciting insights.
Consequences of Protein Sumoylation on Neurological Functions
Protein sumoylation has proven to be an integral part in the regulation of many cellular pathways. Originally, phosphorylation and ubiquitination were considered as main regulators of the nervous system, but now also the emerging SUMO system further enhances the complexity of the system.
Already, the SUMO pathway could be linked to diverse neurologic functions and disorders (described in more detail in other chapters of this issue), though mechanistic details of how SUMO actually contributes to health and disease remain largely elusive. As sumoylation is often difficult to detect and can regulate protein functions in various aspects, it is often hard to identify underlying mechanistic consequences. In this last section, we would like to present examples of how sumoylation impacts specific substrates with implications on neuronal function and dysfunction.
An often observed phenomenon is the alteration of protein localization upon SUMO modification. A nice example is the recent observation that sumoylation of the tyrosyl DNA phosphodiesterase-1 (TDP1) is a prerequisite for efficient DNA repair in neurons (Hudson et al. 2012). TDP1 plays an important role in resealing single-strand DNA breaks (Takashima et al. 2002). Its sumoylation was observed to facilitate appropriate accumulation of TDP1 at DNA damage sites to ensure efficient repair activity (Hudson et al. 2012). Comparable results were obtained with the multifunctional protein DJ-1 that plays a role in Parkinson’s disease (PD). The disease-related mutation L166P, as well as the SUMO acceptor lysine mutant K130R, completely blocked the functionality of DJ-1 (Shinbo et al. 2006). It could be shown that the non-sumoylable K130R mutant impaired the translocation of DJ-1 to the nucleus, thus interfering with its ability to regulate transcription (Fan et al. 2008). Furthermore, sumoylation was found to modulate synaptic transmission and plasticity of neurons, by inducing the endocytotic internalization of the kainate receptor subunit GluR6a (Martin et al. 2007). In another instance, it was observed that hippocampal neuronal excitability could be regulated in a direct and graded manner by staged sumoylation of subunits of the potassium channel Kv2.1 (Plant et al. 2011).
A more indirect effect of protein localization was identified in Alzheimer’s disease (AD). It is widely accepted that the formation of aggregates containing amyloid-β (Aβ) constitutes a primary factor in disease development, as it is the major component of senile plaques (Masters et al. 1985). Already a decade ago, overexpression of SUMO was correlated with reduced targeting of Aβ into aggregates (Dorval et al. 2007; Li et al. 2003). Aβ is a cleavage product generated by β-secretase-mediated proteolysis of the amyloid precursor protein (APP) and its sumoylation reduces Aβ-aggregate formation (Zhang and Sarge 2008). Although it was originally assumed that sumoylation regulates the localization of Aβ, it turned out that the modified lysines of APP are located in close proximity to the β-secretase cleavage site, indicating that sumoylation rather blocks Aβ-production by steric interference with the protease-binding site. Taken together, these results indicate that the SUMO-mediated impairment of APP cleavage indirectly regulates the localization of its product resulting in reduced aggregation. In that sense, APP sumoylation stresses the importance for better understanding of detailed molecular mechanisms.
The SUMO-mediated targeting of proteins into subnuclear structures is a common phenomenon also observed in other neurodegenerative diseases (recently reviewed in Krumova and Weishaupt 2013). An interesting example is the SUMO modification of ataxin (ATX) 1. Mutant ATX1 (also referred to as ATX1-Q82) contains an extended CAG repeat and accumulates in intranuclear inclusion bodies in the course of the polyQ disease spinocerebellar ataxia type (SCA) 1. Wild type ATX1 was found to be sumoylated in a phosphorylation and NLS-dependent manner (Riley et al. 2005). However, the increased polyQ stretch of mutant ATX1-Q82 negatively correlates with its sumoylation (Riley et al. 2005) and additionally was linked with reduced export from the nucleus (Irwin et al. 2005), thus promoting the disease state. Together, these observations suggest that sumoylation facilitates ATX1 nuclear export and thus prevents the local accumulation and subsequent formation of pathogenic nuclear aggregates.
In another example, sumoylation was implicated in axonal trafficking of mRNA in sensory neurons. The La RNA chaperone protein binds to mRNA and mediates both anterograde and retrograde transport in axons. However, upon sumoylation only the anterograde transport is maintained, thus regulating the directionality of axonal mRNA transport (van Niekerk et al. 2007), and it will be exciting to gain further insights in understanding the underlying mechanism.
Apart from changes in spatial protein levels, disease development can also be facilitated by changed protein solubility. For example, this was observed in neurodegenerative synucleinopathies, where the aggregation prone protein α-synuclein is inducing a disease state. Sumoylation of α-synuclein on two lysine residues severely reduces aggregate formation, at least in vitro, while a sumoylation-deficient variant exacerbated aggregation and cytotoxicity in dopaminergic neurons (Krumova et al. 2011). Similarly, sumoylation was observed to attenuate the aggregation propensity of ATX7 (Janer et al. 2010) and the androgen receptor (Mukherjee et al. 2009). Together, these studies suggest a neuroprotective role for SUMO in maintaining the solubility of proteins prone to aggregation.
A pathogenic change of protein stability is another common cause of aggregate formation in neurodegenerative diseases. Just recently, it was reported that in SCA3 pathology, SUMO modification increases the stability of mutant ATX3, leading to higher toxicity and disease pathology (Zhou et al. 2013). However, sumoylation had no influence on the subcellular localization, aggregate formation, or more importantly the ubiquitination of mutant-type ATX3, suggesting the involvement of other processes. Another example how SUMO may regulate substrate stability is described for amyotrophic lateral sclerosis (ALS), a disease associated with the degeneration of motor neurons. Some cases of this disease involve mutations in the superoxide dismutase-1 (SOD1). Sumoylation of SOD1 is reported to modulate its expression level and aggregation ability (Fei et al. 2006) although the exact mechanism remains to be established.
Interestingly, in many neurodegenerative diseases, the inclusion bodies stain positive for SUMO and/or ubiquitin, and evidence suggests that their formation is facilitated by impaired proteasome function (Kim et al. 2011; Riedel et al. 2011). Given the cellular relevance of the ubiquitin-driven proteasomal degradation, it would be crucial to understand the overlapping roles of SUMO and ubiquitin in target regulation. In some cases, SUMO and ubiquitin directly compete for modification of target lysines, thus preventing degradation of proteins (recently summarized in Praefcke et al. 2012). On the other hand, SUMO can function as the exact opposite by delivering the sumoylated target to SUMO-targeted ubiquitin E3 ligases (StUbLs) consequently leading to their degradation (recently reviewed in Praefcke et al. 2012). Detailed studies of the dynamic conjugation/deconjugation cycles of specific proteins modified by both SUMO and ubiquitin are therefore essential to understand their role. In Huntington’s disease (HD), SUMO participates in the disease pathology in multiple ways: by competition for target lysines with ubiquitin and by altering protein stability. The soluble pathogenic fragment of Huntingtin (Httex1p) contains lysines targeted by both, SUMO and ubiquitin (Steffan et al. 2004). Ubiquitination sequesters the potentially toxic Httex1p to aggregates leading to degradation, thus relieving pathogenicity. In contrast, sumoylation rather stabilizes the protein and therefore reduces aggregate formation and increases toxicity. Currently, little is known about the cross talk between sumoylation and ubiquitination; however, lysine mutations blocking both modifications reduced the disease pathology, indicating additional functions (Steffan et al. 2004).
SUMO- and/or proteasome-mediated protein degradation is also implicated in the neuronal intranuclear inclusion disease (NIID) as SUMO1 and ubiquitin completely overlap in such inclusions (Pountney et al. 2003; Terashima et al. 2002; Ueda et al. 2002). However, as in many other known diseases, direct evidence of protein sumoylation is still missing and the role of SUMO in NIID pathology remains obscure. A proteomic approach to characterize SUMO1 positive inclusion bodies revealed some interesting hits including membrane trafficking involved proteins like dynamin-1, NSF, Unc-18-1, and the chaperone HSP90 (Pountney et al. 2008). Now these substrates constitute the foundation for detailed analysis, like if they are SUMO substrates themselves or rather bind to sumoylated proteins, as well as their relation to ubiquitin.
Together, these examples highlight the versatile manner how sumoylation can regulate its substrates underlining the importance to obtain mechanistic insights for each individual substrate.
Conclusion
Next to specific substrates, diverse enzymatic components of the SUMO machinery are prone to regulation themselves, resulting in changed enzymatic activity with consequences on specific or even global SUMO conjugation. For example, in the developing rat brain, the expression levels of the SUMO E2 enzyme Ubc9 and SUMO1 are adapted in a temporal and spatial manner (Loriol et al. 2012). Accordingly, a decrease in SUMO-conjugated substrates and a redistribution of the SUMO enzymes to dendritic sites were observed during maturation of neurons, suggesting that a concerted action of SUMO conjugation plays a role in the development of the central nervous system. Global changes in sumoylation can have positive or negative consequences in disease development. The finding that increased global sumoylation protects from brain damage upon ischemic challenge, but on the other hand appears to accelerate tumorigenesis, highlights the importance of understanding the detailed mechanism and consequences behind such phenomena.
The vast implications of SUMO in disease pathology make the pathway a general target of drug development. However, the global importance of the SUMO pathway for essential cellular processes makes it a difficult target for disease-specific therapeutic interventions, since usually many pathways are affected simultaneously. On the other hand, timed and limited alteration of global sumoylation, for example, to counteract changes in tumorigenesis or in response to ischemic challenge, could be a very interesting therapeutic approach in the future. Less global effects could be obtained by targeting specific E3 ligases or isopeptidases.
In summary, although many studies are now investigating the link between neurologic diseases and the SUMO pathway, mechanistic insights into how SUMO actually contributes to the pathology are still missing. Taking into consideration the amount of pathologic observations that are somehow connected to the SUMO pathway, it is important to increase our basic understanding of SUMO regulation in order to envision therapeutic options with limited ‘side effects’.
References
Andrews, E. A., Palecek, J., Sergeant, J., Taylor, E., Lehmann, A. R., & Watts, F. Z. (2005). Nse2, a component of the Smc5–6 complex, is a SUMO ligase required for the response to DNA damage. Molecular and Cellular Biology, 25(1), 185–196.
Aslanukov, A., Bhowmick, R., Guruju, M., Oswald, J., Raz, D., Bush, R. A., et al. (2006). RanBP2 modulates Cox11 and hexokinase I activities and haploinsufficiency of RanBP2 causes deficits in glucose metabolism. PLoS Genetics, 2(10), e177.
Azuma, Y., Arnaoutov, A., Anan, T., & Dasso, M. (2005). PIASy mediates SUMO-2 conjugation of Topoisomerase-II on mitotic chromosomes. EMBO Journal, 24(12), 2172–2182.
Bachant, J., Alcasabas, A., Blat, Y., Kleckner, N., & Elledge, S. J. (2002). The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Molecular Cell, 9(6), 1169–1182.
Bailey, D., & O’Hare, P. (2002). Herpes simplex virus 1 ICP0 co-localizes with a SUMO-specific protease. Journal of General Virology, 83(Pt 12), 2951–2964.
Baldwin, M. L., Julius, J. A., Tang, X., Wang, Y., & Bachant, J. (2009). The yeast SUMO isopeptidase Smt4/Ulp2 and the polo kinase Cdc5 act in an opposing fashion to regulate sumoylation in mitosis and cohesion at centromeres. Cell Cycle, 8(20), 3406–3419.
Bayer, P., Arndt, A., Metzger, S., Mahajan, R., Melchior, F., Jaenicke, R., et al. (1998). Structure determination of the small ubiquitin-related modifier SUMO-1. Journal of Molecular Biology, 280(2), 275–286.
Bencsath, K. P., Podgorski, M. S., Pagala, V. R., Slaughter, C. A., & Schulman, B. A. (2002). Identification of a multifunctional binding site on Ubc9p required for Smt3p conjugation. Journal of Biological Chemistry, 277(49), 47938–47945.
Bernier-Villamor, V., Sampson, D. A., Matunis, M. J., & Lima, C. D. (2002). Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell, 108(3), 345–356.
Bettermann, K., Benesch, M., Weis, S., & Haybaeck, J. (2012). SUMOylation in carcinogenesis. Cancer Letters, 316(2), 113–125.
Blomster, H. A., Imanishi, S. Y., Siimes, J., Kastu, J., Morrice, N. A., Eriksson, J. E., et al. (2010). In vivo identification of sumoylation sites by a signature tag and cysteine-targeted affinity purification. Journal of Biological Chemistry, 285(25), 19324–19329.
Boggio, R., Colombo, R., Hay, R. T., Draetta, G. F., & Chiocca, S. (2004). A mechanism for inhibiting the SUMO pathway. Molecular Cell, 16(4), 549–561.
Boggio, R., Passafaro, A., & Chiocca, S. (2007). Targeting SUMO E1 to ubiquitin ligases: a viral strategy to counteract sumoylation. Journal of Biological Chemistry, 282(21), 15376–15382.
Bohren, K. M., Nadkarni, V., Song, J. H., Gabbay, K. H., & Owerbach, D. (2004). A M55 V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. Journal of Biological Chemistry, 279(26), 27233–27238.
Bossis, G., & Melchior, F. (2006). Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Molecular Cell, 21(3), 349–357.
Branzei, D., Sollier, J., Liberi, G., Zhao, X., Maeda, D., Seki, M., et al. (2006). Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell, 127(3), 509–522.
Braschi, E., Zunino, R., & McBride, H. M. (2009). MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Reports, 10(7), 748–754.
Bruderer, R., Tatham, M. H., Plechanovova, A., Matic, I., Garg, A. K., & Hay, R. T. (2011). Purification and identification of endogenous polySUMO conjugates. EMBO Reports, 12(2), 142–148.
Burgess, R. C., Rahman, S., Lisby, M., Rothstein, R., & Zhao, X. (2007). The Slx5-Slx8 complex affects sumoylation of DNA repair proteins and negatively regulates recombination. Molecular and Cellular Biology, 27(17), 6153–6162.
Burn, B., Brown, S., & Chang, C. (2011). Regulation of early Xenopus development by the PIAS genes. Developmental Dynamics, 240(9), 2120–2126.
Cai, Q., Verma, S. C., Kumar, P., Ma, M., & Robertson, E. S. (2010). Hypoxia inactivates the VHL tumor suppressor through PIASy-mediated SUMO modification. PLoS ONE, 5(3), e9720.
Capili, A. D., & Lima, C. D. (2007). Structure and analysis of a complex between SUMO and Ubc9 illustrates features of a conserved E2-Ubl interaction. Journal of Molecular Biology, 369(3), 608–618.
Castillo-Lluva, S., Tatham, M. H., Jones, R. C., Jaffray, E. G., Edmondson, R. D., Hay, R. T., et al. (2010). SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nature Cell Biology, 12(11), 1078–1085.
Chan, D. C. (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annual Review of Genetics, 46, 265–287.
Chang, C. C., Naik, M. T., Huang, Y. S., Jeng, J. C., Liao, P. H., Kuo, H. Y., et al. (2011). Structural and functional roles of Daxx SIM phosphorylation in SUMO paralog-selective binding and apoptosis modulation. Molecular Cell, 42(1), 62–74.
Chen, S. F., Gong, C., Luo, M., Yao, H. R., Zeng, Y. J., & Su, F. X. (2011). Ubc9 expression predicts chemoresistance in breast cancer. Chin J Cancer, 30(9), 638–644.
Chen, X. L., Silver, H. R., Xiong, L., Belichenko, I., Adegite, C., & Johnson, E. S. (2007). Topoisomerase I-dependent viability loss in saccharomyces cerevisiae mutants defective in both SUMO conjugation and DNA repair. Genetics, 177(1), 17–30.
Cheng, J., Kang, X., Zhang, S., & Yeh, E. T. (2007). SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell, 131(3), 584–595.
Cheng, C. H., Lo, Y. H., Liang, S. S., Ti, S. C., Lin, F. M., Yeh, C. H., et al. (2006). SUMO modifications control assembly of synaptonemal complex and polycomplex in meiosis of Saccharomyces cerevisiae. Genes & Development, 20(15), 2067–2081.
Chiu, S. Y., Asai, N., Costantini, F., & Hsu, W. (2008). SUMO-specific protease 2 is essential for modulating p53-Mdm2 in development of trophoblast stem cell niches and lineages. PLoS Biology, 6(12), e310.
Cho, K. I., Searle, K., Webb, M., Yi, H., & Ferreira, P. A. (2012). Ranbp2 haploinsufficiency mediates distinct cellular and biochemical phenotypes in brain and retinal dopaminergic and glia cells elicited by the Parkinsonian neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Cellular and Molecular Life Sciences, 69(20), 3511–3527.
Chu, Y., & Yang, X. (2011). SUMO E3 ligase activity of TRIM proteins. Oncogene, 30(9), 1108–1116.
Cimarosti, H., Lindberg, C., Bomholt, S. F., Ronn, L. C., & Henley, J. M. (2008). Increased protein SUMOylation following focal cerebral ischemia. Neuropharmacology, 54(2), 280–289.
Comerford, K. M., Leonard, M. O., Karhausen, J., Carey, R., Colgan, S. P., & Taylor, C. T. (2003). Small ubiquitin-related modifier-1 modification mediates resolution of CREB-dependent responses to hypoxia. Proceedings of National Academy of Sciences of the United States of America, 100(3), 986–991.
Cubenas-Potts, C., & Matunis, M. J. (2013). SUMO: A multifaceted modifier of chromatin structure and function. Developmental Cell, 24(1), 1–12.
Danielsen, J. R., Povlsen, L. K., Villumsen, B. H., Streicher, W., Nilsson, J., Wikstrom, M., et al. (2012). DNA damage-inducible SUMOylation of HERC2 promotes RNF8 binding via a novel SUMO-binding Zinc finger. Journal of Cell Biology, 197(2), 179–187.
Datwyler, A. L., Lattig-Tunnemann, G., Yang, W., Paschen, W., Lee, S. L., Dirnagl, U., et al. (2011). SUMO2/3 conjugation is an endogenous neuroprotective mechanism. Journal of Cerebral Blood Flow and Metabolism, 31(11), 2152–2159.
Dawlaty, M. M., Malureanu, L., Jeganathan, K. B., Kao, E., Sustmann, C., Tahk, S., et al. (2008). Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell, 133(1), 103–115.
Depaux, A., Regnier-Ricard, F., Germani, A., & Varin-Blank, N. (2007). A crosstalk between hSiah2 and Pias E3-ligases modulates Pias-dependent activation. Oncogene, 26(46), 6665–6676.
Desterro, J. M., Rodriguez, M. S., Kemp, G. D., & Hay, R. T. (1999). Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. Journal of Biological Chemistry, 274(15), 10618–10624.
Desterro, J. M., Thomson, J., & Hay, R. T. (1997). Ubch9 conjugates SUMO but not ubiquitin. FEBS Letters, 417(3), 297–300.
Di Bacco, A., Ouyang, J., Lee, H. Y., Catic, A., Ploegh, H., & Gill, G. (2006). The SUMO-specific protease SENP5 is required for cell division. Molecular and Cellular Biology, 26(12), 4489–4498.
Dong, M., Pang, X., Xu, Y., Wen, F., & Zhang, Y. (2013). Ubiquitin-conjugating enzyme 9 promotes epithelial ovarian cancer cell proliferation in vitro. International Journal of Molecular Sciences, 14(6), 11061–11071.
Dorval, V., Mazzella, M. J., Mathews, P. M., Hay, R. T., & Fraser, P. E. (2007). Modulation of Abeta generation by small ubiquitin-like modifiers does not require conjugation to target proteins. Biochemical Journal, 404(2), 309–316.
Duda, D. M., van Waardenburg, R. C., Borg, L. A., McGarity, S., Nourse, A., Waddell, M. B., et al. (2007). Structure of a SUMO-binding-motif mimic bound to Smt3p-Ubc9p: conservation of a non-covalent ubiquitin-like protein-E2 complex as a platform for selective interactions within a SUMO pathway. Journal of Molecular Biology, 369(3), 619–630.
Dutting, E., Schroder-Kress, N., Sticht, H., & Enz, R. (2011). SUMO E3 ligases are expressed in the retina and regulate SUMOylation of the metabotropic glutamate receptor 8b. Biochemical Journal, 435(2), 365–371.
Erker, Y., Neyret-Kahn, H., Seeler, J. S., Dejean, A., Atfi, A., & Levy, L. (2013). Arkadia, a novel SUMO-targeted ubiquitin ligase involved in PML degradation. Molecular and Cellular Biology, 33(11), 2163–2177.
Evdokimov, E., Sharma, P., Lockett, S. J., Lualdi, M., & Kuehn, M. R. (2008). Loss of SUMO1 in mice affects RanGAP1 localization and formation of PML nuclear bodies, but is not lethal as it can be compensated by SUMO2 or SUMO3. Journal of Cell Science, 121(Pt 24), 4106–4113.
Fan, J., Ren, H., Fei, E., Jia, N., Ying, Z., Jiang, P., et al. (2008). Sumoylation is critical for DJ-1 to repress p53 transcriptional activity. FEBS Letters, 582(7), 1151–1156.
Fei, E., Jia, N., Yan, M., Ying, Z., Sun, Q., Wang, H., et al. (2006). SUMO-1 modification increases human SOD1 stability and aggregation. Biochemical and Biophysical Research Communications, 347(2), 406–412.
Flotho, A., & Melchior, F. (2013). Sumoylation: A regulatory protein modification in health and disease. Annual Review of Biochemistry, 82, 357–385.
Galanty, Y., Belotserkovskaya, R., Coates, J., & Jackson, S. P. (2012). RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes & Development, 26(11), 1179–1195.
Galanty, Y., Belotserkovskaya, R., Coates, J., Polo, S., Miller, K. M., & Jackson, S. P. (2009). Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature, 462(7275), 935–939.
Garcia-Dominguez, M., & Reyes, J. C. (2009). SUMO association with repressor complexes, emerging routes for transcriptional control. Biochimica et Biophysica Acta, 1789(6–8), 451–459.
Garcia-Gutierrez, P., Juarez-Vicente, F., Gallardo-Chamizo, F., Charnay, P., & Garcia-Dominguez, M. (2011). The transcription factor Krox20 is an E3 ligase that sumoylates its Nab coregulators. EMBO Reports, 12(10), 1018–1023.
Gareau, J. R., & Lima, C. D. (2010). The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nature Reviews Molecular Cell Biology, 11(12), 861–871.
Gareau, J. R., Reverter, D., & Lima, C. D. (2012). Determinants of small ubiquitin-like modifier 1 (SUMO1) protein specificity, E3 ligase, and SUMO-RanGAP1 binding activities of nucleoporin RanBP2. Journal of Biological Chemistry, 287(7), 4740–4751.
Geiss-Friedlander, R., & Melchior, F. (2007). Concepts in sumoylation: A decade on. Nature Reviews Molecular Cell Biology, 8(12), 947–956.
Geoffroy, M. C., & Hay, R. T. (2009). An additional role for SUMO in ubiquitin-mediated proteolysis. Nature Reviews Molecular Cell Biology, 10(8), 564–568.
Girdwood, D., Bumpass, D., Vaughan, O. A., Thain, A., Anderson, L. A., Snowden, A. W., et al. (2003). P300 transcriptional repression is mediated by SUMO modification. Molecular Cell, 11(4), 1043–1054.
Golebiowski, F., Matic, I., Tatham, M. H., Cole, C., Yin, Y., Nakamura, A., et al. (2009). System-wide changes to SUMO modifications in response to heat shock. Science Signaling, 2(72), ra24.
Gong, L., Kamitani, T., Fujise, K., Caskey, L. S., & Yeh, E. T. (1997). Preferential interaction of sentrin with a ubiquitin-conjugating enzyme, Ubc9. Journal of Biological Chemistry, 272(45), 28198–28201.
Gong, L., Millas, S., Maul, G. G., & Yeh, E. T. (2000). Differential regulation of sentrinized proteins by a novel sentrin-specific protease. Journal of Biological Chemistry, 275(5), 3355–3359.
Gong, L., & Yeh, E. T. (2006). Characterization of a family of nucleolar SUMO-specific proteases with preference for SUMO-2 or SUMO-3. Journal of Biological Chemistry, 281(23), 15869–15877.
Guo, C., Hildick, K. L., Luo, J., Dearden, L., Wilkinson, K. A., & Henley, J. M. (2013). SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO Journal, 32(11), 1514–1528.
Guo, D., Li, M., Zhang, Y., Yang, P., Eckenrode, S., Hopkins, D., et al. (2004). A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nature Genetics, 36(8), 837–841.
Guzzo, C. M., Berndsen, C. E., Zhu, J., Gupta, V., Datta, A., Greenberg, R. A., et al. (2012). RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Science Signaling, 5(253), ra88.
Hamada, M., Haeger, A., Jeganathan, K. B., van Ree, J. H., Malureanu, L., Walde, S., et al. (2011). Ran-dependent docking of importin-beta to RanBP2/Nup358 filaments is essential for protein import and cell viability. Journal of Cell Biology, 194(4), 597–612.
Hang, J., & Dasso, M. (2002). Association of the human SUMO-1 protease SENP2 with the nuclear pore. Journal of Biological Chemistry, 277(22), 19961–19966.
Harder, Z., Zunino, R., & McBride, H. (2004). Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Current Biology, 14(4), 340–345.
Hattersley, N., Shen, L., Jaffray, E. G., & Hay, R. T. (2011). The SUMO protease SENP6 is a direct regulator of PML nuclear bodies. Molecular Biology of the Cell, 22(1), 78–90.
Hay, R. T. (2007). SUMO-specific proteases: A twist in the tail. Trends in Cell Biology, 17(8), 370–376.
Heaton, P. R., Deyrieux, A. F., Bian, X. L., & Wilson, V. G. (2011). HPV E6 proteins target Ubc9, the SUMO conjugating enzyme. Virus Research, 158(1–2), 199–208.
Hecker, C. M., Rabiller, M., Haglund, K., Bayer, P., & Dikic, I. (2006). Specification of SUMO1- and SUMO2-interacting motifs. Journal of Biological Chemistry, 281(23), 16117–16127.
Hickey, C. M., Wilson, N. R., & Hochstrasser, M. (2012). Function and regulation of SUMO proteases. Nature Reviews Molecular Cell Biology, 13(12), 755–766.
Hietakangas, V., Anckar, J., Blomster, H. A., Fujimoto, M., Palvimo, J. J., Nakai, A., et al. (2006). PDSM, a motif for phosphorylation-dependent SUMO modification. Proceedings of National Academy of Sciences of the United States of America, 103(1), 45–50.
Hochstrasser, M. (2001). SP-RING for SUMO: New functions bloom for a ubiquitin-like protein. Cell, 107(1), 5–8.
Hoefer, J., Schafer, G., Klocker, H., Erb, H. H., Mills, I. G., Hengst, L., et al. (2012). PIAS1 is increased in human prostate cancer and enhances proliferation through inhibition of p21. American Journal of Pathology, 180(5), 2097–2107.
Hsieh, Y. L., Kuo, H. Y., Chang, C. C., Naik, M. T., Liao, P. H., Ho, C. C., et al. (2013). Ubc9 acetylation modulates distinct SUMO target modification and hypoxia response. EMBO Journal, 32(6), 791–804.
Huang, C., Han, Y., Wang, Y., Sun, X., Yan, S., Yeh, E. T., et al. (2009). SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. EMBO Journal, 28(18), 2748–2762.
Hudson, J. J., Chiang, S. C., Wells, O. S., Rookyard, C., & El-Khamisy, S. F. (2012). SUMO modification of the neuroprotective protein TDP1 facilitates chromosomal single-strand break repair. Nature Communications, 3, 733.
Ihara, M., Yamamoto, H., & Kikuchi, A. (2005). SUMO-1 modification of PIASy, an E3 ligase, is necessary for PIASy-dependent activation of Tcf-4. Molecular and Cellular Biology, 25(9), 3506–3518.
Irwin, S., Vandelft, M., Pinchev, D., Howell, J. L., Graczyk, J., Orr, H. T., et al. (2005). RNA association and nucleocytoplasmic shuttling by ataxin-1. Journal of Cell Science, 118(Pt 1), 233–242.
Itahana, Y., Yeh, E. T., & Zhang, Y. (2006). Nucleocytoplasmic shuttling modulates activity and ubiquitination-dependent turnover of SUMO-specific protease 2. Molecular and Cellular Biology, 26(12), 4675–4689.
Jackson, S. P., & Durocher, D. (2013). Regulation of DNA damage responses by ubiquitin and SUMO. Molecular Cell, 49(5), 795–807.
Janer, A., Werner, A., Takahashi-Fujigasaki, J., Daret, A., Fujigasaki, H., Takada, K., et al. (2010). SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Human Molecular Genetics, 19(1), 181–195.
Johnson, E. S., & Blobel, G. (1997). Ubc9p is the conjugating enzyme for the ubiquitin-like protein Smt3p. Journal of Biological Chemistry, 272(43), 26799–26802.
Johnson, E. S., & Blobel, G. (1999). Cell cycle-regulated attachment of the ubiquitin-related protein SUMO to the yeast septins. Journal of Cell Biology, 147(5), 981–994.
Johnson, E. S., & Gupta, A. A. (2001). An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell, 106(6), 735–744.
Johnson, E. S., Schwienhorst, I., Dohmen, R. J., & Blobel, G. (1997). The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO Journal, 16(18), 5509–5519.
Joseph, J., Tan, S. H., Karpova, T. S., McNally, J. G., & Dasso, M. (2002). SUMO-1 targets RanGAP1 to kinetochores and mitotic spindles. Journal of Cell Biology, 156(4), 595–602.
Kahyo, T., Nishida, T., & Yasuda, H. (2001). Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Molecular Cell, 8(3), 713–718.
Kim, J. H., & Baek, S. H. (2009). Emerging roles of desumoylating enzymes. Biochimica et Biophysica Acta, 1792(3), 155–162.
Kim, K. I., Baek, S. H., & Chung, C. H. (2002). Versatile protein tag, SUMO: Its enzymology and biological function. Journal of Cellular Physiology, 191(3), 257–268.
Kim, Y. M., Jang, W. H., Quezado, M. M., Oh, Y., Chung, K. C., Junn, E., et al. (2011). Proteasome inhibition induces alpha-synuclein SUMOylation and aggregate formation. Journal of the Neurological Sciences, 307(1–2), 157–161.
Kim, Y. H., Sung, K. S., Lee, S. J., Kim, Y. O., Choi, C. Y., & Kim, Y. (2005). Desumoylation of homeodomain-interacting protein kinase 2 (HIPK2) through the cytoplasmic-nuclear shuttling of the SUMO-specific protease SENP1. FEBS Letters, 579(27), 6272–6278.
Klug, H., Xaver, M., Chaugule, V. K., Koidl, S., Mittler, G., Klein, F., et al. (2013). Ubc9 sumoylation controls SUMO chain formation and meiotic synapsis in Saccharomyces cerevisiae. Molecular Cell, 50(5), 625–636.
Knipscheer, P., Flotho, A., Klug, H., Olsen, J. V., van Dijk, W. J., Fish, A., et al. (2008). Ubc9 sumoylation regulates SUMO target discrimination. Molecular Cell, 31(3), 371–382.
Knipscheer, P., van Dijk, W. J., Olsen, J. V., Mann, M., & Sixma, T. K. (2007). Noncovalent interaction between Ubc9 and SUMO promotes SUMO chain formation. EMBO Journal, 26(11), 2797–2807.
Kolli, N., Mikolajczyk, J., Drag, M., Mukhopadhyay, D., Moffatt, N., Dasso, M., et al. (2010). Distribution and paralogue specificity of mammalian deSUMOylating enzymes. Biochemical Journal, 430(2), 335–344.
Komander, D., & Rape, M. (2012). The ubiquitin code. Annual Review of Biochemistry, 81, 203–229.
Kotaja, N., Karvonen, U., Janne, O. A., & Palvimo, J. J. (2002). PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Molecular and Cellular Biology, 22(14), 5222–5234.
Krumova, P., Meulmeester, E., Garrido, M., Tirard, M., Hsiao, H. H., Bossis, G., et al. (2011). Sumoylation inhibits alpha-synuclein aggregation and toxicity. Journal of Cell Biology, 194(1), 49–60.
Krumova, P., & Weishaupt, J. H. (2013). Sumoylation in neurodegenerative diseases. Cellular and Molecular Life Sciences, 70(12), 2123–2138.
Kuo, M. L., den Besten, W., Thomas, M. C., & Sherr, C. J. (2008). Arf-induced turnover of the nucleolar nucleophosmin-associated SUMO-2/3 protease Senp3. Cell Cycle, 7(21), 3378–3387.
Lallemand-Breitenbach, V., Jeanne, M., Benhenda, S., Nasr, R., Lei, M., Peres, L., et al. (2008). Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nature Cell Biology, 10(5), 547–555.
Lamoliatte, F., Bonneil, E., Durette, C., Caron-Lizotte, O., Wildemann, D., Zerweck, J., et al. (2013). Targeted identification of SUMOylation sites in human proteins using affinity enrichment and paralog-specific reporter ions. Molecular & Cellular Proteomics. doi:10.1074/mcp.M112.025569.
Lee, M. H., Mabb, A. M., Gill, G. B., Yeh, E. T., & Miyamoto, S. (2011a). NF-kappaB induction of the SUMO protease SENP2: A negative feedback loop to attenuate cell survival response to genotoxic stress. Molecular Cell, 43(2), 180–191.
Lee, Y. J., Miyake, S., Wakita, H., McMullen, D. C., Azuma, Y., Auh, S., et al. (2007). Protein SUMOylation is massively increased in hibernation torpor and is critical for the cytoprotection provided by ischemic preconditioning and hypothermia in SHSY5Y cells. Journal of Cerebral Blood Flow and Metabolism, 27(5), 950–962.
Lee, Y. J., Mou, Y., Maric, D., Klimanis, D., Auh, S., & Hallenbeck, J. M. (2011b). Elevated global SUMOylation in Ubc9 transgenic mice protects their brains against focal cerebral ischemic damage. PLoS ONE, 6(10), e25852.
Lee, J. H., Park, S. M., Kim, O. S., Lee, C. S., Woo, J. H., Park, S. J., et al. (2009). Differential SUMOylation of LXRalpha and LXRbeta mediates transrepression of STAT1 inflammatory signaling in IFN-gamma-stimulated brain astrocytes. Molecular Cell, 35(6), 806–817.
Leitao, B. B., Jones, M. C., & Brosens, J. J. (2011). The SUMO E3-ligase PIAS1 couples reactive oxygen species-dependent JNK activation to oxidative cell death. FASEB J, 25(10), 3416–3425.
Li, W., Bengtson, M. H., Ulbrich, A., Matsuda, A., Reddy, V. A., Orth, A., et al. (2008). Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS ONE, 3(1), e1487.
Li, S. J., & Hochstrasser, M. (1999). A new protease required for cell-cycle progression in yeast. Nature, 398(6724), 246–251.
Li, S. J., & Hochstrasser, M. (2000). The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Molecular and Cellular Biology, 20(7), 2367–2377.
Li, S. J., & Hochstrasser, M. (2003). The Ulp1 SUMO isopeptidase: Distinct domains required for viability, nuclear envelope localization, and substrate specificity. Journal of Cell Biology, 160(7), 1069–1081.
Li, Z., Hu, Q., Zhou, M., Vandenbrink, J., Li, D., Menchyk, N., et al. (2013a). Heterologous expression of OsSIZ1, a rice SUMO E3 ligase, enhances broad abiotic stress tolerance in transgenic creeping bentgrass. Plant Biotechnology Journal, 11(4), 432–445.
Li, H., Niu, H., Peng, Y., Wang, J., & He, P. (2013b). Ubc9 promotes invasion and metastasis of lung cancer cells. Oncology Reports, 29(4), 1588–1594.
Li, Y., Wang, H., Wang, S., Quon, D., Liu, Y. W., & Cordell, B. (2003). Positive and negative regulation of APP amyloidogenesis by sumoylation. Proceedings of National Academy of Sciences of the United States of America, 100(1), 259–264.
Lima, C. D., & Reverter, D. (2008). Structure of the human SENP7 catalytic domain and poly-SUMO deconjugation activities for SENP6 and SENP7. Journal of Biological Chemistry, 283(46), 32045–32055.
Liu, B., Mink, S., Wong, K. A., Stein, N., Getman, C., Dempsey, P. W., et al. (2004). PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nature Immunology, 5(9), 891–898.
Liu, B., & Shuai, K. (2001). Induction of apoptosis by protein inhibitor of activated Stat1 through c-Jun NH2-terminal kinase activation. Journal of Biological Chemistry, 276(39), 36624–36631.
Liu, B., Yang, Y., Chernishof, V., Loo, R. R., Jang, H., Tahk, S., et al. (2007). Proinflammatory stimuli induce IKKalpha-mediated phosphorylation of PIAS1 to restrict inflammation and immunity. Cell, 129(5), 903–914.
Liu, B., Yang, R., Wong, K. A., Getman, C., Stein, N., Teitell, M. A., et al. (2005). Negative regulation of NF-kappaB signaling by PIAS1. Molecular and Cellular Biology, 25(3), 1113–1123.
Loftus, L. T., Gala, R., Yang, T., Jessick, V. J., Ashley, M. D., Ordonez, A. N., et al. (2009). Sumo-2/3-ylation following in vitro modeled ischemia is reduced in delayed ischemic tolerance. Brain Research, 1272, 71–80.
Loriol, C., Parisot, J., Poupon, G., Gwizdek, C., & Martin, S. (2012). Developmental regulation and spatiotemporal redistribution of the sumoylation machinery in the rat central nervous system. PLoS ONE, 7(3), e33757.
Mahajan, R., Gerace, L., & Melchior, F. (1998). Molecular characterization of the SUMO-1 modification of RanGAP1 and its role in nuclear envelope association. Journal of Cell Biology, 140(2), 259–270.
Martin, S., Nishimune, A., Mellor, J. R., & Henley, J. M. (2007). SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature, 447(7142), 321–325.
Masters, C. L., Multhaup, G., Simms, G., Pottgiesser, J., Martins, R. N., & Beyreuther, K. (1985). Neuronal origin of a cerebral amyloid: Neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO Journal, 4(11), 2757–2763.
Matic, I., Macek, B., Hilger, M., Walther, T. C., & Mann, M. (2008a). Phosphorylation of SUMO-1 occurs in vivo and is conserved through evolution. Journal of Proteome Research, 7(9), 4050–4057.
Matic, I., Schimmel, J., Hendriks, I. A., van Santen, M. A., van de Rijke, F., van Dam, H., et al. (2010). Site-specific identification of SUMO-2 targets in cells reveals an inverted SUMOylation motif and a hydrophobic cluster SUMOylation motif. Molecular Cell, 39(4), 641–652.
Matic, I., van Hagen, M., Schimmel, J., Macek, B., Ogg, S. C., Tatham, M. H., et al. (2008b). In vivo identification of human small ubiquitin-like modifier polymerization sites by high accuracy mass spectrometry and an in vitro to in vivo strategy. Molecular and Cellular Proteomics, 7(1), 132–144.
Meulmeester, E., Kunze, M., Hsiao, H. H., Urlaub, H., & Melchior, F. (2008). Mechanism and consequences for paralog-specific sumoylation of ubiquitin-specific protease 25. Molecular Cell, 30(5), 610–619.
Mo, Y. Y., & Moschos, S. J. (2005). Targeting Ubc9 for cancer therapy. Expert Opinion of Therapeutic Targets, 9(6), 1203–1216.
Mohideen, F., Capili, A. D., Bilimoria, P. M., Yamada, T., Bonni, A., & Lima, C. D. (2009). A molecular basis for phosphorylation-dependent SUMO conjugation by the E2 UBC9. Nature Structural & Molecular Biology, 16(9), 945–952.
Morris, J. R., Boutell, C., Keppler, M., Densham, R., Weekes, D., Alamshah, A., et al. (2009). The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature, 462(7275), 886–890.
Moschos, S. J., Jukic, D. M., Athanassiou, C., Bhargava, R., Dacic, S., Wang, X., et al. (2010). Expression analysis of Ubc9, the single small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, in normal and malignant tissues. Human Pathology, 41(9), 1286–1298.
Moschos, S. J., Smith, A. P., Mandic, M., Athanassiou, C., Watson-Hurst, K., Jukic, D. M., et al. (2007). SAGE and antibody array analysis of melanoma-infiltrated lymph nodes: Identification of Ubc9 as an important molecule in advanced-stage melanomas. Oncogene, 26(29), 4216–4225.
Mossessova, E., & Lima, C. D. (2000). Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Molecular Cell, 5(5), 865–876.
Mukherjee, S., Thomas, M., Dadgar, N., Lieberman, A. P., & Iniguez-Lluhi, J. A. (2009). Small ubiquitin-like modifier (SUMO) modification of the androgen receptor attenuates polyglutamine-mediated aggregation. Journal of Biological Chemistry, 284(32), 21296–21306.
Mukhopadhyay, D., Arnaoutov, A., & Dasso, M. (2010). The SUMO protease SENP6 is essential for inner kinetochore assembly. Journal of Cell Biology, 188(5), 681–692.
Mukhopadhyay, D., Ayaydin, F., Kolli, N., Tan, S. H., Anan, T., Kametaka, A., et al. (2006). SUSP1 antagonizes formation of highly SUMO2/3-conjugated species. Journal of Cell Biology, 174(7), 939–949.
Mukhopadhyay, D., & Dasso, M. (2007). Modification in reverse: The SUMO proteases. Trends in Biochemical Sciences, 32(6), 286–295.
Mullen, J. R., & Brill, S. J. (2008). Activation of the Slx5-Slx8 ubiquitin ligase by poly-small ubiquitin-like modifier conjugates. Journal of Biological Chemistry, 283(29), 19912–19921.
Nacerddine, K., Lehembre, F., Bhaumik, M., Artus, J., Cohen-Tannoudji, M., Babinet, C., et al. (2005). The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Developmental Cell, 9(6), 769–779.
Novatchkova, M., Bachmair, A., Eisenhaber, B., & Eisenhaber, F. (2005). Proteins with two SUMO-like domains in chromatin-associated complexes: The RENi (Rad60-Esc2-NIP45) family. BMC Bioinformatics, 6, 22.
Okuma, T., Honda, R., Ichikawa, G., Tsumagari, N., & Yasuda, H. (1999). In vitro SUMO-1 modification requires two enzymatic steps, E1 and E2. Biochemical and Biophysical Research Communications, 254(3), 693–698.
Ouyang, J., Shi, Y., Valin, A., Xuan, Y., & Gill, G. (2009). Direct binding of CoREST1 to SUMO-2/3 contributes to gene-specific repression by the LSD1/CoREST1/HDAC complex. Molecular Cell, 34(2), 145–154.
Owerbach, D., McKay, E. M., Yeh, E. T., Gabbay, K. H., & Bohren, K. M. (2005). A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochemical and Biophysical Research Communications, 337(2), 517–520.
Panse, V. G., Kuster, B., Gerstberger, T., & Hurt, E. (2003). Unconventional tethering of Ulp1 to the transport channel of the nuclear pore complex by karyopherins. Nature Cell Biology, 5(1), 21–27.
Pedrioli, P. G., Raught, B., Zhang, X. D., Rogers, R., Aitchison, J., Matunis, M., et al. (2006). Automated identification of SUMOylation sites using mass spectrometry and SUMmOn pattern recognition software. Nature Methods, 3(7), 533–539.
Pelisch, F., Gerez, J., Druker, J., Schor, I. E., Munoz, M. J., Risso, G., et al. (2010). The serine/arginine-rich protein SF2/ASF regulates protein sumoylation. Proceedings of National Academy of Sciences of the United States of America, 107(37), 16119–16124.
Perry, J., Kleckner, N., & Borner, G. V. (2005). Bioinformatic analyses implicate the collaborating meiotic crossover/chiasma proteins Zip2, Zip3, and Spo22/Zip4 in ubiquitin labeling. Proceedings of National Academy of Sciences of the United States of America, 102(49), 17594–17599.
Picard, N., Caron, V., Bilodeau, S., Sanchez, M., Mascle, X., Aubry, M., et al. (2012). Identification of estrogen receptor beta as a SUMO-1 target reveals a novel phosphorylated sumoylation motif and regulation by glycogen synthase kinase 3beta. Molecular and Cellular Biology, 32(14), 2709–2721.
Pichler, A., Gast, A., Seeler, J. S., Dejean, A., & Melchior, F. (2002). The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell, 108(1), 109–120.
Pichler, A., Knipscheer, P., Oberhofer, E., van Dijk, W. J., Korner, R., Olsen, J. V., et al. (2005). SUMO modification of the ubiquitin-conjugating enzyme E2-25K. Nature Structural & Molecular Biology, 12(3), 264–269.
Pichler, A., Knipscheer, P., Saitoh, H., Sixma, T. K., & Melchior, F. (2004). The RanBP2 SUMO E3 ligase is neither HECT- nor RING-type. Nature Structural & Molecular Biology, 11(10), 984–991.
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annual Review of Biochemistry, 70, 503–533.
Pilla, E., Moller, U., Sauer, G., Mattiroli, F., Melchior, F., & Geiss-Friedlander, R. (2012). A novel SUMO1-specific interacting motif in dipeptidyl peptidase 9 (DPP9) that is important for enzymatic regulation. Journal of Biological Chemistry, 287(53), 44320–44329.
Pinto, M. P., Carvalho, A. F., Grou, C. P., Rodriguez-Borges, J. E., Sa-Miranda, C., & Azevedo, J. E. (2012). Heat shock induces a massive but differential inactivation of SUMO-specific proteases. Biochimica et Biophysica Acta, 1823(10), 1958–1966.
Plant, L. D., Dowdell, E. J., Dementieva, I. S., Marks, J. D., & Goldstein, S. A. (2011). SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. Journal of General Physiology, 137(5), 441–454.
Plechanovova, A., Jaffray, E. G., Tatham, M. H., Naismith, J. H., & Hay, R. T. (2012). Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature, 489(7414), 115–120.
Potts, P. R. (2009). The Yin and Yang of the MMS21-SMC5/6 SUMO ligase complex in homologous recombination. DNA Repair (Amst), 8(4), 499–506.
Potts, P. R., & Yu, H. (2005). Human MMS21/NSE2 is a SUMO ligase required for DNA repair. Molecular and Cellular Biology, 25(16), 7021–7032.
Poulsen, S. L., Hansen, R. K., Wagner, S. A., van Cuijk, L., van Belle, G. J., Streicher, W., et al. (2013). RNF111/Arkadia is a SUMO-targeted ubiquitin ligase that facilitates the DNA damage response. Journal of Cell Biology, 201(6), 797–807.
Pountney, D. L., Huang, Y., Burns, R. J., Haan, E., Thompson, P. D., Blumbergs, P. C., et al. (2003). SUMO-1 marks the nuclear inclusions in familial neuronal intranuclear inclusion disease. Experimental Neurology, 184(1), 436–446.
Pountney, D. L., Raftery, M. J., Chegini, F., Blumbergs, P. C., & Gai, W. P. (2008). NSF, Unc-18-1, dynamin-1 and HSP90 are inclusion body components in neuronal intranuclear inclusion disease identified by anti-SUMO-1-immunocapture. Acta Neuropathologica, 116(6), 603–614.
Praefcke, G. J., Hofmann, K., & Dohmen, R. J. (2012). SUMO playing tag with ubiquitin. Trends in Biochemical Sciences, 37(1), 23–31.
Prudden, J., Pebernard, S., Raffa, G., Slavin, D. A., Perry, J. J., Tainer, J. A., et al. (2007). SUMO-targeted ubiquitin ligases in genome stability. EMBO Journal, 26(18), 4089–4101.
Prudden, J., Perry, J. J., Arvai, A. S., Tainer, J. A., & Boddy, M. N. (2009). Molecular mimicry of SUMO promotes DNA repair. Nature Structural & Molecular Biology, 16(5), 509–516.
Prudden, J., Perry, J. J., Nie, M., Vashisht, A. A., Arvai, A. S., Hitomi, C., et al. (2011). DNA repair and global sumoylation are regulated by distinct Ubc9 noncovalent complexes. Molecular and Cellular Biology, 31(11), 2299–2310.
Psakhye, I., & Jentsch, S. (2012). Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell, 151(4), 807–820.
Qu, J., Liu, G. H., Wu, K., Han, P., Wang, P., Li, J., et al. (2007). Nitric oxide destabilizes Pias3 and regulates sumoylation. PLoS ONE, 2(10), e1085.
Rajendra, R., Malegaonkar, D., Pungaliya, P., Marshall, H., Rasheed, Z., Brownell, J., et al. (2004). Topors functions as an E3 ubiquitin ligase with specific E2 enzymes and ubiquitinates p53. Journal of Biological Chemistry, 279(35), 36440–36444.
Rawlings, N. D., Barrett, A. J., & Bateman, A. (2012). MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res, 40(Database issue), D343–D350.
Reverter, D., & Lima, C. D. (2004). A basis for SUMO protease specificity provided by analysis of human Senp2 and a Senp2-SUMO complex. Structure, 12(8), 1519–1531.
Reverter, D., & Lima, C. D. (2005). Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature, 435(7042), 687–692.
Reynolds, A., Qiao, H., Yang, Y., Chen, J. K., Jackson, N., Biswas, K., et al. (2013). RNF212 is a dosage-sensitive regulator of crossing-over during mammalian meiosis. Nature Genetics, 45(3), 269–278.
Ribet, D., Hamon, M., Gouin, E., Nahori, M. A., Impens, F., Neyret-Kahn, H., et al. (2010). Listeria monocytogenes impairs SUMOylation for efficient infection. Nature, 464(7292), 1192–1195.
Riedel, M., Goldbaum, O., Wille, M., & Richter-Landsberg, C. (2011). Membrane lipid modification by docosahexaenoic acid (DHA) promotes the formation of alpha-synuclein inclusion bodies immunopositive for SUMO-1 in oligodendroglial cells after oxidative stress. Journal of Molecular Neuroscience, 43(3), 290–302.
Riley, B. E., Zoghbi, H. Y., & Orr, H. T. (2005). SUMOylation of the polyglutamine repeat protein, ataxin-1, is dependent on a functional nuclear localization signal. Journal of Biological Chemistry, 280(23), 21942–21948.
Ronen, O., Malone, J. P., Kay, P., Bivens, C., Hall, K., Paruchuri, L. P., et al. (2009). Expression of a novel marker, Ubc9, in squamous cell carcinoma of the head and neck. Head and Neck, 31(7), 845–855.
Roscioli, E., Di Francesco, L., Bolognesi, A., Giubettini, M., Orlando, S., Harel, A., et al. (2012). Importin-beta negatively regulates multiple aspects of mitosis including RANGAP1 recruitment to kinetochores. Journal of Cell Biology, 196(4), 435–450.
Ross, S., Best, J. L., Zon, L. I., & Gill, G. (2002). SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Molecular Cell, 10(4), 831–842.
Roth, W., Sustmann, C., Kieslinger, M., Gilmozzi, A., Irmer, D., Kremmer, E., et al. (2004). PIASy-deficient mice display modest defects in IFN and Wnt signaling. Journal of Immunology, 173(10), 6189–6199.
Rytinki, M. M., Kaikkonen, S., Pehkonen, P., Jaaskelainen, T., & Palvimo, J. J. (2009). PIAS proteins: Pleiotropic interactors associated with SUMO. Cellular and Molecular Life Sciences, 66(18), 3029–3041.
Ryu, H., & Azuma, Y. (2010). Rod/Zw10 complex is required for PIASy-dependent centromeric SUMOylation. Journal of Biological Chemistry, 285(42), 32576–32585.
Sachdev, S., Bruhn, L., Sieber, H., Pichler, A., Melchior, F., & Grosschedl, R. (2001). PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes & Development, 15(23), 3088–3103.
Saitoh, H., & Hinchey, J. (2000). Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. Journal of Biological Chemistry, 275(9), 6252–6258.
Santti, H., Mikkonen, L., Anand, A., Hirvonen-Santti, S., Toppari, J., Panhuysen, M., et al. (2005). Disruption of the murine PIASx gene results in reduced testis weight. Journal of Molecular Endocrinology, 34(3), 645–654.
Sapetschnig, A., Rischitor, G., Braun, H., Doll, A., Schergaut, M., Melchior, F., et al. (2002). Transcription factor Sp3 is silenced through SUMO modification by PIAS1. EMBO Journal, 21(19), 5206–5215.
Schmidt, D., & Muller, S. (2002). Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proceedings of National Academy of Sciences of the United States of America, 99(5), 2872–2877.
Schmidt, D., & Muller, S. (2003). PIAS/SUMO: New partners in transcriptional regulation. Cellular and Molecular Life Sciences, 60(12), 2561–2574.
Schulz, S., Chachami, G., Kozaczkiewicz, L., Winter, U., Stankovic-Valentin, N., Haas, P., et al. (2012). Ubiquitin-specific protease-like 1 (USPL1) is a SUMO isopeptidase with essential, non-catalytic functions. EMBO Reports, 13(10), 930–938.
Sekiyama, N., Arita, K., Ikeda, Y., Hashiguchi, K., Ariyoshi, M., Tochio, H., et al. (2010). Structural basis for regulation of poly-SUMO chain by a SUMO-like domain of Nip45. Proteins, 78(6), 1491–1502.
Shao, R., Zhang, F. P., Tian, F., Anders Friberg, P., Wang, X., Sjoland, H., et al. (2004). Increase of SUMO-1 expression in response to hypoxia: Direct interaction with HIF-1alpha in adult mouse brain and heart in vivo. FEBS Letters, 569(1–3), 293–300.
Sharrocks, A. D. (2006). PIAS proteins and transcriptional regulation–more than just SUMO E3 ligases? Genes & Development, 20(7), 754–758.
Shen, L. N., Geoffroy, M. C., Jaffray, E. G., & Hay, R. T. (2009). Characterization of SENP7, a SUMO-2/3-specific isopeptidase. Biochemical Journal, 421(2), 223–230.
Shen, L., Tatham, M. H., Dong, C., Zagorska, A., Naismith, J. H., & Hay, R. T. (2006). SUMO protease SENP1 induces isomerization of the scissile peptide bond. Nature Structural & Molecular Biology, 13(12), 1069–1077.
Shin, E. J., Shin, H. M., Nam, E., Kim, W. S., Kim, J. H., Oh, B. H., et al. (2012). DeSUMOylating isopeptidase: A second class of SUMO protease. EMBO Reports, 13(4), 339–346.
Shinbo, Y., Niki, T., Taira, T., Ooe, H., Takahashi-Niki, K., Maita, C., et al. (2006). Proper SUMO-1 conjugation is essential to DJ-1 to exert its full activities. Cell Death and Differentiation, 13(1), 96–108.
Silver, H. R., Nissley, J. A., Reed, S. H., Hou, Y. M., & Johnson, E. S. (2011). A role for SUMO in nucleotide excision repair. DNA Repair (Amst), 10(12), 1243–1251.
Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C., Slepko, N., et al. (2004). SUMO modification of Huntingtin and Huntington’s disease pathology. Science, 304(5667), 100–104.
Stehmeier, P., & Muller, S. (2009). Phospho-regulated SUMO interaction modules connect the SUMO system to CK2 signaling. Molecular Cell, 33(3), 400–409.
Su, Y. F., Yang, T., Huang, H., Liu, L. F., & Hwang, J. (2012). Phosphorylation of Ubc9 by Cdk1 enhances SUMOylation activity. PLoS ONE, 7(4), e34250.
Subramaniam, S., Sixt, K. M., Barrow, R., & Snyder, S. H. (2009). Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science, 324(5932), 1327–1330.
Suh, H. Y., Kim, J. H., Woo, J. S., Ku, B., Shin, E. J., Yun, Y., et al. (2012). Crystal structure of DeSI-1, a novel deSUMOylase belonging to a putative isopeptidase superfamily. Proteins, 80(8), 2099–2104.
Swaminathan, S., Kiendl, F., Korner, R., Lupetti, R., Hengst, L., & Melchior, F. (2004). RanGAP1*SUMO1 is phosphorylated at the onset of mitosis and remains associated with RanBP2 upon NPC disassembly. Journal of Cell Biology, 164(7), 965–971.
Tago, K., Chiocca, S., & Sherr, C. J. (2005). Sumoylation induced by the Arf tumor suppressor: A p53-independent function. Proceedings of National Academy of Sciences of the United States of America, 102(21), 7689–7694.
Takahashi, Y., Iwase, M., Konishi, M., Tanaka, M., Toh-e, A., & Kikuchi, Y. (1999). Smt3, a SUMO-1 homolog, is conjugated to Cdc3, a component of septin rings at the mother-bud neck in budding yeast. Biochemical and Biophysical Research Communications, 259(3), 582–587.
Takashima, H., Boerkoel, C. F., John, J., Saifi, G. M., Salih, M. A., Armstrong, D., et al. (2002). Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nature Genetics, 32(2), 267–272.
Tatham, M. H., Geoffroy, M. C., Shen, L., Plechanovova, A., Hattersley, N., Jaffray, E. G., et al. (2008). RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nature Cell Biology, 10(5), 538–546.
Tatham, M. H., Jaffray, E., Vaughan, O. A., Desterro, J. M., Botting, C. H., Naismith, J. H., et al. (2001). Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. Journal of Biological Chemistry, 276(38), 35368–35374.
Tatham, M. H., Kim, S., Jaffray, E., Song, J., Chen, Y., & Hay, R. T. (2005). Unique binding interactions among Ubc9, SUMO and RanBP2 reveal a mechanism for SUMO paralog selection. Nature Structural & Molecular Biology, 12(1), 67–74.
Terashima, T., Kawai, H., Fujitani, M., Maeda, K., & Yasuda, H. (2002). SUMO-1 co-localized with mutant atrophin-1 with expanded polyglutamines accelerates intranuclear aggregation and cell death. NeuroReport, 13(17), 2359–2364.
Tomasi, M. L., Tomasi, I., Ramani, K., Pascale, R. M., Xu, J., Giordano, P., et al. (2012). S-adenosyl methionine regulates ubiquitin-conjugating enzyme 9 protein expression and sumoylation in murine liver and human cancers. Hepatology, 56(3), 982–993.
Truong, K., Lee, T. D., & Chen, Y. (2012). Small ubiquitin-like modifier (SUMO) modification of E1 Cys domain inhibits E1 Cys domain enzymatic activity. Journal of Biological Chemistry, 287(19), 15154–15163.
Ueda, H., Goto, J., Hashida, H., Lin, X., Oyanagi, K., Kawano, H., et al. (2002). Enhanced SUMOylation in polyglutamine diseases. Biochemical and Biophysical Research Communications, 293(1), 307–313.
Ullmann, R., Chien, C. D., Avantaggiati, M. L., & Muller, S. (2012). An acetylation switch regulates SUMO-dependent protein interaction networks. Molecular Cell, 46(6), 759–770.
Uzunova, K., Gottsche, K., Miteva, M., Weisshaar, S. R., Glanemann, C., Schnellhardt, M., et al. (2007). Ubiquitin-dependent proteolytic control of SUMO conjugates. Journal of Biological Chemistry, 282(47), 34167–34175.
van Niekerk, E. A., Willis, D. E., Chang, J. H., Reumann, K., Heise, T., & Twiss, J. L. (2007). Sumoylation in axons triggers retrograde transport of the RNA-binding protein La. Proceedings of National Academy of Sciences of the United States of America, 104(31), 12913–12918.
Vyas, R., Kumar, R., Clermont, F., Helfricht, A., Kalev, P., Sotiropoulou, P., et al. (2013). RNF4 is required for DNA double-strand break repair in vivo. Cell Death and Differentiation, 20(3), 490–502.
Walde, S., Thakar, K., Hutten, S., Spillner, C., Nath, A., Rothbauer, U., et al. (2012). The nucleoporin Nup358/RanBP2 promotes nuclear import in a cargo- and transport receptor-specific manner. Traffic, 13(2), 218–233.
Wang, J. (2011). Cardiac function and disease: Emerging role of small ubiquitin-related modifier. Wiley Interdisciplinary Reviews: Systems Biology and Medicine, 3(4), 446–457.
Wang, L., & Banerjee, S. (2004). Differential PIAS3 expression in human malignancy. Oncology Reports, 11(6), 1319–1324.
Wang, C. Y., & She, J. X. (2008). SUMO4 and its role in type 1 diabetes pathogenesis. Diabetes Metabolism Research and Reviews, 24(2), 93–102.
Wang, Q., Wang, Y., Chen, L., He, L., Li, W., & Jiang, H. (2012). Expression characteristics of the SUMOylation genes SUMO-1 and Ubc9 in the developing testis and ovary of Chinese mitten crab, Eriocheir sinensis. Gene, 501(2), 135–143.
Weger, S., Hammer, E., & Heilbronn, R. (2005). Topors acts as a SUMO-1 E3 ligase for p53 in vitro and in vivo. FEBS Letters, 579(22), 5007–5012.
Wei, W., Yang, P., Pang, J., Zhang, S., Wang, Y., Wang, M. H., et al. (2008). A stress-dependent SUMO4 sumoylation of its substrate proteins. Biochemical and Biophysical Research Communications, 375(3), 454–459.
Weisshaar, S. R., Keusekotten, K., Krause, A., Horst, C., Springer, H. M., Gottsche, K., et al. (2008). Arsenic trioxide stimulates SUMO-2/3 modification leading to RNF4-dependent proteolytic targeting of PML. FEBS Letters, 582(21–22), 3174–3178.
Werner, A., Flotho, A., & Melchior, F. (2012). The RanBP2/RanGAP1*SUMO1/Ubc9 complex is a multisubunit SUMO E3 ligase. Molecular Cell, 46(3), 287–298.
Wilkinson, K. A., & Henley, J. M. (2010). Mechanisms, regulation and consequences of protein SUMOylation. Biochemistry Journal, 428(2), 133–145.
Wong, K. A., Kim, R., Christofk, H., Gao, J., Lawson, G., & Wu, H. (2004). Protein inhibitor of activated STAT Y (PIASy) and a splice variant lacking exon 6 enhance sumoylation but are not essential for embryogenesis and adult life. Molecular and Cellular Biology, 24(12), 5577–5586.
Woods, Y. L., Xirodimas, D. P., Prescott, A. R., Sparks, A., Lane, D. P., & Saville, M. K. (2004). p14 Arf promotes small ubiquitin-like modifier conjugation of Werners helicase. Journal of Biological Chemistry, 279(48), 50157–50166.
Wotton, D., & Merrill, J. C. (2007). Pc2 and SUMOylation. Biochemical Society Transactions, 35(Pt 6), 1401–1404.
Wu, J., Matunis, M. J., Kraemer, D., Blobel, G., & Coutavas, E. (1995). Nup358, a cytoplasmically exposed nucleoporin with peptide repeats, Ran-GTP binding sites, zinc fingers, a cyclophilin A homologous domain, and a leucine-rich region. Journal of Biological Chemistry, 270(23), 14209–14213.
Wu, F., Zhu, S., Ding, Y., Beck, W. T., & Mo, Y. Y. (2009). MicroRNA-mediated regulation of Ubc9 expression in cancer cells. Clinical Cancer Research, 15(5), 1550–1557.
Xie, Y., Kerscher, O., Kroetz, M. B., McConchie, H. F., Sung, P., & Hochstrasser, M. (2007). The yeast Hex3.Slx8 heterodimer is a ubiquitin ligase stimulated by substrate sumoylation. Journal of Biological Chemistry, 282(47), 34176–34184.
Xirodimas, D. P., Chisholm, J., Desterro, J. M., Lane, D. P., & Hay, R. T. (2002). P14ARF promotes accumulation of SUMO-1 conjugated (H)Mdm2. FEBS Letters, 528(1–3), 207–211.
Xu, Z., & Au, S. W. (2005). Mapping residues of SUMO precursors essential in differential maturation by SUMO-specific protease, SENP1. Biochemistry Journal, 386(Pt 2), 325–330.
Xu, Z., Lam, L. S., Lam, L. H., Chau, S. F., Ng, T. B., & Au, S. W. (2008). Molecular basis of the redox regulation of SUMO proteases: a protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB Journal, 22(1), 127–137.
Xu, Y., Zuo, Y., Zhang, H., Kang, X., Yue, F., Yi, Z., et al. (2010). Induction of SENP1 in endothelial cells contributes to hypoxia-driven VEGF expression and angiogenesis. Journal of Biological Chemistry, 285(47), 36682–36688.
Yan, S., Sun, X., Xiang, B., Cang, H., Kang, X., Chen, Y., et al. (2010). Redox regulation of the stability of the SUMO protease SENP3 via interactions with CHIP and Hsp90. EMBO Journal, 29(22), 3773–3786.
Yang, S. H., Galanis, A., Witty, J., & Sharrocks, A. D. (2006). An extended consensus motif enhances the specificity of substrate modification by SUMO. EMBO Journal, 25(21), 5083–5093.
Yang, S. H., Jaffray, E., Hay, R. T., & Sharrocks, A. D. (2003). Dynamic interplay of the SUMO and ERK pathways in regulating Elk-1 transcriptional activity. Molecular Cell, 12(1), 63–74.
Yang, X., Li, H., Zhou, Z., Wang, W. H., Deng, A., Andrisani, O., et al. (2009). Plk1-mediated phosphorylation of Topors regulates p53 stability. Journal of Biological Chemistry, 284(28), 18588–18592.
Yang, K., Moldovan, G. L., Vinciguerra, P., Murai, J., Takeda, S., & D’Andrea, A. D. (2011a). Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes & Development, 25(17), 1847–1858.
Yang, S. H., & Sharrocks, A. D. (2006). PIASxalpha differentially regulates the amplitudes of transcriptional responses following activation of the ERK and p38 MAPK pathways. Molecular Cell, 22(4), 477–487.
Yang, W., Sheng, H., Homi, H. M., Warner, D. S., & Paschen, W. (2008). Cerebral ischemia/stroke and small ubiquitin-like modifier (SUMO) conjugation—A new target for therapeutic intervention? Journal of Neurochemistry, 106(3), 989–999.
Yang, W., Thompson, J. W., Wang, Z., Wang, L., Sheng, H., Foster, M. W., et al. (2012). Analysis of oxygen/glucose-deprivation-induced changes in SUMO3 conjugation using SILAC-based quantitative proteomics. Journal of Proteome Research, 11(2), 1108–1117.
Yang, Y., Tse, A. K., Li, P., Ma, Q., Xiang, S., Nicosia, S. V., et al. (2011b). Inhibition of androgen receptor activity by histone deacetylase 4 through receptor SUMOylation. Oncogene, 30(19), 2207–2218.
Yang, W., Wang, L., Roehn, G., Pearlstein, R. D., Ali-Osman, F., Pan, H., et al. (2013). Small ubiquitin-like modifier 1-3 conjugation [corrected] is activated in human astrocytic brain tumors and is required for glioblastoma cell survival. Cancer Science, 104(1), 70–77.
Yeh, E. T., Gong, L., & Kamitani, T. (2000). Ubiquitin-like proteins: new wines in new bottles. Gene, 248(1–2), 1–14.
Yin, Y., Seifert, A., Chua, J. S., Maure, J. F., Golebiowski, F., & Hay, R. T. (2012). SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes & Development, 26(11), 1196–1208.
Yokoyama, N., Hayashi, N., Seki, T., Pante, N., Ohba, T., Nishii, K., et al. (1995). A giant nucleopore protein that binds Ran/TC4. Nature, 376(6536), 184–188.
Youle, R. J., & van der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science, 337(6098), 1062–1065.
Yunus, A. A., & Lima, C. D. (2009). Structure of the Siz/PIAS SUMO E3 ligase Siz1 and determinants required for SUMO modification of PCNA. Molecular Cell, 35(5), 669–682.
Zhang, F. P., Mikkonen, L., Toppari, J., Palvimo, J. J., Thesleff, I., & Janne, O. A. (2008). Sumo-1 function is dispensable in normal mouse development. Molecular and Cellular Biology, 28(17), 5381–5390.
Zhang, H., Saitoh, H., & Matunis, M. J. (2002). Enzymes of the SUMO modification pathway localize to filaments of the nuclear pore complex. Molecular and Cellular Biology, 22(18), 6498–6508.
Zhang, Y. Q., & Sarge, K. D. (2008). Sumoylation of amyloid precursor protein negatively regulates Abeta aggregate levels. Biochemical and Biophysical Research Communications, 374(4), 673–678.
Zhang, C., Yuan, X., Yue, L., Fu, J., Luo, L., & Yin, Z. (2010). PIASy interacts with p73alpha and regulates cell cycle in HEK293 cells. Cellular Immunology, 263(2), 235–240.
Zhao, X., & Blobel, G. (2005). A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proceedings of National Academy of Sciences of the United States of America, 102(13), 4777–4782.
Zhao, X., Sternsdorf, T., Bolger, T. A., Evans, R. M., & Yao, T. P. (2005). Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Molecular and Cellular Biology, 25(19), 8456–8464.
Zhao, Z., Tan, X., Zhao, A., Zhu, L., Yin, B., Yuan, J., et al. (2012). microRNA-214-mediated UBC9 expression in glioma. BMB Rep, 45(11), 641–646.
Zhou, Y. F., Liao, S. S., Luo, Y. Y., Tang, J. G., Wang, J. L., Lei, L. F., et al. (2013). SUMO-1 modification on K166 of polyQ-expanded ataxin-3 strengthens its stability and increases its cytotoxicity. PLoS ONE, 8(1), e54214.
Zhu, S., Sachdeva, M., Wu, F., Lu, Z., & Mo, Y. Y. (2010). Ubc9 promotes breast cell invasion and metastasis in a sumoylation-independent manner. Oncogene, 29(12), 1763–1772.
Zhu, J., Zhu, S., Guzzo, C. M., Ellis, N. A., Sung, K. S., Choi, C. Y., et al. (2008). Small ubiquitin-related modifier (SUMO) binding determines substrate recognition and paralog-selective SUMO modification. Journal of Biological Chemistry, 283(43), 29405–29415.
Zlatanou, A., & Stewart, G. S. (2010). A PIAS-ed view of DNA double strand break repair focuses on SUMO. DNA Repair (Amst), 9(5), 588–592.
Zunino, R., Braschi, E., Xu, L., & McBride, H. M. (2009). Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. Journal of Biological Chemistry, 284(26), 17783–17795.
Zunino, R., Schauss, A., Rippstein, P., Andrade-Navarro, M., & McBride, H. M. (2007). The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. Journal of Cell Science, 120(Pt 7), 1178–1188.
Acknowledgments
We thank all Pichler laboratory members for critical reading of the manuscript. The work in the Pichler laboratory is funded by the Max Planck Society, the Deutsche Forschungsgemeinschaft (DFG, PI 917/1-1, DFG-SPP1365 PI 917/2-1) and the Fritz Thyssen Stiftung (10.11.1.210).
Conflict of interest
The authors declare that there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Droescher, M., Chaugule, V.K. & Pichler, A. SUMO Rules: Regulatory Concepts and Their Implication in Neurologic Functions. Neuromol Med 15, 639–660 (2013). https://doi.org/10.1007/s12017-013-8258-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-013-8258-6