
Abstract
Sjögren’s syndrome (SjS) is a systemic autoimmune disease marked by xerostomia (dry mouth), keratoconjunctivitis sicca (eye dryness), and other systematic disorders. Its pathogenesis involves an inflammatory process that is characterized by lymphocytic infiltration into exocrine glands and other tissues. Although the development of ectopic lymphoid tissue and overproduction of autoantibodies by hyperactive B cells suggest that they may promote SjS development, treatment directed towards them fails to induce significant laboratory or clinical improvement. T cells are overwhelming infiltrators in most phases of the disease, and the involvement of multiple T cell subsets of suggests the extraordinary complexity of SjS pathogenesis. The factors, including various cellular subtypes and molecules, regulate the activation and suppression of T cells. T cell activation induces inflammatory cell infiltration, B cell activation, tissue damage, and metabolic changes in SjS. Knowledge of the pathways that link these T cell subtypes and regulation of their activities are not completely understood. This review comprehensively summarizes the research progress and our understanding of T cells in SjS, including CD4+ T cells, CD8+ TRM cells, and innate T cells, to provide insights into for clinical treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sjögren’s syndrome (SjS) is a relatively common systemic autoimmune disease, manifested by the destruction and dysfunction of exocrine glands due to the infiltration of activated mononuclear cells. The hallmarks of this disease are dryness of the mouth and eyes, fatigue, and joint pain. Primary SjS may present by itself without any other autoimmune diathesis. However, approximately 60% of SjS patients have coexisting autoimmune diseases, such as rheumatoid arthritis (RA), autoimmune thyroid disease, primary biliary cholangitis, and systemic lupus erythematosus (SLE) [1]. The majority of SjS patients are perimenopausal women, and the female/male ratio ranges from 9:1 to 20:1 [2]. Beyond severe organ involvement, ectopic lymphoid-like structures (ELS) are identified in situ due to abnormal proliferation and activation of T and B lymphocytes. The prevalence of B lymphoma, one of the most severe complications of SjS, is estimated at about 5%, which is 15–20 times higher than that of the general population [3].
However, the pathogenesis of SjS remains to be established. Current evidence suggests that three factors are important for the development of SjS. The first is the loss of immune tolerance. This is supported by elevated type I interferon (IFN) levels in the serum and salivary gland (SG) tissues, putatively related to an antecedent viral infection. The second factor is increased acinar cell and SG epithelial cell (SGEC) apoptosis, which evokes activation of T and nature killer (NK) cells through pro-inflammatory cytokines [4,5,6,7]. Thereafter, lymphocytic foci are established with the recruitment of B cells and antigen-presenting cells (APCs). This leads to an increased the production of interleukin 21 (IL-21), B cell activating factor (BAFF), and other cytokines that promote the activation of B cells and production of autoantibodies [8]. The third and final factor is that exocrine gland dysfunction occurs in conjunction with the presence of autoantibodies.
Although B cells produce autoantibodies, SjS is dominated by T cells in the early stages of disease. T cell activation leads to tissue damage and secretory dysfunction [9]. In 1983, it was noted that more than 75% of the infiltrating cells in the SGs of SjS patients were CD4+ T cells [10]. Recently, several researches have described the unique phenotype and potential pathogenic effects of CD8+ T cells in SjS [11,12,13,14]. In this review, the activation of T cells and the outcomes of their activation, as well as the possible biological treatments strategies, will be discussed.
T Cell Subsets in SjS
CD4+T Cells
In SjS, there are decreased CD4+ T cells in blood. In contrast, lymphocyte infiltration into SGs is increased, supporting the hypothesis that the lymphopenia in the periphery is due to the migration of CD4+ T cells into tissues [13, 15]. Previous studies have revealed that CD4+ T cells are crucial contributors to autoantibody production, lymphocytic infiltration, and even lymphoma development through releasing multiple cytokines [3, 13, 16]. Meanwhile, CD4+T cells can be divided into several subsets, and each of them may play different roles in SjS, which will be introduced comprehensively in the following sections.
Th1 and Th2 Cells
Increased levels of IL-1β, IL-6, tumor necrosis factor alpha (TNF-α), and IFN-γ are detected in saliva, with a predominance of IFN-γ+CD4+ T cells in the SGs of SjS patients [9, 17]. Type 1 T helper (Th1) cell-associated transcription factor T-bet and cytokine IFN-γ are enriched in the SG of SjS without germinal center (GC)-like structures (GLS) [16, 18]. Increased Th2 cell-derived cytokines, such as IL-10, in saliva are positively correlated with disease activity [19,20,21]. The presence of Th2 cell-related transcripts in glandular tissue is only found in patients with severe B cell accumulation [17]. For example, GATA binding protein 3 (GATA3), the Th2 cell-associated transcription factor, and its related cytokine IL-4 are almost exclusively detected in GC-positive SjS patients [16]. These data suggest a distinct cellular involvement at different stages during the development of SjS.
Th17 Cells
Th17 cells, expressing IL-17 and IL-22, chemokine receptors (CCR6, CCR4), and transcription factors (retinoic acid-related orphan receptor gamma t [ROR-γt], signal transducer, and activator of transcription 3 [STAT3]) play significant role in many autoimmune diseases, including RA and psoriasis [22,23,24]. In SjS, IL-17 is increased in CD4+ T cell-rich areas around SG ducts [25,26,27]. The frequency of Th17 cells is also increased in the periphery of SjS patients with moderate-to-high disease activity manifested as the elevated expression of ROR-γt and IL-17 in circulating memory CD4+ T cells [23].
T Follicular Helper Cells
T follicular helper (Tfh) cells, identified by the phenotype C-X-C chemokine receptor-type 5+-programmed cell death protein 1+ inducible co-stimulator+ B cell lymphoma 6+ (CXCR5+PD-1+ICOS+Bcl-6+), are essential for shaping the effector function and fate of B cells. They play a critical role in GC formation, affinity maturation, and the development of memory B cells, mainly through the secretion of IL-21 [28, 29]. Tfh cells are increased in the peripheral blood and SGs of SjS patients compared with healthy controls, and their level is positively correlated with the EULAR SjS disease activity index (ESSDAI) [30,31,32]. For example, Tfh cell infiltration is strongly enriched in SG of patients with ELS [29]. In addition, increased frequency of Tfh cells in peripheral blood has been suggested to be correlated with the presence of extraglandular manifestations (EGMs) [32]. Although IL-4 and GATA3 are Th2-associated molecules, IL-4-producing T cells within the follicles of B cells have the phenotype of Tfh cells and express high levels of CXCR5, ICOS, PD-1, IL-21, and Bcl-6 [33]. Therefore, Tfh cells may therefore be a source of IL-4 in the SGs of GLS positive patients.
Treg and Tr1 Cells
The role of regulatory T (Treg) cells in SjS is uncertain [34,35,36,37,38,39,40]. There are several reasons for these differences, including the disease state of patients, selected markers of Treg cells, and Treg/Th17 balance. For example, the percentage of CD4+CD25+ Treg cells in peripheral blood is reduced in SjS compared with controls but is elevated in patients with EGMs compared with patients with sicca symptoms only. Cluster of differentiation 25high forkhead box p3+ (CD25highFoxp3+) Treg cell is increased in type I IFN-positive patients with primary SjS (pSjS), along with enhancement of indoleamine 2,3-dioxygenase activity [41]. In addition, CD25 expression has been used in many studies to identify Treg cells in SjS patients, but CD25 expression can also be upregulated by activated CD4+ T cells [42]. Furthermore, soluble mediators, such as transforming growth factor beta (TGF-β), the level of which is increased in SGs of SjS patients than in controls, is required for both Treg and Th17 cell development [43, 44].
Despite these conflicting data, a specific subset of circulating CD4+ T cells that express low levels of CD25 is detectable in SjS patients. They express glucocorticoid-induced TNF receptor-related protein, a surface molecule on Treg cells in murine models. These T cells also present in SGs and possess a Treg phenotype, expressing Foxp3, TGF-β, and IL-10 but low levels of CD25. They are only expanded in patients with inactive disease and elicit strong inhibitory activity against autoreactive cells [45]. It has been found that the number of these regulatory cells in periphery and SGs is increased in SjS patients compared with healthy controls [39].
The percentage of Helios+ Treg cell is also increased in SjS patients compared with healthy controls. The frequency of them in SjS is inversely correlated with the ESSDAI and the level of autoantibodies, but is positively correlated with IFN-γ/IFN-α [46,47,48,49]. A prior study noted that the incidence of activated memory Treg cell, but not naïve Treg cell, is increased [50]. Another subset of T cells with regulatory functions, but unlike traditional Treg cells, type 1 regulatory T (Tr1) cells, is induced by IL-27, and is characterized by high expression of IL-10 and the ability to inhibit T cell responses. The frequency of Tr1 cells is significantly decreased in SjS mouse models and in patients with SjS [51, 52]. Regulatory T cell family-mediated negative regulation of the immune system has garnered much attention in SjS. However, the relationship between these regulatory T cells and the pathogenesis of SjS is still not completely understood.
Tfr Cells
It has been reported that Tfh differentiation and germinal center response rely on the regulation of follicular regulatory T (Tfr) cells. Tfr cells, carrying the phenotype CD4+CXCR5highPD-1highFoxp3+Blimp-1+, have been shown to suppress Tfh cell proliferation in vitro and limit Tfh and GC B cell numbers in vivo, thereby controlling GC B cell selection by limiting the proliferation of non-antigen-specific clones [53, 54].
Tfr cells are enriched in the blood and SGs of SjS patients [30, 31]. The Tfr/Tfh ratio in blood is significantly increased in SjS patients compared with non-SjS sicca patients. However, whether this ratio in blood is associated with increased disease activity in SGs remains controversial [30, 31]. Tfr cells, like Tfh cells, can migrate into the circulation from draining secondary lymphoid organs (SLO), and are known as circulating Tfr (cTfr) in both mice and humans [55, 56]. After priming by dendritic cells (DCs), cTfr cells can exit draining SLO even before T-B interaction and GC formation and possess distinct characteristics compared with GC-Tfr cells [57, 58]. Tfr cells undergo a robust proliferation but act as bystanders after immunization with antigens in mice [59]. It has also been shown that human Tfr cells in blood are indicators of ongoing humoral activity [56]. Therefore, increased cTfr cells may be associated with active lymphocyte proliferation and differentiation in SLOs, rather than with glandular inflammation. By contrast, it appears clear that the number of Tfr cells is positively correlated with the presence of ELS in SGs from patients with SjS [29].
CCR9+ Th Cells
A unique subset of T helper cells, which exhibit similar phenotypic characteristics as Tfh cells, including expression of Bcl-6, IL-21, c-Maf, and ICOS is increased in blood and SGs from patients with SjS [60,61,62]. These cells differ from Tfh cells in their specific expression of CCR9, limited expression of CXCR5, and higher cytokines producing capacity. In general, retinoic acid can induce the expression of CCR9 on lymphocytes to allow their migration towards tissues with high levels of the chemokine ligand CCL25 [63]. CCL25 protein and mRNA levels are elevated in the SGs of SjS patients compared with non-SjS sicca controls [62, 64]. Thus, CCL25/CCR9 axis may be involved in inflammatory cell infiltration and promote the development of SjS.
Moreover, CCR9+ T cells in blood, whether from SjS patients or controls, display higher expression of IL-7Rα and secrete higher levels of IL-21, IL-17, and IFN-γ compared with T helper cells expressing high levels of CXCR5+. And both CCR9+ T cells and CXCR5+ T cells are associated with B cell hyperactivity [62]. However, the numbers of CCR9+ T cells in the blood and SGs are much lower than Tfh cells, although they share similar or even a higher capacity to produce inflammatory cytokines. In this context, whether CCR9+ Th cell represents a novel therapeutic target remains to be further studied.
CD8+ T Cells
As histopathological staining has shown that the primary T cells that infiltrate SGs in SjS are CD4+ T cells, the role of CD4+ T cells in SjS has been far more extensively explored than CD8+ T cells. However, recent studies have demonstrated that CD8+T cells are also enriched in the SGs of SjS patients and are positively correlated with multiple disease signatures. Intriguingly, we observed that a very large proportion of T cells in the SGs are TRM cells, manifested as CD103+ and/or CD69+ in both SjS patients and SjS mouse models. Interestingly, the majority of TRM cells in SGs that express CD103 are CD69+CD8+ tissue resident memory T (TRM) cells rather than CD69+CD4+ TRM cells [11].
CD103, also known as αEβ7, is associated with TRM cell phenotype and acts as a molecular tether that attaches to E-cadherin–expressing epithelial cells [65, 66]. Mice infected by murine cytomegalovirus (MCMV) demonstrate that specific CD103+CD8+ TRM cells adopt intraepithelial localization in the SG by connecting with integrin CD103 [67]. Not all of the TRM cells express CD103, but they do express the membrane protein CD69 [68]. The expression of CD69 helps TRM cells resist chemotaxis that is induced by high concentrations of sphingosine-1-phosphate (S1P) in blood or lymphatic vessels and prevents TRM cells from entering circulation [69, 70]. Therefore, CD8+ T cells may reside in SGs and co-localize with acinar cells and salivary duct epithelial cells via the interaction between CD103 on CD8+ TRM cells and E-cadherin on epithelial cells in SjS.
Many studies have demonstrated that TRM cells are functionally active, which can promote an inflammatory response and lymphocytic infiltration via producing various immune molecules [65]. Studies in mice have shown that CD8+ TRM cells in SGs protect against viral infection through degranulation and production of IFN-γ [67]. Taken together, CD8+ TRM cells may critically contribute to in promoting SjS development, which will be discussed in subsequent sections.
CD4−CD8− (Double-Negative) T Cells
As mentioned above, CD4−CD8− double-negative (DN) T cells, whose pathogenic role has been identified in many autoimmune disorders, are IL-17-producing T cells [71]. They spontaneously produce IL-17, undergo expansion in peripheral blood, and infiltrate SGs in patients with SjS [72]. Their expansion and infiltration are associated with disease activity, such as the extent of glandular involvement, presence of GLS, and dryness symptoms [73]. Notably, consistent with the insufficient effect of corticosteroids on SjS patients, DN T cells from SjS patients, but not from controls, do not respond to dexamethasone in vitro [72]. Moreover, DN T cells reportedly increase the production of BAFF and IFN-γ in SjS [74, 75]. Therefore, DN T cells may contribute to the pathogenesis of SjS.
Innate T Cells
Innate T cells, including γδ T, mucosal-associated invariant T (MAIT), and invariant NK T (iNKT) cells, recognize antigen in a non-major histocompatibility complex class (MHC) class I or II restriction manner and rapidly respond upon activation [76, 77]. The frequency of γδ T cells is increased in the blood of SjS patients compared with healthy subjects [78]. However, they cannot be detected by immunohistochemistry in SGs from patients with SjS [79]. The frequency of MAIT cells is decreased in peripheral blood but increased in the SGs of SjS patients [80]. MAIT cells in SjS patients are enriched in CD4+ and naïve subpopulations, with reduce expression of CD69 and CD40L, and lower production of TNF and IFN-γ [80]. iNKT cells from pSjS patients have elevated ability to express IL-21, which may counter-regulate B cells to express granzyme B [81].
Initiation of T Cell Activation
Know yourself and know your enemy—The Art of War
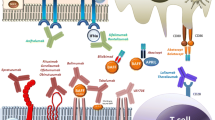
T cell activation is crucial to the initiation and regulation of immune responses. The central tenets of T cell activation are signaling through the T cell receptor (signal 1), co-stimulatory molecules (signal 2), and cytokines (signal 3) [82]. Three signals play a crucial role in optimal effector functions in the development and memory formation of T cells [83, 84]. The immune system is a very subtle machine with sophisticated brake systems to avoid excessive responses. Activating T cells upregulate the expression of co-stimulatory molecules as well as co-inhibitory proteins. These inhibitory molecules deliver negative signals to conventional T cells as well as promote the function of Treg cells to inhibit conventional T cell reactivity [85]. Understanding how the immune system works hopefully provides clues to explore possible treatments in different diseases [85] (Fig. 1).

Immune response in salivary gland. Immune responses in salivary gland during SjS comprise activation of innate and adaptive immune responses. Autoantigens released by apoptotic and damaged salivary gland epithelial cells are presented by antigen-presenting cells including DC, Mφ, and B cells, facilitating activation of CD4+ T and CD8+ T cell subsets. Activated T cells secrete inflammatory cytokines and promote further inflammation. DC, dendritic cell; Mφ, macrophage; Tfh, T follicular helper; ICOS, inducible T cell co-stimulator; ICOSL, ICOS ligand; IFN-γ, interferon gamma; IL, interleukin; TNF-α, tumor necrosis factor alpha; MHC, major histocompatibility complex class; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule; BAFF, B cell activating factor; TLR, Toll-like receptor; TCR, T cell receptor; TGF-β, transforming growth factor beta; S1P, sphingosine 1-phosphate
Antigen Presentation
Genetic risk haplotypes within human leukocyte antigen–DR isotype (HLA-DR) and HLA-DQ regions are positively associated with the pathogenesis of SjS in different ethnic groups, supporting a pathogenic role of class II MHC molecules in SjS. Cathepsin S (Cat S), a cysteine protease which is expressed in lysosomal and endosomal compartments of APCs, cleaves invariant chain, a 10 kDa fragment of the MHC-II-bound invariant chain, to promote the maturation of class II MHC molecules. The level of this protein is elevated in the tears of SjS patients [86, 87]. Treatment with Cat S inhibitors alters autoantigen presentation and significantly improves lymphocyte infiltration into SGs in an SjS mouse model [88]. The participation of class II MHC and the serological presence of various autoantibodies support a pivotal role of antigen presenting in the initiation of SjS.
The levels of professional APCs, such as DCs and macrophages, are also increased in SGs from patients with SjS and are positively correlated with disease severity [89, 90]. HLA-DR+ DCs located between epithelial cells and intercalated ducts are found in 5–10% of the striated and excretory duct walls in healthy human SGs [91]. An increased frequency of CD16+ monocytes is detected in SGs in SjS patients and may develop into DC-lysosome-associated membrane glycoprotein (LAMP)+ and CD83+ DCs, which have a high capacity for antigen presentation [92]. Plasmacytoid DCs (pDCs) are present in the SGs of SjS patients but not controls. Moreover, SGECs are thought as nonprofessional APCs because of their potential to present autoantigens to T cells via upregulating expression of class I and II MHC molecules via the stimulation of inflammatory cytokines.
B cells are also significantly increased in SGs of SjS patients. B effector cells have been described to promote the expansion and development of CD4+ T cells in a positive feedback loop. Activated B cells are capable of internalizing antigens through the B cell receptor (BCR). This is followed by processing and presenting peptides to CD4+ T cells in a class II MHC-independent manner, thereby affecting primary and memory CD4+ T cells responses [93]. HLA-DR15+ B cells in peripheral blood of multiple sclerosis patients rely on HLA molecules to present autoantigen to self-responsive CD4+ T cells to promote the self-proliferation of antigen-specific CD4+ T cells [94]. B cells also activate CD8+ T cells via cross-presentation or through activation of CD4+ T cells and the downstream effects of these CD4+ T cells. Spontaneous B cell-dependent CD8+ T cell activation occurs in systemic autoimmunity. It is likely that B cells promote CD8+ T cells activation first by activating CD4+ T cells, thereafter facilitating activation of CD8+ T cells either through production of cytokines and/or via interaction with APCs [95].
In summary, the accumulation of professional or nonprofessional APCs in SGs may present autoantigens to T cells, consequently inducing activation and aggregating of inflammatory cells in SGs. Indeed, increased expression of apoptotic molecules, such as B cell-2-associated X protein and Fas, in SGECs is initiated by pathogenic infection. Apoptosis of SGECs results in redistribution of the autoantigen La from the nucleus to the cytoplasm. Both Ro and La are found in apoptotic blebs and exosomes, and then are exposed to cell surface [96, 97]. Apoptotic particles are internalized by plasmacytoid dendritic cells (DCs) and stimulate toll-like receptor 7 (TLR-7) and TLR-9 expression, inducing the production of pro-inflammatory cytokines, which leads to generation of autoantibodies against the apoptotic materials [98, 99].
Co-Stimulation
Activated APCs in SGs from SjS patients manifest high expression of ligands for co-stimulatory receptors, such as CD80, CD86 and CD40, which may provide a strong co-stimulatory signal to facilitate T cell activation. For example, B7.2 (CD86) molecules expressed by SGECs prefer to interact with CD28 rather than the negative regulatory receptor-cytotoxic T lymphocyte-associated protein 4 [100]. Moreover, SGECs express functional TLRs − 2, − 3, and − 4 that recognize bacterial peptidoglycans, viral RNA, and lipopolysaccharides, respectively. The activation of TLR signaling leads to the production of inflammatory cytokines and upregulation of co-stimulatory and adhesion molecules which result in the activation of adaptive immune responses. Stimulating SGECs with corresponding TLR agonists induces the upregulation of MHC-I, intercellular adhesion molecule 1 (ICAM-1), CD40, and Fas molecules [101]. Therefore, SGECs are suspected to be involved in the activation of T cells in SjS by upregulating co-stimulatory molecules.
Cytokines
DCs or macrophage-derived IL-7, IL-12, and IL-17, in conjunction with SGEC-derived TNF, IL-1, and IL-6, significantly contribute to the inflammatory milieu, leading to consistent activation of T cells and enhanced survival and effector functions [102,103,104,105]. For example, compared with non-SjS, the expression level of IL-7 and IL-7R in the SGs of SjS patients is significantly increased, and their expression is positively correlated with the degree of inflammatory infiltration in the SGs [106]. Furthermore, IL-7 has the strong ability to promote secretion of Th1 cytokines (IFN-γ) and Th17 cytokines (IL-17) from the T cells of SjS patients in vitro [107]. GWAS data have shown that polymorphisms of IL-12 and STAT4 are risk factors for SjS. Mice that overexpress IL-12 show SjS-like symptoms [108]. APCs are the main sources of IL-12. IL-12 induces IFN-γ secretion, a key pathogenic cytokine in SjS. IL-12 prolongs the duration of the connection between CD8+ T cells and DCs, thereby increasing production of the chemokines CCL1 and CCL17 in APCs. Neutralization of these chemokines results in reduced interaction time and IFN-γ production, illustrating the importance of these APC-derived cytokines and chemokines in T cell activation [109].
Expression of IL-18 by APCs in the SGs of SjS patients is positively correlated with lymphocytic infiltration and the development of lymphoma. IL-18 may reinforce IL-12-induced Th1 responses [90, 110]. It has been demonstrated that SGECs from SjS patients can induce naïve CD4+ T cells differentiation into Tfh cells and maintenance of B cell survival in vitro. Mechanistically, IL-6 and ICOSL expression by SGECs are contributors to Tfh cell formation [111]. TGF-β, in synergy with IL-12 or IL-23, successfully induces the expression of typical Tfh signatures, CXCR5, Bcl-6, and IL-21, by human CD4+ T cells in vitro [112]. Therefore, a TGF-β-enriched milieu, coupled with increased production of IL-6 and IL-12 from SGECs and infiltration of myeloid cells into the SGs of SjS patients, probably gives rise to the activation and differentiation of naïve CD4+ T cells into specific Th cells, such as Tfh cells [43]. Sphingosine 1-phosphate (S1P) receptor, S1P1, and sphingosine kinase 1, which converts sphingosine to S1P, are detected in the cytoplasm of inflammatory mononuclear cells, vascular endothelial cells, and epithelium in all SGs of SjS patients. Interestingly, S1P has been shown to enhance the proliferation and IFN-γ production of CD4+ T cells, which increases apoptosis of SGECs by inducing Fas expression and Fas-mediated caspase-3 activation (Table 1) [125].
Others
In addition to the aforementioned three signals involved in T cell activation, other molecules expressed on APCs, such as vascular cell adhesion molecule-1 (VCAM-1) and ICAM-1, are also closely associated with the progression of various immunological disorders [136]. VCAM-1 (CD106), a glycoprotein that is inducible and predominantly expressed on endothelial cells, is a major regulator of leukocyte adhesion and trans-endothelial migration via interaction with α4β1 integrin [137]. In addition to endothelial cells, VCAM-1 is also expressed on the surface of macrophages, DCs, and activated SGECs. Furthermore, recruitment of macrophage, neutrophil, eosinophil, and mast cell precursors can be attenuated by utilizing anti-VCAM-1 antibody [138, 139]. Enhanced immunological synapse formation has been found in SjS [140]. A recent study reported that α4β1 integrin promoted accumulation of CD8+ TRM cells in SGs. These findings suggest that VCAM-1 may play an important role in T cell accumulation and activation in SGs during the progression of SjS [141].
In summary, the secretion of inflammatory cytokines and the expression of “assistant” molecules together promote the activation and recruitment of T cells, which further amplifies the inflammatory response.
Outcomes of T Cell Activation
Activated T cells continually stimulate the local inflammatory immune responses via activation of the innate immune system. APCs process antigen, which is followed by recruitment of leukocytes into target tissues by chemokines and inflammatory cytokines. This leads to amplification of the inflammatory response and aggravation of tissue damage. SjS is thought to be a T cell-dominated disease, as intralesional infiltrators are comprised principally by T cells at early stages of the disease.
Enhancing Antigen-Presenting
SjS is an autoimmune epithelitis that produces systemic inflammatory injury of epithelial cells. SGECs are not innocent bystanders as they produce large amounts of inflammatory cytokines. They also act as APCs and are stimulated by inflammatory cytokines, such as IFN-γ to upregulate expression of class II MHC [113]. IFN-γ and TNF-α are predominantly produced by Th1 cells and CD8+ cytotoxic T cells in SjS, and stimulate the expression of CD80, CD86, ICAM-1, VCAM, HLA class I antigens, and HLA-DR antigens on SGECs [113, 116, 117].
IFN-γ is a strong inducer of Cat S, whose expression in APCs in SGs from patients with SjS is significantly higher compared with controls. Meanwhile, IFN-γ, TNF-α, and IL-21 induce production of macrophage chemotactic protein and CCL20, which may participate in DC recruitment [123, 142, 143]. Activated CD4+ T cells strongly upregulate transcription levels of CCL3 and CCL4, key chemokines in the recruitment of activated macrophages [144,145,146,147]. Taken together, activated T cells in SG presumably promote the accumulation of APCs and reinforce their antigen presentation capacity during the development of SjS.
Lymphocyte Recruitment
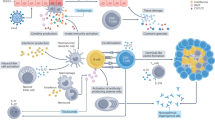
The interactions between chemokines and their receptors play a pivotal role in lymphocyte infiltration in SjS. These interactions lead to recruitment of monocytes and immature DCs to sites of inflammation, delivery of antigens to T cells within the lymphoid structures, and homing of T cells to sites of inflammation (Fig. 2). It has been found that there are decreased levels of chemokines CXCL8, CCL1, CCL4, CCL5, and CCL11 in the peripheral blood of SjS patients. In contrast, there is elevated expression of CCL3, CCL5, CCL17, CCL18, CCL19, CCL21, CCL22, CXCL8, CXCL9, CXCL10, and CXCL11 in SGs [148,149,150].

Crosstalk of salivary gland and periphery. In salivary glands, epithelial cells, DCs, and Mφ secrete chemokines and recruit T cells from periphery. Activated CD8+ T cells directly bind to salivary gland epithelial cells, leading to tissue damage. Activated CD4+ T and CD8+ T cell-derived cytokines further stimulate tissue inflammation. In draining lymph nodes, CD4+ T cells promote germinal center response and generation of autoantibodies from B cells. CCL, chemokine (C-C motif) ligand; CXCL, chemokine (C-X-C motif) ligand; CCR, CXC chemokine receptor; CXCR, CXC chemokine receptor
T cells in the SGs of SjS patients highly express CXCR3. CXCR3 ligands including CXCL9, CXCL10, and CXCL11 are primarily expressed by SGECs in SjS, and can be strongly induced by IFN-γ [151, 152]. Our previous work also showed that knockout of IFN-γ in a SjS mouse model hampered the infiltration of either CD4+ T cells or CD8+ T cells into SGs from draining lymph nodes (dLNs) [11]. IL-7 secreted by APCs may participate in enhancing the expression of CXCR3 ligands in a T cell- and IFN-γ-dependent manner [107]. IFN-γ derived from T cells may, therefore, contribute to lymphocyte infiltration into SGs. The CCR7 ligands CCL19 and CCL21 are induced by IL-1β and TNF-α. They function as chemoattractants for T cells and dendritic cells, and are increased in the SGs of SjS patients [153, 154].
IL-17-induced CXCL12 is found to recruit B cells and induce follicle formation in bronchus-associated lymphoid tissue in the lung [155]. IL-4 from Tfh cells is reported to trigger CXCL12 upregulation in human stromal cell precursors [156]. CXCL12 is associated with the infiltrated malignant B cell component and is possibly involved in the regulation of malignant B cell survival, which may play a key role in SjS [157,158,159]. IL-22 induces CXCL13 secretion in a mouse model of virus-induced lymphoid hyperplasia, and deficiency of IL-22 reduces B cell infiltration in SGs [126]. Moreover, local production of the type I IFNs by pDCs induces CXCL13 production in macrophages. pDC density is correlated with an increase of CXCR5+CD19+ B cells and CXCL13+CD68+ macrophages in the minor SGs of patients with SjS [160].
As previously mentioned, infiltrated T cells in SGs may induce the accumulation of APCs, such as macrophages and DCs. IL-1, TNF-α, and IL-4 promote the production of CCL2 from macrophages, DCs, and endothelial cells, and consequently induce CCR2+ monocyte and T cell aggregation [161, 162]. The expression of Th1 chemokines, thymus, and activation-regulated chemokine (TARC/CCL17) and macrophage-derived chemokine (MDC/CCL22), which are CCR4 ligands, mediate preferential Th2 recruitment. Both CCL17 and CCL22 are also increased significantly in SGs in SjS patients compared with controls and are strongly correlated with lymphocytic infiltration [163]. Furthermore, CCL17 and CCL22 are detected more strongly in and around the ductal epithelial cells in SGs with severe lymphocytic infiltration. CCR4 expression is increased in infiltrating lymphocytes in SGs in SjS [163, 164].
Chemokines may function as a stimulus for lymphocyte activation, activators of adhesion molecules, and drivers of leukocyte migration to inflammatory sites [165]. CXCL8, an inflammatory chemokine, is upregulated by IL18 and IL-17 in the ductal epithelium of SGs [25]. CXCL10 is induced by IFN-γ to recruit CXCR3+ T cells into SGs in SjS, but there is no significant association between CXCL10 and the focus score. Similar to CXCL8, CXCL10 is an inflammatory rather than a homoeostatic chemokine. A recent study demonstrated the role of either CXCL10 or CXCL9 as inflammatory regulators of CD8+ T cells to control tumor growth rather than as chemoattractants [166]. Salivary levels of CXCL8 and CXCL10 are elevated and found to be associated with SG dysfunction [167].
RANTES/CCL5, which is generated predominantly by CD8+ T cells and epithelial cells and is involved in Th1 cell migration, is significantly higher in the SGs of SjS patients compared with controls [163, 168]. Intriguingly, CD8+ T cells in RANTES−/− mice have poor cytokine production and manifest higher expression of inhibitory receptors. This suggests an important role for CCL5 in sustaining a CD8+ T cell response in viral infections [169]. Increased levels of chemokines play a crucial role in sustaining local lymphocyte activation and lymphocytic infiltration (Table 2).
Facilitating B Cell Activation
T cells and B cells can both infiltrate the epithelium and induce hyperplasia of duct cells to form lymphoepithelial lesions. The numbers of T and B cells are both significantly increased in SGs in SjS. T cells are predominant in lymphoepithelial lesions in less severe stages, whereas B cells outnumber T cells in more severe stages and occasionally form GCs, which consequently enhance the risk of B cell lymphoma, particularly mucosa-associated lymphoid tissue lymphomas in SGs [172] (Fig. 3).

Immune response in salivary gland. Stimulated salivary gland epithelial cells activate CD4+ T cells through MHC class II molecules, co-stimulatory molecules, and cytokines. Then, these activated CD4+ T cells can differentiate into CXCR5+ Tfh cells, which increase B cell survival, proliferation, and plasma cell differentiation. Peripheral cTfh cells, including CCR7lowPD-1high and CCR7highPD-1low subsets, also participate in the progress of the SjS. Cytokines and chemokines secreted by Tfh cells, as crucial factors, are involved in the activation of B cells, including B cell activation, isotype switching, migration, and localization of B cells. Activated T cells and B cells form a positive feedback loop, which lead to formation of ectopic germinal centers and plasma cell differentiation. Thereby, a large number of autoantibodies produced by plasma cells bind to autoantigens released by damaged host cells, enhancing tissue damage and gland dysfunction. cTfh, circulating Tfh
Physiologically, GC responses in lymphoid structures require the presence of CXCR5+ Tfh cells, which can migrate into B cell follicles in response to CXCL13. GC Tfh cells interact with B cells through ICOS and ICOS ligand, which is ubiquitously expressed on B cells, and release high amount of IL-21, thereby supporting B cell survival, proliferation, and plasma cell differentiation in synergy with the CD40-CD40L axis and BAFF-BAFFR. Tfh cell markers, including PD-1, CD84, and Bcl-6, are predominantly present in biopsies with higher focus scores, and Tfh cells that co-express CD3 and Bcl-6 localize closely to Bcl-6+ B cells within germinal centers [173]. However, limiting the number of Tfh cells appears to set a threshold to favor the survival of high affinity clones [174]. Hence, overexpansion of Tfh cells in SGs of SjS may lead to the dysregulation of B cell dynamics and autoantibody production as a result of aberrant selection of B cells, thus allowing survival of low affinity or self-reactive clones.
In addition, CXCR5+CD4+ T cells, defined as circulating Tfh (cTfh) cells, are increased in the peripheral blood of SjS patients compared with controls, and are positively correlated with the levels of plasmablasts, plasma cells, and autoantibodies [32, 175, 176]. cTfh cells, comprising the CCR7lowPD-1high and CCR7highPD-1low subsets, are known as precursor Tfh cells and correlate with Tfh cell activity [177]. CCR7lowPD-1high Tfh cells rapidly differentiate into mature Tfh cells to promote antibody production. An increased frequency of CCR7lowPD-1high Tfh cells correlates with elevated autoantibody profiles and more severe disease activity in SLE and RA [177]. Therefore, higher level of cTfh cells in SjS may activate B cell responses in SGs from SjS patients.
The canonical molecules secreted by Tfh cells include CXCL13, IL-4, and IL-21. It has been found that CXCL13 levels in the serum and saliva of SjS patients are significantly increased and positively correlate with many disease parameters, including hyposalivation, anti-SSB, anti-SSA/SSB, hypergammaglobulinemia, and rheumatoid factor [127, 170, 171, 178]. IL-4 plays an essential role in the activation and antibody production of mature B cells. IL-4 from Th2 and Tfh cells, which are increased in SjS, also protects B cells from spontaneous and BCR-mediated apoptosis [179, 180]. Recently, Tfh cell-derived IL-4, in conjunction with CXCL12, has been reported to trigger primary follicular lymphoma B cell activation, migration, and adhesion [156]. IL-4 also provides critical stimulatory signals for the growth and activation of B cells and facilitates the secretion of IgG and IgE in SLE [181]. Increased IL-4 levels have been described in SGs of a portion of SjS patients, especially those with severe B cell aggregation. Deficiency of IL-4 or STAT6 specifically halts the production of IgG1 isotype against M3R [122, 182]. Therefore, IL-4 seems to play an indispensable role in B cell activation and isotype switching, subsequently leading to exocrine dysfunction in an SjS mouse model.
IL-21 has profound effects on B cell proliferation, terminal differentiation into plasma cells, and immunoglobulin class-switching. IL-21 can induce isotype switching to produce IgG1 and IgG3 in combination with activation via CD40. The pathogenic role for IL-21 in SjS has been documented at length [123]. In addition to Tfh cells, IL-21 is secreted by CCR9+ Th cells and Th17 cells, suggesting the potential role of these cells in facilitating B cell activation and antibody production [61, 183]. IL-21 induced by the expression of IL-6 and the transcription factor ROR-γt results in further production of IL-21 from Th17 cells, which may amplify the progression of B cell responses [184]. Adoptive transfer of Th17 cells into IL-17 knockout mice that are immunized with SG protein increases the frequency of GL-7+ GC B cells in dLNs [185].
IL-22 facilitates the formation of ectopic germinal centers in SGs of mice by inducing production of CXCL13 [126]. IL-22 expression is associated with autoimmune B cell activation and correlates with clinical manifestations in SjS, as well as autoantibody production [127]. B cell depletion treatment modulates the expression of IL-22 in SGs of SjS patients [186]. This indicates the potential role of IL-22 in B cell accumulation and local pathology. The IL-22 receptor (IL-22R), composed of IL-22RA1 and IL-10R2, transduces a signal through phosphorylation of tyrosine kinases JAK1 and TYK2, followed by the activation of STAT3. IL-22R1-expressing cells also facilitate phosphorylation of STAT3. Aberrant expression of IL-22R1 has been observed in various lymphoma cells but not in benign lymphoid cells or other cells of hematopoietic origin [187]. Aberrant expression of IL-22R1 is induced by IL-18 and is found in tissue and circulating myeloid cells of SjS patients and macrophages of Sjögren-associated non-Hodgkin lymphoma tissues, but not in non-specific chronic sialoadenitis patients [188]. IL-22 may act on epithelium and myeloid cells which aberrantly express IL-22R1, leading to the production of chemokines and inflammatory cytokines, such as CXCL13, IL-22, and IL-17, which spur B cell infiltration and amplify Th17 polarization and contribute to increased B cell activation and GC formation in SGs. In addition to Th17 cells, NKp44+ NK cells, epithelial cells, and myeloid cells in the SGs of SjS patients may be potential sources of IL-22, and participate in spurring B cell responses in situ in cooperation with Th17 cells [128, 129].
Spontaneously developed GCs in SGs, a fairly common phenomenon in SjS, harbor autoreactive B cells that generate somatically mutated and class-switched pathogenic autoantibodies to promote disease development. B cell intrinsic IFN-γ receptor (IFN-γR) and STAT1 signaling are known to control spontaneously developed GCs and Tfh cell development to favor isotype switching [189]. The IFN-γ-IFN-γR axis in B cells is associated with autoimmune responses. IFN-γR deficiency abrogates autoimmune GCs, class-switching to IgG isotypes and systematic autoimmunity, but does not affect normal GC responses and Tfh cell or antibody production. Mechanistically, IFN-γ in synergy with BCR, TLR, and/or CD40 activation signals to promote Bcl-6 expression [190].
IFN-γR signaling also increases T-bet expression in B cells, which is thought to exert an isolated impact on class-switch recombination to pathogenic autoantibody subclasses without impacting GC development. However, IFN-γ produced by antigen-specific CD8+ T cells has been reported to induce class-switching in responding B cells in T-bet-dependent and T-bet-independent manners, and is required for IFN-γ-dependent class-switching from IgG1 to IgG2a and IgG2b [190]. Our previous work showed that both CD4+ T and CD8+ T cells in SGs and dLNs possess a higher capacity for IFN-γ production [11]. CD8a deficiency decreases the frequency of GC B cells in draining lymph nodes but increases serum levels of IgM and IgG1. GC B cell response and antibody production are present in an SjS mouse model of CD8a deficiency or CD8+ T cell depletion, but absent in a CD4-deficient SjS mouse model. GCs are completely hampered by knocking out IFN-γ in our mouse model, indicating that IFN-γ favors autoimmune GC responses in a SjS mouse model [11]. Autoantigen-specific CD4+ T cells present in a SjS mouse model contribute to autoimmune GC responses and high affinity autoantibody production partly by elevating IFN-γ level.
Skewing Negative Regulation
An altered Th17/Treg cell ratio is frequently linked with autoimmune diseases. The Th17/Treg imbalance also exists in several SjS mouse models, manifested by increased Th17 cells and decreased Treg cells, and these alterations are positively correlated with disease severity [23, 191]. However, imbalance in the proportions of effector Th17 cells and Treg cells in SjS patients requires further investigation. It is noteworthy that IL-2, a necessary cytokine for Treg cell differentiation but that inhibits Th17 cells is decreased in the serum of SjS patients [192]. A lack of IL-2 leads to attenuation of its suppressive capacity on Th17 cells in SjS patients, thus contributing to the increase of Th17 cells [120]. Meanwhile, short-term treatment with low-dose IL-2 reportedly restores the Th17/Treg balance in the periphery of SjS patients [121].
Furthermore, Foxp3+ Treg cells convert to inflammatory IFN-γ+Foxp3+ Treg or IL-17+Foxp3+ Treg cells either upon stimulation by IL-12 or in the absence of exogenous TGF-β [119]. IFN-γ expression does not attenuate the ability of Treg cells to suppress conventional T cell proliferation, but rather decreases Treg numbers and Foxp3 levels in Treg cells. Moreover, IL-12 favors the outgrowth of non-Tregs by increasing the expression of the IL-2 receptor, but diminishes the proliferation of Tregs and decreases IL-2 production by CD8+ T cells [193]. Meanwhile, IL-6 overcomes the suppressive effect on ROR-γt by Foxp3 and induces genetic reprogramming in Foxp3+ Treg cells in synergy with IL-1. These results suggest that Treg cells may be unstable and convertible, and they can possibly lose Foxp3 expression and acquire the expression of pro-inflammatory signatures.
Tr1 cells are significantly decreased in SjS mice and patients and are inversely correlated with disease activity and IL-12 level. Recombinant IL-12 treatment aggravates disease activity in a SjS mouse model, inducing lower saliva flow rates and pronounced inflammation and tissue damage in SGs [51]. IL-27, a member of the IL-12 family, can induce the differentiation Tr1, whose mRNA expression decreases in SjS patients and is lower in patients with extraglandular manifestation compared with those with only glandular infiltration [132, 133, 194]. Therefore, increased levels of IL-12 with decreased levels of IL-27 can exacerbate SjS through inhibiting the differentiation of Tr1 cells.
Mediating Tissue Damage
T lymphocytic infiltrates in SGs are the characteristic of SjS, especially in early disease stages. T cell infiltration and activation induce inflammation, resulting in a loss of glandular structure. The majority of T cells in the SGs of SjS patients are TRM cells. The function of TRM cells has been more extensively studied in infection rather than autoimmune diseases. The ultimate role that TRM cells play is not fully understood. One possibility is that these cells produce cytokines and control downstream immune responses to cause cytotoxic effects. Alternatively, TRM cells can target cells directly. Activated human TRM cells not only lead to IFN-γ production, but they also release granzyme B and perforins [195]. TRM cells co-express CD69 and CD103 and co-localize with tissue cells through E-cadherin, consequently inducing tissue damage. In 1999, Fujihara showed that CD8+ T cells that express CD103 are preferentially localized around acinar epithelial cells with apoptosis in patients with SjS, suggesting that CD8+ T cells participate in inducing epithelial cell apoptosis, leading to secretory dysfunction of exocrine glands [196].
Nearly all of the CD8+ T cells in SGs of SjS patients manifest as the TRM phenotype, and a high proportion of them is CD69 and CD103 double-positive. CD8+ T cells in SGs from patients with SjS outnumber CD4+ T cells in the vicinity of ductal epithelial cells, and may even embed or tightly attach to apoptotic ductal or acinar cells. Targeted depletion of CD8+ T cells appears to significantly reduce tissue damage and restore SG functions even in SjS mouse models with established disease [11]. Similar results have been detected in other SjS mouse models that transfer CD8+ T cells from NOD mice into NOD-SCID mice, inducing epithelial cell damage in the glands regardless of the presence of CD4+ T cells [12].
Recent findings have suggested that CD8+ T cells are just as abundant as CD4+ T cells in SGs and both the frequencies of HLA-DR-expressing activated CD8+ T and CD4+ T cells in blood correlated with ESSDAI scores, all of which support an equally critical role of CD8+ T cells compared with CD4+ T cells in SjS [13]. A multiomics study with blood and tissue samples from patients with SjS indicated that cytotoxic CD8+ T cells are associated with SjS gene signatures [14]. These data suggest that CD8+ TRM cells in SGs play a critical role in the development and progression of SjS. It is conceivable that CD8+ TRM cells are cytotoxic resulting in tissue cell death in SjS patients, but further proof is needed.
CD4+ T cell infiltration in SGs of MCMV-infected mice is suspected to induce tissue damage, glandular dysfunction, and autoantibody production [197]. T cell clustering and the formation of infiltrating foci during the pathogenesis of SjS are suspected to rely on the existence of CD4+ T cells. CD4 knockout leads to a more diffuse-infiltrated pattern of T cells in the submandibular glands of SjS mice. Tissue damage in murine SjS is suspected to be mediated primarily by CD8+ TRM cells rather than by CD4+ T cells. CD4+ T cells may cause tissue damage by secreting cytokines and then amplifying downstream immune responses. Studies of CD4+ T cells in SjS appears to support this hypothesis.
Th17 cells play an important physiological role at mucosal sites in supporting the epithelial barrier integrity by stimulating tight junction protein formation. Activated Th17 cells may cause tissue destruction by inducing MMP and other inflammatory molecule release from tissue cells through the production of pro-inflammatory cytokines. MMPs, especially MMP-9, is increased in epithelial cells upon stimulation with IL-17 in vitro. IL-17 expression is increased and associated with acinar damage in SjS [134, 198, 199].
Cytokines associated with a Th17 response, such as TGF-β, IL- 6, IL-1β, IL-21, IL-17A, and IL-23, are increased in SG, serum, and saliva of patients with SjS [131] (Table 1). The expression of these cytokines and their receptors increase with the progression of lesion severity in the SGs of SjS patients. IL-17 cells play a significant role in the pathogenesis of SjS [23].
Mice are protected from disease development upon IL-17 knockout in different SjS mouse models. For example, in an SjS mouse model in which mice were immunized with SG protein, Lin et al. discovered that large amounts of IL-17+CD4+ T cells infiltrated the SGs and were associated with lymphocytic foci formation and saliva reduction [185]. In fact, as early as 2010, Nguyen et al. have reported that overexpression of IL-17 in mouse SGs by adenovirus can induce SjS-like symptoms in mice [135]. In addition to IL-17, Th17 cells also secrete IL-21 and IL-22. IL-21 can stimulate the secretion of TGF-β, which in turn further induces the differentiation of CD4+ T cells into Th17 cells and the expression of IL-23R [200, 201].
IL-21, a pleiotropic cytokine derived mainly from activated CD4+ T cells, has been reported to inhibit antigen presentation but enhances the proliferation and effector function of antigen-specific CD8+ T cell by inducing IFN-γ, TNF, and co-stimulatory molecules [123, 124, 202]. IL-22 is significantly increased at both the protein and mRNA levels in the inflamed SGs of patients with SjS, accompanied by increased STAT3 mRNA and tyrosine phosphorylation of the corresponding protein [128]. IL-22 treatment of human SG epithelial cells induces STAT3 activation and inhibits cell expansion in vitro [203]. IL-22 treatment also decreases the salivary flow rate and increases the level of caspase-3 in the SG of NOD mice. Neutralization of endogenous IL-22 improves saliva production and reduced caspase-3 activation in the submandibular glands in anti-CD3-treated C57BL/6 mice. Taken together, these results provide strong evidence that IL-22 can directly act on SG epithelial cells and promote damage and dysfunction of SG tissues [130].
Infiltrated T cells that highly express Th1 cytokines correlate with the extent of injury of exocrine glands [204, 205]. IFN-γ, a canonical Th1 cytokine, mainly secreted from CD4+ T and CD8+ T cells in the SGs of SjS patients, is involved in promoting the loss of glandular function. In addition to favoring autoimmune GC responses and facilitating lymphocytic infiltration, IFN-γ can alter tight junction components and increase permeability of epithelium [114]. This alteration is observed in SGs of SjS patients. IFN-γ induces SGEC line Fas-mediated apoptosis in dose- and time-dependent manners in vitro [115]. The acinar epithelial cells undergoing apoptosis in SjS are Fas+ and FasL+ [206]. TUNEL staining has shown that compared with healthy controls, SG epithelial cell apoptosis rate is significantly increased in SjS patients. The apoptosis of epithelial cells can release autoantigens, including Ro/SSA and La/SSB, in great quantities. Knockout of IFN-γ or its receptor alleviates autoimmune disorders in NOD mice which were used as a SjS mouse model [207, 208]. The pathogenesis of a Ro/SSA-immunized SjS mouse model is dependent on the presence of IFN-γ [209]. These results suggest that the accumulation of IFN-γ-producing T cells in the SGs may contribute to epithelial cell damage and diminished saliva secretion. Moreover, TNF-α reportedly augments the surface expression of Ro/SSA and La/SSB on human keratinocytes, which may induces apoptosis and secretory dysfunction of SGs in SjS [118].
Inducing Metabolic Disorders
Chronic inflammatory diseases are known to be associated with metabolic dysfunction. As with other rheumatic diseases (RA, osteoarthritis, SLE), SjS is associated with an increase in the prevalence of metabolic disorders [210,211,212]. Patients with SjS manifest a higher frequency of dyslipidemia, diabetes mellitus, and hyperuricemia in comparison with controls. Dyslipidemia is the most common finding in SjS and correlates with disease severity [212]. SjS patients with metabolic syndrome (MetS) have higher values of insulin, homeostasis model assessment index, low-density lipoprotein-cholesterol, very-low-density lipoprotein-cholesterol, triglycerides, and leptin than non-MetS patients [213]. Increased levels of serum lipid may contribute to a higher rate of subclinical cardiovascular diseases, such as atherosclerosis, and diabetes, in SjS patients. Adipocytes can also occupy a large fraction of SG tissue and seem to be more prominent in the SG of SjS patients [214].
The development of adipogenesis and lipid metabolism is regulated not only by hormones, lipid, sugar metabolites, and adipokines, but also by inflammatory mediators [215]. Increased levels of inflammatory proteins, such as IL-17, IFN-γ, IL-6, and TNF-α, are thought to contribute to insulin resistance, heart disease, and diabetes. Conversely, anti-inflammatory cytokines, including IL-4 and IL-10, play a protective role in insulin sensitivity [216]. Adipose Th17/Treg balance is critical for the control of adipose tissue remodeling and insulin sensitivity [217]. IL-17+/IFN-γ+ T cells enhance macrophage pro-inflammatory function, while IL-4- and IL-13-producing Th2 cells and Treg cells facilitate macrophage differentiation into anti-inflammatory IL-10-secreting, M2, or “alternatively activated” macrophages [218]. Given these observations, it would be tempting to presume that elevated levels of inflammatory mediators produced and/or stimulated by T cells in patients with SjS are associated with metabolic disorders.
Metabolic dysfunction can also regulate immune responses. T cells undergo metabolic transition from oxidative phosphorylation to glycolysis upon activation. The transition from effector cells to memory cells undergoes further metabolic changes since memory cells rely mainly on fatty acid oxidation [219]. Free fatty acid uptake via expression of fatty acid binding and/or transport proteins by CD8+ T cells is linked to memory phenotype maintenance, cell survival, and proliferation. For instance, expression of glycerol channel aquaporin (AQP) 9 induced by IL-7 allows memory CD8+ T cells to import glycerol used for triglyceride synthesis and storage and sustain ATP levels required for long-term metabolic fitness and fast response to reinfection [220]. Similarly, survival of CD8+ TRM cells requires exogenous lipid uptake and metabolism [221]. Fatty acid oxidation in T cells increases the number of antigen-reactive CD8+ T cells and facilitates T cell function [222]. Therefore, dyslipidemia and insulin resistance may affect memory T cell function during SjS development by reducing energy storage. However, high levels of circulating lipids may also damage the endothelium to increase autoantigen release and inflammatory responses [223]. Hypertriglyceridemia and diabetes in SjS are linked with a higher prevalence of extraglandular features, such as renal, liver, and vasculitic involvement [212]. Moreover, adipose tissue infiltration in SGs of patients with SjS is reported to stimulate IL-6 and IL-17 secretion, which may exacerbate SjS [214]. Therefore, abnormal lipid metabolism caused by excessive immune response may further promote the autoimmune response.
Biological Treatments in SjS
One of the major challenges in the clinical treatment of SjS is the lack of specific agents with low side effect profiles which have effective suppression of abnormal immune responses and relief of dry oral and ocular symptoms. Current treatments for SjS involves the following: (1) relieving dry eye and mouth symptoms by inducing tear production or using artificial tears and saliva, respectively; (2) suppressing the immune system by using immunosuppressant and immunomodulatory drugs; and (3) targeting specific immune cells or proteins through biological modifiers. A typical example of the first strategy is the use of muscarinic agonists, including pilocarpine hydrochloride and cevimeline hydrochloride [224]. Immunosuppressant, such as glucocorticoid and cyclosporine, is used to treat SjS, but they should be applied cautiously due to their propensity to induce extensive immunosuppression and increase the risk of infection [225]. Moreover, long-term use of glucocorticoids is associated with severe side effects, including increasing the prevalence of metabolic disorders, resulting in cardiovascular diseases in SjS patients [226]. While immunomodulatory drugs are usually used in severe organ-involved situations according to the therapeutic guidelines for other connective-tissue diseases in SjS, such as SLE, but systematic studies are lacking [225].
Biological agents against abnormal immune components or pathways are widely used in many autoimmune diseases [227]. However, their applications in SjS are challenging (Fig. 4). TNF blockers (infliximab and etanercept) do not produce effective alleviation of dryness symptoms [228]. Rituximab, an anti-CD20 antibody against B cells, has undergone clinical trials in SjS with mixed results [225]. Belimumab, an inhibitor of BAFF approved for the treatment of SLE, is effective in about 60% of SjS patients by improving at least two of five disease indicators, including dryness, pain, fatigue, systemic activity, and B cell biomarkers, but not salivary flow or tear production [229]. We propose that B cells may not be the main pathogenic cells during the development of SjS, at least at the stage when clinical and pathologic changes, such as dryness and lymphocytic infiltration, appear. Several other biologics, such as leniolisib (PI3K inhibitor), petesicatib (Cat S inhibitor), prezalumab (anti-ICOSL), and iscalimab (anti-CD40), have shown limited efficacy in SjS clinical trials [230, 231].

Biological treatments in SjS. Activated infiltrated lymphocytes form a complex signal network with salivary gland cells, leading to whose increased apoptosis and secretion dysfunction. Current treatments to these cells and factors in mice or human are shown in gray boxes. MMP, matrix metalloproteinase; E-cad, E-cadherin; Cat S, cathepsin S; OE, overexpression; KO, knockout; TRM, tissue resident memory T cell
The loss of secretory function of exocrine glands is due mainly to the T cell-mediated destruction of secretory cells. Thus, treatment targeting T cells may provide the best outlook for SjS. Abatacept (CTLA4-Ig), a unique biologic agent that binds CD80/CD86 on APCs to inhibit co-stimulation required for complete T cell activation, seems to be effective for SjS, as assessed by histologic, serologic, and clinical changes [232, 233]. Mechanistically, abatacept reduces cTfh cell number and expression levels of ICOS on T cells [234]. However, the clinical effectiveness of abatacept in SjS needs to be further confirmed, as open label pilot studies have produced equivocal results on the preservation of salivary and lacrimal gland function [232, 235]. The idea that CD4+ T cells are the main pathogenic T cells in SjS is deeply rooted, but recent research suggests that CD8+ T cells may play a more important role [11, 196]. Thus, the therapeutic effect of abatacept may be related to its suppressive effect on the function of both CD8+ T cells and CD4+ T cells. Future research needs to be directed towards critical signaling pathways of CD8+ T cells in SGs with more specific drug delivery strategies, such as using a nanocarrier.
Other reported studies on biological treatments in SjS involve overexpression of specific proteins, such as water channel (AQP5) and Cl− channel, as well as engineering T cells [236,237,238]. But these methods have a long way to go before they can be used clinically.
Conclusion
It has been proved by many studies that T cells may mediate tissue damage and participate in the amplification of immune response in SjS, which suggests that treatment against T cells in SjS may be an effective therapeutic strategy. However, T cells also play critical roles in immune surveillance, such as preventing pathogens from invading and clearance of abnormal cells. Therefore, it is necessary to identify specific targets to inhibit autoreactive T cells or their associated cytokines but involve the least amount of side effects. Identifying these specific therapeutic targets is challenging, because the detailed pathogenesis of SjS is not fully understood. As the second most common systemic autoimmune disease after SLE, SjS is more often understood as a “cold disease” with slow progression and has not received enough attention. Studies on the immunological mechanisms in SjS, particularly involving T cells, are expected to provide new clues for the clinical therapy of SjS.
Abbreviations
- APCs:
-
Antigen-presenting cells
- AQP:
-
Aquaporin
- BAFF:
-
B cell activating factor
- Cat S:
-
Cathepsin S
- cTfh:
-
Circulating follicular helper T cell
- cTfr:
-
Circulating follicular regulatory T cell
- CCL:
-
Chemokine (C-C motif) ligand
- CXCL:
-
Chemokine (C-X-C motif) ligand
- CCR:
-
CXC chemokine receptor
- CXCR:
-
CXC chemokine receptor
- DCs:
-
Dendritic cells
- DN T cells:
-
CD4&CD8 double-negative T cells
- EGMs:
-
Extraglandular manifestations
- ELS:
-
Ectopic lymphoid-like structures
- GC:
-
Germinal center
- GLS:
-
GC-like structures
- GWAS:
-
Genome-wide association studies
- LAMP:
-
Lysosome-associated membrane glycoprotein
- ICAM:
-
Intercellular adhesion molecule
- ICOS:
-
Inducible T cell co-stimulator
- ICOSL:
-
ICOS ligand
- IFN:
-
Interferon
- IFN-γR:
-
IFN-γ receptor
- IL:
-
Interleukin
- IL-22R:
-
IL-22 receptor
- iNKT cell:
-
Invariant natural killer T cell
- MAIT cell:
-
Mucosal-associated invariant T cell
- MDC:
-
Macrophage-derived chemokine
- MetS:
-
Metabolic syndrome
- MHC:
-
Major histocompatibility complex class
- MMP:
-
Matrix metalloproteinase
- NK:
-
Natural killer
- NOD:
-
Non-obese diabetic mouse
- pDCs:
-
Plasmacytoid DC cells
- ROR:
-
RAR-related orphan receptor
- SGEC:
-
Salivary gland epithelial cell
- SjS:
-
Sjögren’s syndrome
- SLE:
-
Systemic lupus erythematosus
- SLO:
-
Secondary lymphoid organs
- S1P:
-
Sphingosine 1-phosphate
- TARC:
-
Thymus and activation-regulated chemokine
- Tfh:
-
Follicular helper T cell
- Tfr:
-
Follicular regulatory T cell
- TGF-β:
-
Transforming growth factor beta
- Th:
-
Helper T cell
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor alpha
- Treg:
-
Regulatory T cell
- TRM:
-
Tissue resident memory T cell
- Tr1:
-
Type 1 regulatory T cell
- VCAM-1:
-
Vascular cell adhesion molecule-1.
References
Anaya JM et al (2019) Sjogren’s syndrome and autoimmune thyroid disease: two sides of the same coin. Clin Rev Allergy Immunol 56(3):362–374
Qin B et al (2015) Epidemiology of primary Sjogren’s syndrome: a systematic review and meta-analysis. Ann Rheum Dis 74(11):1983–1989
Nocturne G, Mariette X (2015) Sjogren syndrome-associated lymphomas: an update on pathogenesis and management. Br J Haematol 168(3):317–327
Katsiougiannis S, Tenta R, Skopouli FN (2019) Autoimmune epithelitis (Sjogren’ syndrome); the impact of metabolic status of glandular epithelial cells on auto-immunogenicity. J Autoimmun 104:102335
Pacheco Y et al (2019) Bystander activation and autoimmunity. J Autoimmun 103:102301
Vakrakou AG et al (2018) Impaired anti-inflammatory activity of PPARgamma in the salivary epithelia of Sjogren’s syndrome patients imposed by intrinsic NF-kappaB activation. J Autoimmun 86:62–74
Sisto M et al (2009) Tumor necrosis factor inhibitors block apoptosis of human epithelial cells of the salivary glands. Ann N Y Acad Sci 1171:407–414
Long D et al (2019) Clinical significance and immunobiology of IL-21 in autoimmunity. J Autoimmun 99:1–14
Singh N, Cohen PL (2012) The T cell in Sjogren’s syndrome: force majeure, not spectateur. J Autoimmun 39(3):229–233
Adamson TC 3rd et al (1983) Immunohistologic analysis of lymphoid infiltrates in primary Sjogren’s syndrome using monoclonal antibodies. J Immunol 130(1):203–208
Gao CY et al (2019) Tissue-resident memory CD8+ T cells acting as mediators of salivary gland damage in a murine model of Sjogren’s syndrome. Arthritis Rheumatol 71(1):121–132
Barr JY et al (2017) CD8 T cells contribute to lacrimal gland pathology in the nonobese diabetic mouse model of Sjogren syndrome. Immunol Cell Biol 95(8):684–694
Mingueneau M et al (2016) Cytometry by time-of-flight immunophenotyping identifies a blood Sjogren’s signature correlating with disease activity and glandular inflammation. J Allergy Clin Immunol 137(6):1809–1821 e12
Tasaki S et al (2017) Multiomic disease signatures converge to cytotoxic CD8 T cells in primary Sjogren’s syndrome. Ann Rheum Dis 76(8):1458–1466
Mandl T et al (2004) CD4+ T-lymphocytopenia--a frequent finding in anti-SSA antibody seropositive patients with primary Sjogren’s syndrome. J Rheumatol 31(4):726–728
Maehara T et al (2012) Selective localization of T helper subsets in labial salivary glands from primary Sjogren’s syndrome patients. Clin Exp Immunol 169(2):89–99
van Woerkom JM et al (2005) Salivary gland and peripheral blood T helper 1 and 2 cell activity in Sjogren’s syndrome compared with non-Sjogren’s sicca syndrome. Ann Rheum Dis 64(10):1474–1479
Mitsias DI et al (2002) The Th1/Th2 cytokine balance changes with the progress of the immunopathological lesion of Sjogren’s syndrome. Clin Exp Immunol 128(3):562–568
Bertorello R et al (2004) Increased levels of interleukin-10 in saliva of Sjogren’s syndrome patients. Correlation with disease activity. Clin Exp Med 4(3):148–151
Hulkkonen J et al (2001) Genetic association between interleukin-10 promoter region polymorphisms and primary Sjogren’s syndrome. Arthritis Rheum 44(1):176–179
Anaya JM et al (2002) Interleukin 10 (IL-10) influences autoimmune response in primary Sjogren’s syndrome and is linked to IL-10 gene polymorphism. J Rheumatol 29(9):1874–1876
Blauvelt A, Chiricozzi A (2018) The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol 55(3):379–390
Verstappen GM et al (2018) Th17 cells in primary Sjogren’s syndrome: pathogenicity and plasticity. J Autoimmun 87:16–25
Azizi G, Jadidi-Niaragh F, Mirshafiey A (2013) Th17 cells in immunopathogenesis and treatment of rheumatoid arthritis. Int J Rheum Dis 16(3):243–253
Sakai A et al (2008) Identification of IL-18 and Th17 cells in salivary glands of patients with Sjogren’s syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J Immunol 181(4):2898–2906
Nguyen CQ et al (2008) Salivary gland tissue expression of interleukin-23 and interleukin-17 in Sjogren’s syndrome: findings in humans and mice. Arthritis Rheum 58(3):734–743
Xiao F et al (2017) Proteasome inhibition suppresses Th17 cell generation and ameliorates autoimmune development in experimental Sjogren’s syndrome. Cell Mol Immunol
Crotty S (2014) T follicular helper cell differentiation, function, and roles in disease. Immunity 41(4):529–542
Pontarini E et al (2017) OP0300 enrichment of T follicular-helper cells (TFH) and exclusion of t follicular-regulatory cells (TFR) from ectoPIC germinal centers in salivary glands of Sjogren’s syndrome patients. Ann Rheum Dis 76(Suppl 2):180–180
Fonseca VR et al (2018) The ratio of blood T follicular regulatory cells to T follicular helper cells marks ectopic lymphoid structure formation while activated follicular helper T cells indicate disease activity in primary Sjogren’s syndrome. Arthritis Rheumatol 70(5):774–784
Verstappen GM et al (2018) Is the T follicular regulatory:follicular helper T cell ratio in blood a biomarker for ectopic lymphoid structure formation in Sjogren’s syndrome? Comment on the article by Fonseca et al. Arthritis Rheumatol 70(8):1354–1355
Szabo K et al (2013) Follicular helper T cells may play an important role in the severity of primary Sjogren’s syndrome. Clin Immunol 147(2):95–104
King IL, Mohrs M (2009) IL-4-producing CD4+ T cells in reactive lymph nodes during helminth infection are T follicular helper cells. J Exp Med 206(5):1001–1007
Gottenberg JE et al (2005) CD4 CD25high regulatory T cells are not impaired in patients with primary Sjogren’s syndrome. J Autoimmun 24(3):235–242
Li X et al (2007) T regulatory cells are markedly diminished in diseased salivary glands of patients with primary Sjogren’s syndrome. J Rheumatol 34(12):2438–2445
Christodoulou MI et al (2008) Foxp3+ T-regulatory cells in Sjogren’s syndrome: correlation with the grade of the autoimmune lesion and certain adverse prognostic factors. Am J Pathol 173(5):1389–1396
Liu MF et al (2008) Decreased CD4+CD25+bright T cells in peripheral blood of patients with primary Sjogren’s syndrome. Lupus 17(1):34–39
Sarigul M et al (2010) The numbers of Foxp3 + Treg cells are positively correlated with higher grade of infiltration at the salivary glands in primary Sjogren’s syndrome. Lupus 19(2):138–145
Alunno A et al (2013) Characterization of a new regulatory CD4+ T cell subset in primary Sjogren’s syndrome. Rheumatology (Oxford) 52(8):1387–1396
Furuzawa-Carballeda J et al (2013) Peripheral regulatory cells immunophenotyping in primary Sjogren’s syndrome: a cross-sectional study. Arthritis Res Ther 15(3):R68
Maria NI et al (2016) Association of increased Treg cell levels with elevated indoleamine 2,3-dioxygenase activity and an imbalanced kynurenine pathway in interferon-positive primary Sjogren’s syndrome. Arthritis Rheumatol 68(7):1688–1699
Raimondi G et al (2006) Regulated compartmentalization of programmed cell death-1 discriminates CD4+CD25+ resting regulatory T cells from activated T cells. J Immunol 176(5):2808–2816
Mason GI et al (2003) Salivary gland expression of transforming growth factor beta isoforms in Sjogren’s syndrome and benign lymphoepithelial lesions. Mol Pathol 56(1):52–59
Noack M, Miossec P (2014) Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev 13(6):668–677
Alunno A et al (2012) Expansion of CD4+CD25-GITR+ regulatory T-cell subset in the peripheral blood of patients with primary Sjogren’s syndrome: correlation with disease activity. Reumatismo 64(5):293–298
Liu C et al (2017) Elevated level of circulating CD4(+)Helios(+)FoxP3(+) cells in primary Sjogren’s syndrome patients. Mod Rheumatol 27(4):630–637
Yao Y et al (2013) Type I interferons in Sjogren’s syndrome. Autoimmun Rev 12(5):558–566
Niewold TB et al (2008) Serum type I interferon activity is dependent on maternal diagnosis in anti-SSA/Ro-positive mothers of children with neonatal lupus. Arthritis Rheum 58(2):541–546
Gottenberg JE et al (2006) Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren’s syndrome. Proc Natl Acad Sci U S A 103(8):2770–2775
Miyara M et al (2015) Sialyl Lewis x (CD15s) identifies highly differentiated and most suppressive FOXP3high regulatory T cells in humans. Proc Natl Acad Sci U S A 112(23):7225–7230
Yao G, Qi J, Sun L (2018) SAT0016 Il-12 suppress tr1 cells in the sjÖgren’s syndrome. Ann Rheum Dis 77(Suppl 2):876–876
Chaudhry A et al (2011) Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34(4):566–578
Linterman MA et al (2011) Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med 17(8):975–982
Xie MM, Dent AL (2018) Unexpected help: follicular regulatory T cells in the germinal center. Front Immunol 9:1536
Liu C et al (2018) Increased circulating CD4(+)CXCR5(+)FoxP3(+) follicular regulatory T cells correlated with severity of systemic lupus erythematosus patients. Int Immunopharmacol 56:261–268
Fonseca VR et al (2017) Human blood Tfr cells are indicators of ongoing humoral activity not fully licensed with suppressive function. Sci Immunol 2(14)
Maceiras AR et al (2017) T follicular regulatory cells in mice and men. Immunology 152(1):25–35
Sage PT et al (2014) Circulating T follicular regulatory and helper cells have memory-like properties. J Clin Invest 124(12):5191–5204
Ritvo PG et al (2018) High-resolution repertoire analysis reveals a major bystander activation of Tfh and Tfr cells. Proc Natl Acad Sci U S A 115(38):9604–9609
Cosorich I et al (2018) CCR9 expressing T helper and T follicular helper cells exhibit site-specific identities during inflammatory disease. Front Immunol 9:2899
McGuire HM et al (2011) A subset of interleukin-21+ chemokine receptor CCR9+ T helper cells target accessory organs of the digestive system in autoimmunity. Immunity 34(4):602–615
Blokland SLM et al (2017) Increased CCL25 and T helper cells expressing CCR9 in the salivary glands of patients with primary Sjogren’s syndrome: potential new axis in lymphoid neogenesis. Arthritis Rheumatol 69(10):2038–2051
Mora JR et al (2006) Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314(5802):1157–1160
Blokland S et al (2018) Ccr9+pathogenic Thelper cells from primary Sjogren’s syndrome patients are characterised by deregulated pathways associated with effector T cell function. Ann Rheum Dis 77:1273–1273
Mueller SN, Mackay LK (2016) Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol 16(2):79–89
Mackay LK et al (2013) The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 14(12):1294–1301
Thom JT et al (2015) The salivary gland acts as a sink for tissue-resident memory CD8(+) T cells, facilitating protection from local cytomegalovirus infection. Cell Rep 13(6):1125–1136
Kumar BV et al (2017) Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep 20(12):2921–2934
Cyster JG, Schwab SR (2012) Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol 30:69–94
Skon CN et al (2013) Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol 14(12):1285–1293
Brandt D, Hedrich CM (2018) TCRalphabeta(+)CD3(+)CD4(−)CD8(−) (double negative) T cells in autoimmunity. Autoimmun Rev 17(4):422–430
Alunno A et al (2013) IL-17-producing CD4-CD8- T cells are expanded in the peripheral blood, infiltrate salivary glands and are resistant to corticosteroids in patients with primary Sjogren’s syndrome. Ann Rheum Dis 72(2):286–292
Alunno A et al (2014) CD4(-)CD8(-) T-cells in primary Sjogren’s syndrome: association with the extent of glandular involvement. J Autoimmun 51:38–43
Renauer PA, Coit P, Sawalha AH (2015) The DNA methylation signature of human TCRalphabeta+CD4-CD8- double negative T cells reveals CG demethylation and a unique epigenetic architecture permissive to a broad stimulatory immune response. Clin Immunol 156(1):19–27
Crispin JC, Tsokos GC (2009) Human TCR-alpha beta+ CD4- CD8- T cells can derive from CD8+ T cells and display an inflammatory effector phenotype. J Immunol 183(7):4675–4681
Gao Y, Williams AP (2015) Role of innate T cells in anti-bacterial immunity. Front Immunol 6:302
Casetti R, Martino A (2008) The plasticity of gamma delta T cells: innate immunity, antigen presentation and new immunotherapy. Cell Mol Immunol 5(3):161–170
Brennan F et al (1989) Coordinate expansion of ‘fetal type’ lymphocytes (TCR gamma delta+T and CD5+B) in rheumatoid arthritis and primary Sjogren’s syndrome. Clin Exp Immunol 77(2):175–178
Caretto A et al (1995) An immunohistochemical study of immunological phenomena in minor salivary glands in patients with Sjogren’s syndrome. Rheumatol Int 15(2):51–55
Wang JJ et al (2016) Mucosal-associated invariant T cells are reduced and functionally immature in the peripheral blood of primary Sjogren’s syndrome patients. Eur J Immunol 46(10):2444–2453
Papp G et al (2016) Increased IL-21 expression induces granzyme B in peripheral CD5(+) B cells as a potential counter-regulatory effect in primary Sjogren’s syndrome. Mediat Inflamm 2016:4328372
Curtsinger JM, Mescher MF (2010) Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol 22(3):333–340
Mescher MF et al (2006) Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev 211:81–92
Bevington SL et al (2017) T cell receptor and cytokine signaling can function at different stages to establish and maintain transcriptional memory and enable T helper cell differentiation. Front Immunol 8:204
Chen L, Flies DB (2013) Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13(4):227–242
Edman MC et al (2018) Increased cathepsin S activity associated with decreased protease inhibitory capacity contributes to altered tear proteins in Sjogren’s syndrome patients. Sci Rep 8(1):11044
Hamm-Alvarez SF et al (2014) Tear cathepsin S as a candidate biomarker for Sjogren’s syndrome. Arthritis Rheumatol 66(7):1872–1881
Hargreaves P et al (2018) FRI0295 inhibition of cathepsin s leads to suppression of antigen specific t cells from patients with primary sjÖgren syndrome. Ann Rheum Dis 77(Suppl 2):684–684
Zhou D, McNamara NA (2014) Macrophages: important players in primary Sjogren’s syndrome? Expert Rev Clin Immunol 10(4):513–520
Manoussakis MN et al (2007) Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjogren’s syndrome: correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum 56(12):3977–3988
Hillen MR et al (2014) Dendritic cells, T-cells and epithelial cells: a crucial interplay in immunopathology of primary Sjogren’s syndrome. Expert Rev Clin Immunol 10(4):521–531
Wildenberg ME et al (2009) Increased frequency of CD16+ monocytes and the presence of activated dendritic cells in salivary glands in primary Sjogren syndrome. Ann Rheum Dis 68(3):420–426
Hurdayal R et al (2017) IL-4-producing B cells regulate T helper cell dichotomy in type 1- and type 2-controlled diseases. Proc Natl Acad Sci U S A 114(40):E8430–E8439
Jelcic I et al (2018) Memory B cells activate brain-homing, autoreactive CD4(+) T cells in multiple sclerosis. Cell
Chan OT, Shlomchik MJ (2000) Cutting edge: B cells promote CD8+ T cell activation in MRL-Fas(lpr) mice independently of MHC class I antigen presentation. J Immunol 164(4):1658–1662
McArthur C et al (2002) Intracellular trafficking and surface expression of SS-A (Ro), SS-B (La), poly(ADP-ribose) polymerase and alpha-fodrin autoantigens during apoptosis in human salivary gland cells induced by tumour necrosis factor-alpha. Arch Oral Biol 47(6):443–448
Kapsogeorgou EK et al (2005) Salivary gland epithelial cell exosomes: a source of autoantigenic ribonucleoproteins. Arthritis Rheum 52(5):1517–1521
Ainola M et al (2018) Activation of plasmacytoid dendritic cells by apoptotic particles - mechanism for the loss of immunological tolerance in Sjogren’s syndrome. Clin Exp Immunol 191(3):301–310
Hunziker L et al (2003) Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections. Nat Immunol 4(4):343–349
Kapsogeorgou EK, Moutsopoulos HM, Manoussakis MN (2001) Functional expression of a costimulatory B7.2 (CD86) protein on human salivary gland epithelial cells that interacts with the CD28 receptor, but has reduced binding to CTLA4. J Immunol 166(5):3107–3113
Spachidou MP et al (2007) Expression of functional toll-like receptors by salivary gland epithelial cells: increased mRNA expression in cells derived from patients with primary Sjogren’s syndrome. Clin Exp Immunol 147(3):497–503
Brito-Zeron P et al (2016) Sjogren syndrome. Nat Rev Dis Primers 2:16047
Yamada A et al (2013) Targeting IL-1 in Sjogren’s syndrome. Expert Opin Ther Targets 17(4):393–401
Nocturne G, Mariette X (2013) Advances in understanding the pathogenesis of primary Sjogren’s syndrome. Nat Rev Rheumatol 9(9):544–556
Kawanami T et al (2012) Skewed production of IL-6 and TGFbeta by cultured salivary gland epithelial cells from patients with Sjogren’s syndrome. PLoS One 7(10):e45689
Bikker A et al (2012) Increased interleukin (IL)-7Ralpha expression in salivary glands of patients with primary Sjogren’s syndrome is restricted to T cells and correlates with IL-7 expression, lymphocyte numbers and activity. Ann Rheum Dis 71(6):1027–1033
Jin JO et al (2013) Interleukin-7 enhances the Th1 response to promote the development of Sjogren’s syndrome-like autoimmune exocrinopathy in mice. Arthritis Rheum 65(8):2132–2142
Vosters JL et al (2009) Interleukin-12 induces salivary gland dysfunction in transgenic mice, providing a new model of Sjogren’s syndrome. Arthritis Rheum 60(12):3633–3641
Henry CJ et al (2008) IL-12 produced by dendritic cells augments CD8+ T cell activation through the production of the chemokines CCL1 and CCL17. J Immunol 181(12):8576–8584
Youinou P, Pers JO (2011) Disturbance of cytokine networks in Sjogren’s syndrome. Arthritis Res Ther 13(4):227
Gong YZ et al (2014) Differentiation of follicular helper T cells by salivary gland epithelial cells in primary Sjogren’s syndrome. J Autoimmun 51:57–66
Schmitt N et al (2014) The cytokine TGF-beta co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol 15(9):856–865
Tsunawaki S et al (2002) Possible function of salivary gland epithelial cells as nonprofessional antigen-presenting cells in the development of Sjogren’s syndrome. J Rheumatol 29(9):1884–1896
Baker OJ (2010) Tight junctions in salivary epithelium. J Biomed Biotechnol 2010:278948
Abu-Helu RF et al (2001) Induction of salivary gland epithelial cell injury in Sjogren’s syndrome: in vitro assessment of T cell-derived cytokines and Fas protein expression. J Autoimmun 17(2):141–153
Zhou J, Kawai T, Yu Q (2017) Pathogenic role of endogenous TNF-alpha in the development of Sjogren’s-like sialadenitis and secretory dysfunction in non-obese diabetic mice. Lab Investig 97(4):458–467
Limaye A et al (2019) Targeted TNF-alpha overexpression drives salivary gland inflammation. J Dent Res 98(6):713–719
Dorner T et al (1995) Enhanced membrane expression of the 52 kDa Ro(SS-A) and La(SS-B) antigens by human keratinocytes induced by TNF alpha. Ann Rheum Dis 54(11):904–909
Esposito M et al (2010) IL-17- and IFN-gamma-secreting Foxp3+ T cells infiltrate the target tissue in experimental autoimmunity. J Immunol 185(12):7467–7473
Luo J et al (2018) IL-2 inhibition of Th17 generation rather than induction of Treg cells is impaired in primary Sjogren’s syndrome patients. Front Immunol 9:1755
Miao M et al (2018) Short-term and low-dose IL-2 therapy restores the Th17/Treg balance in the peripheral blood of patients with primary Sjogren’s syndrome. Ann Rheum Dis 77(12):1838–1840
Gao J et al (2006) Sjogren’s syndrome in the NOD mouse model is an interleukin-4 time-dependent, antibody isotype-specific autoimmune disease. J Autoimmun 26(2):90–103
Kwok SK et al (2015) A pathogenetic role for IL-21 in primary Sjogren syndrome. Nat Rev Rheumatol 11(6):368–374
Liu H et al (2012) Local suppression of IL-21 in submandibular glands retards the development of Sjogren’s syndrome in non-obese diabetic mice. J Oral Pathol Med 41(10):728–735
Sekiguchi M et al (2008) Role of sphingosine 1-phosphate in the pathogenesis of Sjogren’s syndrome. J Immunol 180(3):1921–1928
Barone F et al (2015) IL-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs. Proc Natl Acad Sci U S A 112(35):11024–11029
Lavoie TN et al (2011) Expression of interleukin-22 in Sjogren’s syndrome: significant correlation with disease parameters. Scand J Immunol 74(4):377–382
Ciccia F et al (2012) Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren’s syndrome. Ann Rheum Dis 71(2):295–301
Ciccia F et al (2014) The role of innate and lymphoid IL-22-producing cells in the immunopathology of primary Sjogren’s syndrome. Expert Rev Clin Immunol 10(4):533–541
Yoo B, Zhou J, Yu Q (2018) A detrimental effect of interleukin-22 on salivary gland tissue integrity and function. J Immunol 200(1 Supplement):41.15–41.15
Katsifis GE et al (2009) Systemic and local interleukin-17 and linked cytokines associated with Sjogren’s syndrome immunopathogenesis. Am J Pathol 175(3):1167–1177
Yao G, Shi B., Qi J, Wang Y, Chen W, Tang X, Wang D, Feng X, Sun L (2015) Deficiency of IL-27 exacerbate Sjögren’s syndrome through inhibiting differentiation of type 1 regulatory T cells. Arthritis Rheumatol 67
Lee BH et al (2012) Gene therapy using IL-27 ameliorates Sjogren’s syndrome-like autoimmune exocrinopathy. Arthritis Res Ther 14(4):R172
Zhang LW et al (2016) Interleukin-17 impairs salivary tight junction integrity in Sjogren’s syndrome. J Dent Res 95(7):784–792
Nguyen CQ et al (2010) Pathogenic effect of interleukin-17A in induction of Sjogren’s syndrome-like disease using adenovirus-mediated gene transfer. Arthritis Res Ther 12(6):R220
Kong DH et al (2018) Emerging roles of vascular cell adhesion molecule-1 (VCAM-1) in immunological disorders and cancer. Int J Mol Sci 19(4)
Cerutti C, Ridley AJ (2017) Endothelial cell-cell adhesion and signaling. Exp Cell Res 358(1):31–38
Lee JH et al (2013) A novel human anti-VCAM-1 monoclonal antibody ameliorates airway inflammation and remodelling. J Cell Mol Med 17(10):1271–1281
Abonia JP et al (2006) Alpha-4 integrins and VCAM-1, but not MAdCAM-1, are essential for recruitment of mast cell progenitors to the inflamed lung. Blood 108(5):1588–1594
Liu Z et al (2019) Elevated CCL19/CCR7 expression during the disease process of primary Sjogren’s syndrome. Front Immunol 10:795
Woyciechowski S, Hofmann M, Pircher H (2017) alpha4 beta1 integrin promotes accumulation of tissue-resident memory CD8(+) T cells in salivary glands. Eur J Immunol 47(2):244–250
Iizuka M et al (2015) A crucial role of RORgammat in the development of spontaneous Sialadenitis-like Sjogren’s syndrome. J Immunol 194(1):56–67
Blokland SLM et al (2017) Decreased circulating CXCR3+CCR9+Th cells coincides with elevated levels of their ligands CXCL10 and CCL25 in the salivary gland of Sjogren’s syndrome patients which synergistically facilitate Th cell migration. Arthritis & Rheumatology 69
Castellino F et al (2006) Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature 440(7086):890–895
Wildenberg ME et al (2008) Lack of CCR5 on dendritic cells promotes a proinflammatory environment in submandibular glands of the NOD mouse. J Leukoc Biol 83(5):1194–1200
Reksten TR et al (2009) Cytokine and autoantibody profiling related to histopathological features in primary Sjogren’s syndrome. Rheumatology (Oxford) 48(9):1102–1106
Hjelmervik TO et al (2005) Gene expression profiling of minor salivary glands clearly distinguishes primary Sjogren’s syndrome patients from healthy control subjects. Arthritis Rheum 52(5):1534–1544
Hernandez-Molina G et al (2011) Chemokine saliva levels in patients with primary Sjogren’s syndrome, associated Sjogren’s syndrome, pre-clinical Sjogren’s syndrome and systemic autoimmune diseases. Rheumatology (Oxford) 50(7):1288–1292
Xanthou G et al (2001) “Lymphoid” chemokine messenger RNA expression by epithelial cells in the chronic inflammatory lesion of the salivary glands of Sjogren’s syndrome patients: possible participation in lymphoid structure formation. Arthritis Rheum 44(2):408–418
Tandon M et al (2017) Laser microdissection coupled with RNA-seq reveal cell-type and disease-specific markers in the salivary gland of Sjogren’s syndrome patients. Clin Exp Rheumatol 35(5):777–785
Ogawa N et al (2002) Involvement of the interferon-gamma-induced T cell-attracting chemokines, interferon-gamma-inducible 10-kd protein (CXCL10) and monokine induced by interferon-gamma (CXCL9), in the salivary gland lesions of patients with Sjogren’s syndrome. Arthritis Rheum 46(10):2730–2741
Ogawa N et al (2004) Expression of interferon-inducible T cell alpha chemoattractant (CXCL11) in the salivary glands of patients with Sjogren’s syndrome. Clin Immunol 112(3):235–238
Barone F et al (2005) Association of CXCL13 and CCL21 expression with the progressive organization of lymphoid-like structures in Sjogren’s syndrome. Arthritis Rheum 52(6):1773–1784
Salomonsson S et al (2003) Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjogren’s syndrome. Arthritis Rheum 48(11):3187–3201
Fleige H et al (2014) IL-17-induced CXCL12 recruits B cells and induces follicle formation in BALT in the absence of differentiated FDCs. J Exp Med 211(4):643–651
Pandey S et al (2017) IL-4/CXCL12 loop is a key regulator of lymphoid stroma function in follicular lymphoma. Blood 129(18):2507–2518
Barone F et al (2008) CXCL13, CCL21, and CXCL12 expression in salivary glands of patients with Sjogren’s syndrome and MALT lymphoma: association with reactive and malignant areas of lymphoid organization. J Immunol 180(7):5130–5140
Szyszko EA et al (2011) Salivary glands of primary Sjogren’s syndrome patients express factors vital for plasma cell survival. Arthritis Res Ther 13(1):R2
Amft N et al (2001) Ectopic expression of the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjogren’s syndrome. Arthritis Rheum 44(11):2633–2641
Zhao J et al (2016) Association of plasmacytoid dendritic cells with B cell infiltration in minor salivary glands in patients with Sjogren’s syndrome. Mod Rheumatol 26(5):716–724
Luther SA, Cyster JG (2001) Chemokines as regulators of T cell differentiation. Nat Immunol 2(2):102–107
Iwamoto N et al (2010) Regulation of disease susceptibility and mononuclear cell infiltration into the labial salivary glands of Sjogren’s syndrome by monocyte chemotactic protein-1. Rheumatology (Oxford) 49(8):1472–1478
Moriyama M et al (2012) Cytokine/chemokine profiles contribute to understanding the pathogenesis and diagnosis of primary Sjogren’s syndrome. Clin Exp Immunol 169(1):17–26
Akpek EK et al (2004) Chemokines in autoimmune lacrimal gland disease in MRL/MpJ mice. Invest Ophthalmol Vis Sci 45(1):185–190
Karin N, Wildbaum G (2015) The role of chemokines in shaping the balance between CD4(+) T cell subsets and its therapeutic implications in autoimmune and cancer diseases. Front Immunol 6:609
Chow MT et al (2019) Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity
Lee YJ et al (2010) Salivary chemokine levels in patients with primary Sjogren’s syndrome. Rheumatology (Oxford) 49(9):1747–1752
Appay V, Rowland-Jones SL (2001) RANTES: a versatile and controversial chemokine. Trends Immunol 22(2):83–87
Crawford A et al (2011) A role for the chemokine RANTES in regulating CD8 T cell responses during chronic viral infection. PLoS Pathog 7(7):e1002098
Blokland SLM et al (2019) Emerging roles for chemokines and cytokines as orchestrators of immunopathology in Sjögren’s syndrome. Rheumatology
Kramer JM, Klimatcheva E, Rothstein TL (2013) CXCL13 is elevated in Sjogren’s syndrome in mice and humans and is implicated in disease pathogenesis. J Leukoc Biol 94(5):1079–1089
Van Ginkel MS et al (2018) AB0164 numbers of b-lymphocytes increase when formation of lymphoepithelial lesions progresses in salivary glands of primary sjögren’s syndrome patients. Ann Rheum Dis 77(Suppl 2):1270–1271
Szabo K et al (2014) The histopathology of labial salivary glands in primary Sjogren’s syndrome: focusing on follicular helper T cells in the inflammatory infiltrates. Mediat Inflamm 2014:631787
Pratama A, Vinuesa CG (2014) Control of TFH cell numbers: why and how? Immunol Cell Biol 92(1):40–48
Brokstad KA, et al (2018) T follicular-like helper cells in the peripheral blood of patients with primary Sjogren’s syndrome. Scand J Immunol e12679
Szabo K et al (2016) A comprehensive investigation on the distribution of circulating follicular T helper cells and B cell subsets in primary Sjogren’s syndrome and systemic lupus erythematosus. Clin Exp Immunol 183(1):76–89
He J et al (2013) Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 39(4):770–781
Nocturne G et al (2015) CXCL13 and CCL11 serum levels and lymphoma and disease activity in primary Sjogren’s syndrome. Arthritis Rheumatol 67(12):3226–3233
Granato A et al (2014) IL-4 regulates Bim expression and promotes B cell maturation in synergy with BAFF conferring resistance to cell death at negative selection checkpoints. J Immunol 192(12):5761–5775
Wurster AL et al (2002) Interleukin-4-mediated protection of primary B cells from apoptosis through Stat6-dependent up-regulation of Bcl-xL. J Biol Chem 277(30):27169–27175
Singh RR et al (2003) Differential contribution of IL-4 and STAT6 vs STAT4 to the development of lupus nephritis. J Immunol 170(9):4818–4825
Nguyen CQ et al (2007) IL-4-STAT6 signal transduction-dependent induction of the clinical phase of Sjogren’s syndrome-like disease of the nonobese diabetic mouse. J Immunol 179(1):382–390
Wei L et al (2007) IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem 282(48):34605–34610
Deenick EK, Tangye SG (2007) Autoimmunity: IL-21: a new player in Th17-cell differentiation. Immunol Cell Biol 85(7):503–505
Lin X et al (2015) Th17 cells play a critical role in the development of experimental Sjogren’s syndrome. Ann Rheum Dis 74(6):1302–1310
Ciccia F et al (2013) Rituximab modulates the expression of IL-22 in the salivary glands of patients with primary Sjogren’s syndrome. Ann Rheum Dis 72(5):782–783
Bard JD et al (2008) Aberrant expression of IL-22 receptor 1 and autocrine IL-22 stimulation contribute to tumorigenicity in ALK+ anaplastic large cell lymphoma. Leukemia 22(8):1595–1603
Ciccia F et al (2015) Interleukin (IL)-22 receptor 1 is over-expressed in primary Sjogren’s syndrome and Sjogren-associated non-Hodgkin lymphomas and is regulated by IL-18. Clin Exp Immunol 181(2):219–229
Domeier PP et al (2016) IFN-gamma receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med 213(5):715–732
Mohr E et al (2010) IFN-{gamma} produced by CD8 T cells induces T-bet-dependent and -independent class switching in B cells in responses to alum-precipitated protein vaccine. Proc Natl Acad Sci U S A 107(40):17292–17297
Matsui K, Sano H (2017) T helper 17 cells in primary Sjogren’s syndrome. J Clin Med 6(7)
Ross SH, Cantrell DA (2018) Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol 36:411–433
Zhao J, Zhao J, Perlman S (2012) Differential effects of IL-12 on Tregs and non-Treg T cells: roles of IFN-gamma, IL-2 and IL-2R. PLoS One 7(9):e46241
Pot C et al (2011) Induction of regulatory Tr1 cells and inhibition of T(H)17 cells by IL-27. Semin Immunol 23(6):438–445
Cheuk S et al (2017) CD49a expression defines tissue-resident CD8(+) T cells poised for cytotoxic function in human skin. Immunity 46(2):287–300
Fujihara T et al (1999) Preferential localization of CD8+ alpha E beta 7+ T cells around acinar epithelial cells with apoptosis in patients with Sjogren’s syndrome. J Immunol 163(4):2226–2235
Schuster IS et al (2014) TRAIL+ NK cells control CD4+ T cell responses during chronic viral infection to limit autoimmunity. Immunity 41(4):646–656
Perez P et al (2005) Increased acinar damage of salivary glands of patients with Sjogren’s syndrome is paralleled by simultaneous imbalance of matrix metalloproteinase 3/tissue inhibitor of metalloproteinases 1 and matrix metalloproteinase 9/tissue inhibitor of metalloproteinases 1 ratios. Arthritis Rheum 52(9):2751–2760
Fogli LK et al (2013) T cell-derived IL-17 mediates epithelial changes in the airway and drives pulmonary neutrophilia. J Immunol 191(6):3100–3111
Zhu J, Paul WE (2008) CD4 T cells: fates, functions, and faults. Blood 112(5):1557–1569
Mangan PR et al (2006) Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441(7090):231–234
Kim SH et al (2019) Delayed IL-21 treatment preferentially expands peptide-specific CD8(+) T cells by reducing bystander activation of T cells. Immunotherapy 11(6):497–513
Lavoie TN et al (2016) IL-22 regulation of functional gene expression in salivary gland cells. Genom Data 7:178–184
Roescher N, Tak PP, Illei GG (2009) Cytokines in Sjogren’s syndrome. Oral Dis 15(8):519–526
Borchers AT et al (2003) Immunopathogenesis of Sjogren’s syndrome. Clin Rev Allergy Immunol 25(1):89–104
Kong L et al (1997) Fas and Fas ligand expression in the salivary glands of patients with primary Sjogren’s syndrome. Arthritis Rheum 40(1):87–97
Cha S et al (2004) A dual role for interferon-gamma in the pathogenesis of Sjogren’s syndrome-like autoimmune exocrinopathy in the nonobese diabetic mouse. Scand J Immunol 60(6):552–565
Savinov AY, Wong FS, Chervonsky AV (2001) IFN-gamma affects homing of diabetogenic T cells. J Immunol 167(11):6637–6643
Yin H et al (2011) Location of immunization and interferon-gamma are central to induction of salivary gland dysfunction in Ro60 peptide immunized model of Sjogren’s syndrome. PLoS One 6(3):e18003
Dessein PH et al (2005) Traditional and nontraditional cardiovascular risk factors are associated with atherosclerosis in rheumatoid arthritis. J Rheumatol 32(3):435–442
Mok CC et al (2013) High-sensitivity C-reactive protein, disease activity, and cardiovascular risk factors in systemic lupus erythematosus. Arthritis Care & Research 65(3):441–447
Ramos-Casals M et al (2007) High prevalence of serum metabolic alterations in primary Sjogren’s syndrome: influence on clinical and immunological expression. J Rheumatol 34(4):754–761
Augusto KL et al (2016) Metabolic syndrome in Sjogren’s syndrome patients: a relevant concern for clinical monitoring. Clin Rheumatol 35(3):639–647
Aqrawi LA et al (2018) Signalling pathways identified in salivary glands from primary Sjogren’s syndrome patients reveal enhanced adipose tissue development. Autoimmunity 51(3):135–146
Stern JH, Rutkowski JM, Scherer PE (2016) Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab 23(5):770–784
McLaughlin T et al (2017) Role of innate and adaptive immunity in obesity-associated metabolic disease. J Clin Invest 127(1):5–13
Gilleron J et al (2018) Rab4b deficiency in T cells promotes adipose Treg/Th17 imbalance, adipose tissue dysfunction, and insulin resistance. Cell Rep 25(12):3329–3341 e5
Jiang Z, Zhu L (2016) Update on the role of alternatively activated macrophages in asthma. J Asthma Allergy 9:101–107
van der Windt GJ, Pearce EL (2012) Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev 249(1):27–42
Cui G et al (2015) IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell 161(4):750–761
Pan Y et al (2017) Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543(7644):252–256
Chowdhury PS et al (2018) PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8(+) T cells and facilitates anti-PD-1 therapy. Cancer Immunol Res 6(11):1375–1387
Kim JA et al (2012) Role of lipotoxicity in endothelial dysfunction. Heart Fail Clin 8(4):589–607
Ramos-Casals M et al (2010) Treatment of primary Sjogren syndrome: a systematic review. JAMA 304(4):452–460
Mariette X, Criswell LA (2018) Primary Sjogren’s syndrome. N Engl J Med 378(10):931–939
Wong SK et al (2016) Animal models of metabolic syndrome: a review. Nutr Metab (Lond) 13:65
Huang C et al (2019) Immune checkpoint molecules. Possible future therapeutic implications in autoimmune diseases. J Autoimmun 104:102333
Ramos-Casals M et al (2012) Topical and systemic medications for the treatment of primary Sjogren’s syndrome. Nat Rev Rheumatol 8(7):399–411
Mariette X et al (2015) Efficacy and safety of belimumab in primary Sjogren’s syndrome: results of the BELISS open-label phase II study. Ann Rheum Dis 74(3):526–531
Baer AN (2020) Oasis or mirage—a new drug shows promise for Sjögren's syndrome. Lancet Rheumatol
Fisher BA, et al Assessment of the anti-CD40 antibody iscalimab in patients with primary Sjögren’s syndrome: a multicentre, randomised, double-blind, placebo-controlled, proof-of-concept study. Lancet Rheumatol
Adler S et al (2013) Evaluation of histologic, serologic, and clinical changes in response to abatacept treatment of primary Sjogren’s syndrome: a pilot study. Arthritis Care Res (Hoboken) 65(11):1862–1868
van Nimwegen JF, et al Abatacept treatment for patients with early active primary Sjögren’s syndrome: a single-centre, randomised, double-blind, placebo-controlled, phase 3 trial (ASAP-III study). Lancet Rheumatol
Verstappen GM et al (2017) Attenuation of follicular helper T cell-dependent B cell hyperactivity by abatacept treatment in primary Sjogren’s syndrome. Arthritis Rheumatol 69(9):1850–1861
Meiners PM et al (2014) Abatacept treatment reduces disease activity in early primary Sjogren’s syndrome (open-label proof of concept ASAP study). Ann Rheum Dis 73(7):1393–1396
Motegi K et al (2005) Expression of aquaporin-5 in and fluid secretion from immortalized human salivary gland ductal cells by treatment with 5-aza-2′-deoxycytidine: a possibility for improvement of xerostomia in patients with Sjogren’s syndrome. Lab Investig 85(3):342–353
Lai Z et al (2016) Aquaporin gene therapy corrects Sjogren’s syndrome phenotype in mice. Proc Natl Acad Sci U S A 113(20):5694–5699
Zeng M et al (2017) Restoration of CFTR activity in ducts rescues acinar cell function and reduces inflammation in pancreatic and salivary glands of mice. Gastroenterology 153(4):1148–1159
Authorship
Yuan Yao, Jin-Fen Ma, Ting Xu, and Cai-Yue Gao wrote the manuscript. Yuan Yao, JIn-Fen Ma, and Cai-Yue Gao designed and drew the figures; Christopher Chang, Patrick M. Eric Gershwin, and Zhe-Xiong Lian wrote and edited the manuscript.
Funding
This work is supported by the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2017ZT07S054), the National Key R&D Program of China (2017YFA0205600), and the National Natural Science Foundation of China (81901653, 81701597).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yao, Y., Ma, JF., Chang, C. et al. Immunobiology of T Cells in Sjögren’s Syndrome. Clinic Rev Allerg Immunol 60, 111–131 (2021). https://doi.org/10.1007/s12016-020-08793-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-020-08793-7