
Abstract
There is evidence that rheumatoid arthritis (RA) is associated with higher overall and cardiovascular (CV) morbidity and mortality as compared with general population. Increased prevalence of traditional risk factors and chronic inflammation, that has been recognized as independent CV risk factor, may play an important role in atherosclerosis and subsequently ischemic heart disease development. However, myocardial dysfunction as a result of chronic inflammation and secondarily myocardial fibrosis markedly participates on heart failure development. Proinflammatory cytokines, such as C-reactive protein, tumor necrosis factor alpha (TNFα), interleukins 1 and 6, that are markedly increased in RA, play a role in the acceleration of atherosclerosis as well as myocardial fibrosis development. Several studies documented that increased CV risk was associated with seropositivity, disease activity score, citrullination, and duration of RA. Early detection of heart dysfunction is based on echocardiographic detection of diastolic dysfunction resulting from myocardial inflammation and fibrosis. Some studies showed also higher prevalence of left ventricular systolic dysfunction and increased prevalence of cardiac arrhythmias as compared to non-RA population. There are still controversies on the impact of NT-proBNP in predicting cardiac impairment in RA patients. Some authors consider it to be a sensitive noninvasive predictor of subclinical CV disease in these patients and also a predictor of all-cause mortality independently on traditional CV risk factors. However, the correlation with parameters of cardiac function was confirmed only in a few studies. The impact of biological treatment on progression of atherosclerosis and heart failure is still controversial and seems to be not harmful in young patients with normal left ventricular function. The effect of biologics, especially anti-TNFα drugs, is probably related to the cardiac function before treatment. Larger prospective clinical, echocardiographic, and magnetic resonance studies are needed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disease affecting approximately 1 % of adults in the general population and resulting in impairment of various organs and tissues [1]. It is associated with higher overall and cardiovascular (CV) morbidity and mortality as compared with population without RA [2, 3]. Excess mortality in RA has been seen in CV disease (31 %), pulmonary fibrosis (4 %), and lymphoma (2.3 %) [4]. Baseline predictors of mortality in patients with RA have been reported male gender, older age, poor function, lower socio-economic status, extra-articular manifestations, rheumatoid factor (RF) and anti-cyclic citrullinated peptide antibodies (anti-CCP) positivity, higher inflammatory parameters, and glucocorticoid therapy [4–8].
Cardiovascular diseases (CVD) have been recognized as the main cause of mortality among RA patients, and recent data confirmed this trend also in earlier stages of RA. The risk of developing CVD is estimated at least 50 % greater than compared with the general population. Increased CV morbidity and mortality in RA patients is mainly due to accelerated atherosclerosis [9–11]. Two major factors have been evoked as determinants of higher CV morbidity and mortality due to atherosclerosis, i.e., elevated prevalence of traditional CV risk factors and chronic systemic inflammation, that has been recognized to be an independent risk factor for CVD in RA. This finding is supported by the evidence that optimal control of the disease activity by conventional or targeted disease modifying antirheumatic drugs (DMARDs) decreases the risk of CV morbidity and mortality [10]. Besides coronary atherosclerosis, myocardial dysfunction itself as a result of chronic systemic inflammation leading to myocardial edema and fibrosis may also markedly participate in CV morbidity, especially in heart failure (HF) development [12, 13].
Three most important pathogenic mechanisms are supposed to participate in cardiac impairment in RA:
-
1.
Genetic factors (HLA DRB1 04, gene polymorphism for TNFα, MTHFR, CCR5, etc.) that seem to be associated with a higher risk of CVD in RA [14]
-
2.
Traditional risk factors for atherosclerosis and CVD (obesity, dyslipidemia, diabetes mellitus, arterial hypertension, smoking, hyperuricemia, etc.) [15]
-
3.
Proinflammatory cytokines (CRP, TNFα, IL-1, IL-6) that are markedly increased in RA play a role in the acceleration of atherosclerosis as well as myocardial fibrosis development [16]
In this article, we review current data on mechanisms participating in CV morbidity and mortality in RA and also the influence of anti-inflammatory treatment.
Atherosclerosis and coronary heart disease in patients with RA
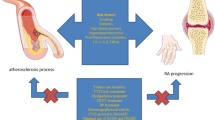
As was mentioned above, increased CV mortality in RA patients is mainly due to atherosclerosis. Several studies over last 20 years showed that patients with RA have significantly higher risk of coronary heart disease (CHD) when compared with non-RA subjects. Patients with RA are more likely to experience unrecognized myocardial infarction and sudden death and, they also have poorer long-term outcomes than those with myocardial infarction without RA [10, 17]. The influence of traditional risk factors and chronic systemic inflammation is described in this article. Figure 1 shows proven or potential mediators and their effects on the cells and tissues resulting in endothelial and myocardial dysfunction in RA. Low-grade inflammation that is typical for the disease results in platelet dysfunction and the formation of a denser clot structure and hypofibrinolysis. Following activation or apoptosis, various cell types release microparticles which contain procoagulants. Activation of neutrophils causes the release of neutrophil extracellular traps (NETs), which display procoagulant properties including platelet activation [3].

Endothelial and myocardial dysfunction resulting from different effects of proven or potential mediators and effects on cells and tissues in chronic inflammatory disease as RA (direction of small arrows by mediator and effect mean up or downregulation; adapted from Schuett et al. [3], Ahmed et al. [40], Maijer et al. [43]; TNFα tumor necrosis factor alpha, IL-1 interleukin 1, IL-6 interleukin 6, IL-10 interleukin 10, TGFβ transforming growth factor beta, PAI-1 plasminogen activator inhibitor 1)
Traditional risk factors
Metabolic syndrome
The metabolic syndrome (MetS) is a cluster of cardiometabolic disorders that result from the increasing prevalence of obesity. Several studies have shown that MetS may provide an additional link between accelerated atherosclerosis and inflammation in RA. However, their results are inconsistent, probably due to differences in the definition of the MetS, study populations and also treatment of RA. The prevalence of MetS in the study of Rostom et al. was mildly but significantly higher in RA patients. Increased systemic inflammatory markers and use of glucocorticoids (GC) were independent predictors associated with the presence of MetS. Approximately 40 % of RA women had MetS and almost all of these patients had abdominal obesity [18]. On the other side, it was not confirmed in Korean and Mexican women. In the Mexican study, the prevalence of MetS in patients with early RA was lower than that in controls [19]. In Korean women, the group who received RA treatment showed significantly lower prevalence of MetS as compared to untreated group. The control of inflammation and disease activity can reduce the frequency of MetS in RA patients. Moreover, treatment with methotrexate (MTX) also may be a protective factor against MetS [20]. Despite this different results, a metaanalysis of Zhang et al. clearly identified a significant association between RA and the risk of MetS with an overall odds ratio of 1.24 (95 % CI, 1.03–1.50) [21]. Conversely, some studies showed that among patients with RA, low body mass index was associated with a significantly increased risk of CV death, probably due to development of “rheumatoid cachexia” [9]. It seems that both obesity and cachexia may represent an increased cardiovascular risk in these patients.
Diabetes mellitus
Recent data showed that RA is associated with increased risk of diabetes mellitus (DM), including type 1 (T1DM), and type 2 (T2DM). Oppositely, patients with T2DM also demonstrated an elevated risk of RA [22, 23]. It is consistent with the hypothesis that chronic low-grade inflammation in T2DM may elicit the development of RA in genetically susceptible individuals. Both types of DM participate on higher risk of CV morbidity and mortality in RA. The risk of CVD in RA could be as high as that conferred by T2DM [22].
In the recent Italian study, increasing age and RA duration were both associated with an increased likelihood of being classified as prediabetes or T2DM. The authors conclude that RA seems to be characterized by increased prevalence of undiagnosed DM, especially in patients with longer disease duration [24].
Insulin resistance and glucose intolerance have been associated with an increased risk of CHD in RA patients on the long-term treatment with GC that can markedly potentiate development of insulin resistance or impaired glucose tolerance. GC use is a clinically important and quantifiable risk factor for DM. The risk is influenced by dose and treatment duration, although only within the last 6 months. Recent metaanalytic study showed that RA patients with T2DM were almost two times more likely to experience any CV event compared to non-diabetic patients with RA [25].
Arterial hypertension
Studies on the prevalence of arterial hypertension (AH) in RA yielded conflicting results, with that reporting lower, equivalent, or elevated mean systolic and/or diastolic blood pressure compared to non-RA population [26–28]. AH was not associated with systemic inflammation or insulin resistance in RA, but it was showed to be associated with increased concentrations of homocysteine and leptin [29]. Higher activity of RA may negatively influence nocturnal fall in blood pressure [30]. Additionally, other factors such as the chronic use of nonsteroidal anti-inflammatory drugs or GC are known to increase blood pressure. If present, AH may participate in increased CV morbidity and mortality as compared with non-hypertensive RA patients. Carotid intima-media thickness (IMT) was associated with age and systolic blood pressure in RA patients [25]. Moreover, anti-tumor necrosis factor alpha (anti-TNFα) therapy was associated with a significantly increased risk of developing AH in these subjects [31].
Dyslipidemia
Accelerated atherosclerosis in RA may be related also to lipid profile changes [32]. Dyslipidemia, commonly observed in patients with active RA, appears to be present very early in the disease development. Majority of studies detected lower total cholesterol (TC) levels as well as lower levels of high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein cholesterol (LDL-C) in RA patients as compared with non-RA subjects, although overall incidence of dyslipidemia was higher in RA [27, 33–35]. Only some studies demonstrated typical atherogenic lipid profile in these patients [36, 37].
In the Japan multicenter observational study of 488 patients with RA, the prevalence of hyper-LDL-C, hypertriacylglycerolemia, and hypo-HDL-C were 29.3, 24.2, and 10.2 %, respectively, and the overall prevalence of dyslipidemia was 56.5 %. The level of HDL-C was in inverse correlation with the disease activity expressing as DAS28 score. Patients treated with low-dose GC showed significantly higher levels of HDL-C and lower TC/HDL-C ratio compared with those GC-untreated. On the other side, patients treated with biological agents showed significantly higher levels of LDL-C, lower levels of HDL-C and higher TC/HDL-C ratio. These results suggest that controlling RA disease activity might improve lipid profiles and eventually lower CV risk [33]. Low-dose atorvastatin was effective for the treatment of dyslipidemia in RA but had no apparent effect on RA disease activity [34].
The impact of dyslipidemia in RA and its influence on CVD risk is still unknown. The complex relationship between LDL-C and HDL-C levels and risk of CV events was not significantly different from that in age and sex-matched non-RA cohort [38]. According to some authors, the association between lipid profile and the risk of CVD seems to be paradoxical, whereby lower levels of TC and LDL-C are associated with higher CVD risk. This may be due to lipid-lowering effects of systemic inflammation. Adipose tissue in RA is quantitatively increased, but it may be more inflamed resulting in metabolic dysfunction of adipose tissue and to lower HDL-C [34].
Inflammatory markers
Over the years, multiple studies evidenced increasing numbers of cytokines involved in RA pathophysiology, further to those used as target of cytokine-blocking therapies. In fact, some experimental and human studies showed that inflammatory pathways of RA may initiate and/or accelerate atherosclerosis and this effect can be ameliorated by anti-inflammatory treatment. In RA, inflammation and atherosclerosis are closely linked. Inflammation mediates its effects on atherosclerosis both through modulation of traditional risk factors and through direct affecting the vessel wall and contributing to plaque formation [39, 40]. This systemic inflammation may result in impaired insulin sensitivity and subsequently enhance the impact of MetS. Studies on IMT in RA patients also demonstrated that systemic inflammation and CV risk factors were associated with rapid IMT progression. MTX and anti-TNFα agents may influence IMT progression by reducing the effect of the systemic inflammation on the IMT [41]. C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) were similarly associated with higher risk of myocardial infarction as well as stroke, reflecting the prominent role of inflammation in CHD risk in RA [42]. In the study of Innala et al., 4.3 % of patients with RA had a new myocardial infarction and 5.1 % of all developed stroke during the follow-up period. In this study, the authors found inflammatory activity to be harmful in terms of new CV events and inflammation potentiates the effect of traditional CV risk factors [8].
The role of anti-CCP positivity in atherosclerosis is supported by some studies showing that patients anti-CCP positive had increased risk of CV death that correlated with higher levels of proinflammatory cytokines such as TNFα and interleukin 6 (IL-6) [6]. If anti-CCP positivity plays a direct role in the development of atherosclerosis or if it represents only marker of proinflammatory state resulting in atherosclerotic process in these patients is still unknown.
Angiotensin II that has been demonstrated to be elevated in RA patients is also closely related to AH, endothelial dysfunction, and atherosclerosis. The role of adipokines, such as leptin, adiponectin, vaspin, resistin, omentin, and others in the prevention or acceleration of atherosclerosis in RA patients is not fully recognized [43]. Treatment modalities such as TNFα inhibition might have a beneficial effect on CV risk (see below). However, whether this benefit is attributable to effective control of inflammation or whether targeting specific cytokines, implicated in atherosclerosis, provides additional risk reduction, is unclear. Further knowledge of the predictors of CV risk, the effects of early control of inflammation and of drug-specific effects are likely to improve the recognition and management of CV risk in RA patients. Cytokines and biomarkers of endothelial dysfunction and heart failure in RA are demonstrated in the Table 1.
Rheumatoid arthritis and heart failure
Heart failure (HF) and inflammation are important contributors to the excess of overall morbidity and mortality in patients with RA. HF rather than ischemic heart disease appears to participate in the increased mortality in these patients [44]. Nicola et al. demonstrated significant excess risk of congestive HF in RA population as compared with non-RA, yielding hazard ratio 1.87 (95 % CI, 1.47–2.39) after 30 years of follow-up [45].
,Similarly in the study of Wolfe and Michaud, HF was more common among patients with RA (3.9 %, n = 461) than in those with osteoarthritis (2.3 %, n = 87). Patients with RA had the same risk factors for HF (e.g., AH, prior myocardial infarction, DM, advanced age) as persons in population-based studies. HF was significantly less common in anti-TNFα-treated patients than in the remaining patients, even after adjusting for baseline differences. In the absence of pre-existing CVD, the risk of HF was low and was not related to anti-TNFα therapy [16]. Factors associated with higher prevalence of congestive HF in RA were similar to that typically associated with HF in the non-RA population (male gender, age, AH, CHD, DM, and smoking). Additionally, RA-related parameters (patient’s disability, pain, severity of RA, RF positivity) were associated with prevalent HF [16, 45]. Disease activity is also considered to be an independent risk factor of HF as well as systemic inflammation (ESR, CRP) or duration of RA [46, 47]. Besides several RA characteristics, the use of GC was also associated with HF after adjustment for CV risk factors and CHD. On the other side, MTX use appeared to be protective against HF [48]. In the study of Listing et al., an increased risk of HF developing after adjustment for traditional CV factors was found in patients who had a higher disease activity score DAS28 at follow-up with hazard ratio 1.47 (95 % CI, 1.07–2.02) [49].
Mechanisms resulting in developing or worsening of heart failure in RA patients are shown in the Table 2. Increased risk of HF in RA may be mediated in part by vascular disease including endothelial dysfunction and coronary atherosclerosis. In addition to atherosclerosis, RA appears to be associated with subclinical structural heart disease. HF in RA typically presents with occult clinical symptomatology and is mainly associated with structural and functional left ventricle (LV) abnormalities leading to diastolic dysfunction, while systolic performance remains well preserved longer time [12, 50].
Causes of heart dysfunction in RA patients include several morphologic changes, such as valvular diseases or myocardial fibrosis and edema. RA is known to cause rheumatoid nodules leading to valvular disease, particularly regurgitation. The most commonly reported valvular diseases include aortic stenosis and regurgitation, mitral regurgitation, tricuspidal regurgitation, or combined valvular lesions [47, 51].
Experimental, histopathologic, and clinical echocardiographic and magnetic resonance studies demonstrated subclinical myocardial fibrosis and dysfunction in RA subjects without clinical symptoms of HF. This association remained significant after adjustment for CV risk factors and comorbidities [52]. In the study of Ntusi et al., myocardial fibrosis was found in 46 % of patients with RA. These patients had larger areas of myocardial edema, but LV mass and ejection fraction (EF) were similar to controls [13]. Both myocardial fibrosis and inflammation were associated with the disease activity. Thus, the chronic low-grade myocardial inflammation resulting in fibrosis may predispose RA patients to diastolic dysfunction that is common in RA and was confirmed by several echocardiographic studies [53–55]. Most studies demonstrated a significant inverse relationship between the E/A ratio and disease duration [54, 56]. In the study of Liang et al., IL-6 levels were independently associated with diastolic dysfunction even after adjustment for CV risk factors. TNFα levels were also in inverse relationship with E/A ratio [54].
LV systolic dysfunction in RA was reported only in a few studies [55–58]. In the study of Bhatia et al., the prevalence of systolic dysfunction was three times more common as compared to general population [57]. In some studies, HF in RA has been associated with concentric LV hypertrophy and reduced longitudinal strain [46, 47].
RA was also associated with an increased incidence of cardiac arrythmias, in particular atrial fibrilation and also other tachyarrythmias. RA patients were twice as likely to experience sudden cardiac death compared with non-RA subjects, pointing to an increased propensity to develop malignant ventricular arrhythmias. Indeed, ventricular repolarization (QT interval) abnormalities and cardiovascular autonomic nervous system dysfunction, representing two well-recognized risk factors for life-threatening ventricular arrhythmias in the general population, are commonly observed in RA. Moreover, large population-based studies seem to indicate that also the prevalence of atrial fibrillation is significantly higher in RA subjects than in the general population, suggesting that these patients are characterized by an abnormal diffuse myocardial electrical instability. Although the underlying mechanisms accounting for the pro-arrhythmogenic substrate in RA are probably intricate, the leading role seems to be played by chronic systemic inflammatory activation which is able to promote arrhythmias both indirectly, by accelerating the development of ischemic heart disease and congestive heart failure, and directly, by affecting cardiac electrophysiology [59, 60].
Biomarkers associated with heart failure
B-type natriuretic peptide (BNP) or N-terminal fragment of proBNP (NT-proBNP)
There are still controversies on the impact of NT-proBNP in predicting cardiac impairment in RA patients. Some authors consider it to be a sensitive noninvasive predictor of subclinical CVD in these patients and a predictor of all-cause mortality independent of traditional CV risk factors [61]. There is evidence that NT-proBNP is increased in RA patients and it is associated with inflammatory markers [35, 61]. However, some studies did not demonstrate relationship between NT-proBNP levels and LV function in RA patients. In our study, RA patients had significantly higher plasma NT-proBNP as compared with controls. A significantly higher levels were detected in RF positive patients. TNFα rather than NT-proBNP was in negative correlation with systolic and diastolic LV function. Thus, we concluded that TNFα appears to be a better marker of heart impairment caused by chronic inflammation in RA patients than NT-proBNP itself [55].
Homocystein
Elevated serum homocysteine have been independently linked to an increased risk of HF, particularly in women. In RA patients, serum homocysteine levels were shown to be significantly higher than in non-RA controls and were associated with both inflammatory markers and MTX therapy. Folic acid treatment reduced homocysteine levels in RA patients. It has been proven recently that a combination therapy with MTX and folic acid was associated with a reduced incidence of CVD in RA patients [29, 62].
Inflammatory cytokines
In RA, the inflamed synovium of the rheumatoid joint as well as peripheral blood mononuclear cells are a major source of macrophage-derived cytokines, such as TNFα, interleukin 1 (IL-1), and IL-6 that are markedly expressed in serum of these patients [63]. These cytokines are in non-RA patients associated with an increased prevalence of congestive HF. However, in RA, the relationship between systolic or diastolic HF and serum interleukin levels is poorly documented [55].
Influence of biological treatment
HF is generally characterized by the elevation of proinflammatory cytokines and mediators that contribute to disease progression and may serve as relevant markers of the disease severity and HF progression. Major proinflammatory cytokines include TNFα, IL-1, and IL-6, which circulating levels are substantially elevated in HF [63–66]. Thus, it was hypothesized that inhibition of these cytokines may improve heart function in patients with HF. Unfortunately, the results of large studies yielded disappointing results.
The effect of infliximab on the heart function was evaluated in a randomized placebo-controlled, double-blind pilot study ATTACH in patients with chronic HF in NYHA classes II–IV. This study did not show any beneficial effect of infliximab over placebo in terms of efficacy. On the contrary, its higher dose was associated with an increase in both all-cause mortality and the number of hospitalizations due to HF [67]. Similar results were observed in two studies RENAISSANCE and RECOVER (design similar to ATTACH) evaluating the influence of etanercept on the heart function. Both studies showed a trend toward increased mortality or hospitalizations due to HF in the etanercept group. The results of the studies with etanercept and infliximab suggest a deleterious effect, especially of higher doses of TNFα blockers in patients with NYHA classes III and IV [68].
How do TNFα blockers influence heart function in patients with RA?
TNFα promotes inflammatory response that is essential in the pathogenesis of RA [69]. A disease activity and systemic inflammation in RA represent an increased risk of CV mortality and HF. Therefore, it is suggested that TNFα inhibitors should reduce the risk of HF in RA group. On the other side, there is a question whether RA patients with heart dysfunction are contraindicated for treatment with TNFα blockers. Studies that were performed so far yielded controversial results. Some of them were stopped prematurely due to lack of efficacy and because of worsening HF in the anti-TNFα-treated groups suggesting that TNFα inhibitors may promote and/or worsen HF in RA patients. Treatment with infliximab led to an increase in LV EF in patients with RA. Unfortunately, this was detected in only patients with normal ejection fraction (EF). This treatment also resulted in a significant reduction of endothelin 1, IL-6, and NT-proBNP levels [70, 71]. In our study, no significant changes in echocardiographic structural and functional parameters of the LV were detected after 6 and 12 months of infliximab administration. We conclude that long-term infliximab administration does not deteriorate both cardiac morphology and function [72]. In other studies, it was also demonstrated protective role of infliximab on the myocardium or no significant changes of HF incidence in young RA patients [16, 73, 74]. On the other side, acute infliximab administration decreased cardiac output due to low stroke volume in RA patients without heart disease [75]. In our study, acute infliximab administration led to significant increase in NT-proBNP levels but long-term treatment showed tendency to decrease the NT-proBNP levels [72]. Similar, but significant, positive effects of long-term TNFα inhibitors treatment (infliximab, adalimumab) were confirmed by other authors in RA patients without previous HF [71, 76].
Based on these data and also the study of Setoguchi et al., TNFα blockade may pose a greater risk of HF hospitalization in elderly patients with RA compared to MTX use. Anti-TNFα therapy may also pose a greater risk of death in those with previous HF [77]. Controversially, recently published papers showed that anti-TNFα therapy was not associated with a risk of HF hospital admissions compared with DMARDs in the RA population [74, 78]. The effects are probably related to heart function before treatment.
RA patients with no history of congestive HF and concomitant indication for TNFα blockers do not need a baseline echocardiography to screen for HF. Patients with well compensated, mild HF (NYHA classes I and II) before starting of TNFα blockers should undergo echocardiography. TNFα blockers should be avoided in patients with decreased EF or in NYHA classes III and IV of HF [48, 70].
In the network metaanalysis of Singh et al., the adverse affects of biologics were evaluated summarizing data from Cochrane Library, Medline, and Embase. Although biologics were associated with significantly higher rates of adverse events and withdrawals due to adverse events, the rate of congestive HF was not significantly higher between biologics and control treatment [79].
There are also few studies with IL-6 inhibition by tocilizumab (TCZ) in patients with RA and HF. In one study, treatment with TCZ for 5 years led to an improvement of inflammatory activity without adverse effects on the heart function [80]. However, in the recent study, TCZ was associated with LV dysfunction in RA patients or resulted in a decrease of EF in RA patients with normal heart function [81].
The most protective effects on the heart function in RA patients have been described in IL-1 inhibition. After anakinra treatment, there was significant improvement of LV systolic and diastolic function. The improvements in biomarkers and vascular and LV function were greater in anakinra group than in the prednisolone group after 30 days of treatment. The possible mechanism is the improvement of myocardial deformation in RA that was shown in some studies [82–84]. Studies comparing TNFα blockers with anakinra showed a safety of anakinra in RA patients on progression of HF in comparison with TNFα blockers [85]. IL-1 blockade led to a lower incidence of HF in RA patients [86].
Conclusion
Summarizing all reported data, we can conclude that patients with RA, who constitute approximately 1 % of the population, are at increased risk of CV morbidity and mortality. Higher prevalence of traditional risk factors and chronic inflammation plays an important role in atherosclerosis and subsequently ischemic heart disease development. However, myocardial dysfunction as a result of myocardial edema and fibrosis participates markedly on HF development. Proinflammatory cytokines, such as CRP, TNFα, IL-1, and IL-6, that are increased markedly in RA, play a role in acceleration of atherosclerosis as well as myocardial fibrosis development. Additional evaluation is needed to clarify the effect of RA as well as therapeutic strategies on the myocardium at all stages of the disease in order to early detect cardiac impairment or to prevent the development of HF in these patients.
References
Alamanos Y, Drosos AA (2005) Epidemiology of adult rheumatoid arthritis. Autoimmun Rev 4(3):130–136
Norton S, Sacker A, Dixey J, Done J, Williams P, Young A (2013) Early rheumatoid arthritis study. Trajectories of functional limitation in early rheumatoid arthritis and their association with mortality. Rheumatology (Oxford) 52(11):2016–2024
Schuett KA, Lehrke M, Marx N, Burgmaier M (2015) High risk cardiovascular patients: clinical features, comorbidities and interconnecting mechanisms. Front Immunol 6:1–9
Young A, Koduri G, Batley M, Kulinskaya E, Gough A, Norton S et al (2007) Mortality in rheumatoid arthritis. Increased in the early course of disease, in ischaemic heart disease and in pulmonary fibrosis. Rheumatol (Oxford) 46(2):350–357
Wallberg-Johnsson S, Ohman ML, Dahlqvist SR (1997) Cardiovascular morbidity and mortality in patients with seropositive rheumatoid arthritis in northern Sweden. J Rheumatol 24:445–451
Ajeganova S, Humphreys JH, Verheul MK, van Steenbergen HW, van Nies JA, Hafstrom I et al. (2016) Anticitrullinated protein antibodies and rheumatoid factor are associated with increased mortality but different cause of death in patients with rheumatoid arthritis: a longitudinal study in three European cohort. Ann Rheum Dis. doi: 10.1136/annrheumdis-2015-208579.
Crepaldi G, Scire CA, Carrara G, Sakellariou G, Caporali R, Hmamouchi I et al (2016) Cardiovascular comorbidities relate more than others with disease activity in rheumatoid arthritis. PLoS One 11(1):e0146991. doi:10.1371/journal.pone.0146991
Innala L, Sjoberg C, Moller B, Ljung L, Smedby T, Sodergren A et al (2016) Co-morbidity in patients with early rheumatoid arthritis-inflammation matters. Arthritis Res Ther 18(1):33. doi:10.1186/s13075-016-0928-y
Maradit-Kremers H, Nicola PJ, Crowson CS, Ballman KV, Gabriel SE (2004) Prognostic importance of low body mass index in relation to cardiovascular mortality in rheumatoid arthritis. Arthritis Rheumatism 50:3450–3456
Maradit-Kremers H, Crowson CS, Nicola PJ, Ballman KV, Roger VL, Jacobsen SJ, Gabriel SE (2005) Increased unrecognized coronary heart disease and sudden death in rheumatoid arthritis. Arthritis Rheum 52:402–410
Kobayashi H, Giles JT, Polak JF, Blumenthal RS, Leffell MS, Szklo M et al (2010) Increased prevalence of carotid artery atherosclerosis in rheumatoid arthritis is artery specific. J Rheumatol 37:730–739
Giles JT, Fernandes V, Lima JAC, Bathon JM (2005) Myocardial dysfunction in rheumatoid arthritis: epidemiology and pathogenesis. Arthritis Res Ther 7:195–207
Ntusi NA, Piechnik SK, Francis JM, Ferreira VM, Matthews PM, Robson MD et al (2015) Diffuse myocardial fibrosis and inflammation in rheumatoid arthritis: insights from CMR T1 mapping. JACC Cardiovasc Imaging 8(5):526–536
Rodriguez-Rodriguez L, Lopez-Mejias R, Fernandez-Gutierez B, Balsa A, Gonzales-Gay MA, Martin J (2015) Rheumatoid arthritis: genetic variants as biomarkers of cardiovascular disease. Curr Pharm Des 21(2):182–201
Kumar N, Marshall NJ, Hammal DM, Pearce MS, Parker L, Furniss SS et al (2007) Causes of death in patients with rheumatoid arthritis: comparison with siblings and matched osteoarthritis controls. J Rheumatol 34:1695–1698
Wolfe F, Michaud K (2004) Heart failure in rheumatoid arthritis: rates, predictors and the effects of anti tumor necrosis factor therapy. Am J Med 116:305–311
McCoy SS, Crowson CS, Maradit-Kremers H, Therneau TM, Roger V, Matteson EL, Gabriel SE (2013) Long-term outcomes and treatment after myocardial infarction in patients with rheumatoid arthritis. J Rheumatol 40(5):605–610. doi:10.3899/jrheum.120941
Rostom S, Mengat M, Lahlou R, Hari A, Bahim R, Hassouni NH (2013) Metabolic syndrome in rheumatoid arthritis: case control study. BMC Musculosceletal Dis 14:147
Parra-Salcedo F, Contreras-Yáñez I, Elías-López D, Aguilar-Salinas CA, Pascual-Ramos V (2015) Prevalence, incidence and characteristics of the metabolic syndrome (MetS) in a cohort of Mexican Mestizo early rheumatoid arthritis patients treated with conventional disease modifying anti-rheumatic drugs: the complex relationship between MetS and disease activity. Arthritis Res Ther 17:34. doi:10.1186/s13075-015-0549-x
Lee SH, Choi H, Cho BL, An AR, Seo YG, Jin HS et al (2016) Relationship between metabolic syndrome and rheumatoid arthritis. Korean J Fam Med 37(1):44–50. doi:10.4082/kjfm.2016.37.1.44
Zhang J, Fu L, Shi J, Chen X, Li Y, Ma B, Zhang Y (2013) The risk of metabolic syndrome in patients with rheumatoid arthritis: a meta-analysis of observational studies. PLoS One 8(10):e78151. doi:10.1371/journal.pone.0078151
Jiang P, Li H, Li X (2015) Diabetes mellitus risk factors in rheumatoid arthritis: a systematic review and meta-analysis. Clin Exp Rheumatol 33:115–121
Lu MC, Yan ST, Yin WY, Koo M, Lai NS (2014) Risk of rheumatoid arthritis in patients with type 2 diabetes: a nationwide population-based case-control study. PLoS One 9(7):e101528. doi:10.1371/journal.pone.0101528
Ursini F, Russo E, D’Angelo S, Arturi F, Hribal ML, D’Antona L et al (2016) Prevalence of undiagnosed diabetes in rheumatoid arthritis: an OGTT study. Medicine (Baltimore) 95(7):e2552. doi:10.1097/MD.0000000000002552
Baghdadi LR, Woodman RJ, Shanahan EM, Mangoni AA (2015) The impact of traditional cardiovascular risk factors on cardiovascular outcomes in patients with rheumatoid arthritis: a systematic review and meta-analysis. PLoS One 10(2):e0117952. doi:10.1371/journal.pone.0117952
Gabriel SE, Crowson CS, O’Fallon WM (1999) Comorbidity in arthritis. J Rheumatol 26:2475–2479
Dessein PH, Stanwix AE, Joffe BI (2002) Cardiovascular risk in rheumatoid arthritis versus osteoarthritis: acute phase response related decreased insulin sensitivity and high-density lipoprotein cholesterol as well as clustering of metabolic syndrome features in rheumatoid arthritis. Arthritis Res 4(5):R5
Solomon DH, Karlson EW, Rimm EB, Cannuscio CC, Mandl LA, Manson JE et al (2003) Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation 107(9):1303–1309
Yang X, Gao F, Liu Y (2015) Association of homocysteine with immunological-inflammatory and metabolic laboratory markers and factors in relation to hyperhomocysteinaemia in rheumatoid arthritis. Clin Exp Rheumatol 33(6):900–903
Hamamoto K, Yamada S, Yasumoto M, Yoda M, Yoda K, Tsuda A et al (2016) Association of nocturnal hypertension with disease activity in rheumatoid arthritis. Am J Hypertens 29(3):340–347. doi:10.1093/ajh/hpv119
Zhao Q, Hong D, Zhang Y, Sang Y, Yang Z, Zhang X (2015) Association between anti-TNF therapy for rheumatoid arthritis and hypertension: a meta-analysis of randomized controlled trials. Medicine (Baltimore) 94(14):e731. doi:10.1097/MD.0000000000000731
Jonsson SW, Backman C, Johnson O, Karp K, Lundstrom E, Sundqvist KG, Dahlqvist SR (2011) Increased prevalence of atherosclerosis in patients with medium term rheumatoid arthritis. J Rheumatol 28:2597–2602
Sakai R, Hirano F, Kihara M, Yokoyama W, Yamazaki H, Harada S et al (2015) High prevalence of cardiovascular comorbidities in patients with rheumatoid arthritis from a population-based cross-sectional study of a Japanese health insurance database. Mod Rheumatol 14:1–7
Myasoedova E, Gabriel SE, Green AB, Matteson EL, Crowson CS (2013) Impact of statin use on lipid levels in statin-naive patients with rheumatoid arthritis versus non-rheumatoid arthritis subjects: results from a population-based study. Arthritis Care Res (Hoboken) 65(10):1592–1599
Provan SA, Semb AG, Hisdal J, Stranden E, Aqewall S, Dagfinud H et al (2011) Remission is the goal for cardiovascular risk management in patients with rheumatoid arthritis: a cross-sectional comparative study. Ann Rheum Dis 70:812–817
Hurt-Camejo E, Paredes S, Masana L, Camejo G, Sartipy P, Rosengren B et al (2001) Elevated levels of small, low-density lipoprotein with high affinity for arterial matrix components in patients with rheumatoid arthritis: possible contribution of phospholipase A2 to this atherogenic profile. Arthritis Rheum 44:2761–2767
Chavan VU, Ramavataram D, Patel PA, Rupani MP (2015) Evaluation of serum magnesium, lipid profile and various biochemical parameters as risk factors of cardiovascular diseases in patients with rheumatoid arthritis. J Clin Diagn Res 9(4):BC01–BC05
Liao KP, Liu J, Lu B, Solomon DH, Kim SC (2015) Association between lipid levels and major adverse cardiovascular events in rheumatoid arthritis compared to non-rheumatoid arthritis patients. Arthritis Rheumatol 67(8):2004–2010
Södergren A, Karp K, Bengtsson C, Möller B, Rantapää-Dahlqvist S, Wållberg-Jonsson S (2015) The extent of subclinical atherosclerosis is partially predicted by the inflammatory load: a prospective study over 5 years in patients with rheumatoid arthritis and matched controls. J Rheumatol 42(6):935–942
Ahmed A, Hollan I, Curran SA, Kitson SM, Riggio MP, Mikkelsen K et al. (2016) Rheumatoid arthritis patients have a pro-atherogenic cytokine microenvironment in the aortic adventitia. Arthritis Rheumatol. doi: 10.1002/art.39574
del Rincón I, Polak JF, O’Leary DH, Battafarano DF, Erikson JM, Restrepo JF et al (2015) Systemic inflammation and cardiovascular risk factors predict rapid progression of atherosclerosis in rheumatoid arthritis. Ann Rheum Dis 74(6):1118–1123
Kuo D, Crowson CS, Gabriel SE, Matteson EL (2014) Hyperuricemia and incident cardiovascular disease and noncardiac vascular events in patients with rheumatoid arthritis. Int J Rheumatol 2014:523897. doi:10.1155/2014/523897
Maijer KI, Neumann E, Müller-Lander U, Drop DACAD, Ramwadhdoeve TH, Choi IYK et al (2015) Serum Vaspin levels are associated with the development of clinically manifest arthritis in autoantibody-positive individuals. PLoS One 10(12):e0144932
Nicola PJ, Crowson CS, Maradit-Kremers H, Ballman KV, Roger VL, Jacobsen SJ, Gabriel SE (2006) Contribution of congestive heart failure and ischemic heart disease to excess mortality in rheumatoid arthritis. Arthritis Rheum 54(1):60–67
Nicola PJ, Maradit-Kremers H, Roger VL, Jacobsen SJ, Crowson CS, Ballman KV, Gabriel SE (2005) The risk of congestive heart failure in rheumatoid arthritis: a population-based study over 46 years. Arthritis Rheum 52(2):412–420
Schau T, Gottwald M, Arbach O, Seifert M, Schopp M, Neuss M et al (2015) Increased prevalence of diastolic heart failure in patients with rheumatoid arthritis correlates with active disease, but not with treatment type. J Rheumatol 42:2029–2037
Guedes C, Bianchi-Fior P, Cormier B, Barthelemy B, Rat AC, Boissier MC (2001) Cardiac manifestation of rheumatoid arthritis: a case-control transesophageal echocardiography study in 30 patients. Arthritis Rheum 45(2):129–135
Myasoedova E, Crowson CS, Nicola PJ, Maradit-Kremers H, Davis JM 3rd, Roger VL et al (2011) The influence of rheumatoid arthritis disease characteristics on heart failure. J Rheumatol 38(8):1601–1606
Listing J, Strangfeld A, Kekow J, Schneider M, Kapelle A, Wassenberg S, Zink A (2008) Does tumor necrosis factor-α inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum 58:667–677
Sarzi-Puttini P, Atzueni F, Doria A, Iaccarino L, Turiel M (2005) Tumor necrosis factor-alpha, biologic agents and cardiovascular risk. Lupus 14(9):780–784
Roldan AC, DeLong C, Qualls CR, Crawford MC (2007) Characterization of valvular heart disease in rheumatoid arthritis by transesophageal echocardiography and clinical correlates. Am J Cardiol 100:496–502
Giles JT, Fert-Bober J, Park JK, Bingham CO 3rd, Andrade F, Fox-Talbot K et al (2012) Myocardial citrullination in rheumatoid arthritis: a correlative histopathologic study. Arthritis Res Ther 14:R39
Di Franco M, Paradiso M, Mammarella A, Paoletti V, Labbadia G, Coppotelli L et al (2000) Diastolic function abnormalities in rheumatoid arthritis. Evaluation by echo Doppler transmitral flow and pulmonary venous flow: relation with duration of disease. Ann Rheum Dis 59:227–229
Liang KP, Myasoedova E, Crowson CS, Davis JM, Roger VL, Karon BL et al (2010) Increased prevalence of diastolic dysfunction in rheumatoid arthritis. Ann Rheum Dis 69(9):1665–1670
Tomáš L, Lazúrová I, Oetterová M, Pundová L, Petrášová D, Studenčan M (2013) Left ventricular morphology and function in patients with rheumatoid arthritis. Wien Klin Wochenschr 125(9-10):233–238
Sakthiswary R, Das S (2015) Predictors of diastolic dysfunction in rheumatoid arthritis. Saudi Med J 36(5):525–529
Bhatia GS, Sosin MD, Patel JV, Grindulis KA, Khattak FH, Huighes EA et al (2006) Left ventricular systolic dysfunction in rheumatoid disease. An unrecognized burden? J Am Coll Cardiol 47:1169–1174
Cioffi G, Viapiana O, Ognibeni F, Dalbeni A, Gatti D, Adami S et al (2015) Prevalence and factors related to left ventricular systolic dysfunction in asymptomatic patients with rheumatoid arthritis. A prospective tissue Doppler echocardiography study. Herz 40(7):989–996
Lazzerini PE, Capecchi PL, Acampa M, Galeazzi M, Laghi-Pasini F (2014) Arrhythmic risk in rheumatoid arthritis: the driving role of systemic inflammation. Autoimmun Rev 13(9):936–944
Lindhardsen J, Ahlehoff O, Gislason GH, Madsen OR, Olesen JB, Svendsen JH et al (2012) Risk of atrial fibrillation and stroke in rheumatoid arthritis: Danish nationwide cohort study. BMJ 344:e1257
Solus J, Chung CP, Oeser A, Avalos I, Gebretsadik T, Shintani A et al (2008) Amino-terminal fragment of the prohormone brain-type natriuretic peptide in rheumatoid arthritis. Arthritis Rheum 58:2662–2669
Yxfeldt A, Wallberg-Jonsson S, Hultdin J, Rantapää-Dahlqvist S (2003) Homocysteine in patients with rheumatoid arthritis in relation to inflammation and B-vitamin treatment. Scand J Rheumatol 32:205–210
Bozkurt B, Mann DL, Deswal A (2009) Biomarkers of inflammation in heart failure. Heart Fail Rev 15(4):331–341
Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C et al (2000) Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation 102(25):3060–3067
Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL (2001) Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 103(16):2055–2059
Bozkurt B, Torre-Amione G, Smith Warren M, Soran OZ, Feldman AM, Mann DL (2001) Results of targeted anti-tumor necrosis factor therapy with etanercept (ENBREL) in patients with advanced heart failure. Am J Assoc 103:1044–1047
Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT, Anti-TNF Therapy Against Congestive Heart Failure Investigators (2003) Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF therapy against congestive heart failure (ATTACH) trial. Circulation 107:3133–3140
Coletta AP, Clark AL, Banarjee P, Cleland JG (2002) Clinical trials update: RENEWAL (RENAISSANCE and RECOVER) and ATTACH. Eur J Heart Fail 4:243–247
Gabriel SE (2008) Tumor necrosis factor inhibition: a part of the Solution or a part of the problem of heart failure in rheumatoid arthritis. Arthritis Rheum 58:637–640
Khanna D, McMahon M, Furst DE (2004) Anti tumor necrosis factor-α therapy and heart failure. Arthritis Rheum 50:1040–1048
Kotyla PJ, Owczarek A, Rakoczy J, Lewicki M, Kucharz EJ, Emery P (2012) Infliximab treatment increases left ventricular ejection fraction in patients with rheumatoid arthritis: assessment of heart function by echocardiography, endothelin-1, interleukin 6 and NT-probrain natriuretic peptide. J Rheumatol 39:4701–4706
Tomáš L, Lazúrová I, Pundová L, Oetterová M, Zakuciová M, Petrášová D, Studenčan M (2013) Acute and long-term effect of infliximab on humoral and echocardiographic parameters in patients with chronic inflammatory diseases. Clin Rheumatol 32(1):61–66
Curtis JR, Kramer JM, Martin C, Saag KG, Patkar N, Shatin D et al (2007) Heart failure among younger rheumatoid arthritis and Crohn’s patients exposed to TNF-α antagonists. Rheumatol 46:1688–1693
Al-Aly Z, Pan H, Zeringue A, Xian H, McDonald JR, El-Achkar TM, Eisen S (2011) Tumor necrosis factor-α blockade, cardiovascular outcomes, and survival in rheumatoid arthritis. Transl Res 157:10–16
Santos RC, Figueiredo VN, Martinz LC, Haro Moraes C, Quinaglia T, Boer-Martins L et al (2012) Infliximab reduces cardiac output in rheumatoid arthritis patients without heart failure. Rev Assoc Med Bras 58:698–702
Peters MJ, Welsh P, McInnes IB, Wolbink G, Dijkmans BA, Sattar N, Nurmohamed MT (2010) Tumour necrosis factor alpha blockade reduces circulating N-terminal pro-brain natriuretic peptide levels in patients with active rheumatoid arthritis: results from a prospective cohort study. Ann Rheum Dis 69(7):1281–1285
Setoguchi S, Schneeweiss S, Avorn J, Katz JN, Weinblatt M, Levin R et al (2008) Tumor necrosis factor-α antagonist use and heart failure in elderly patients with rheumatoid arthritis. Am Heart J 156:336–341
Solomon DH, Rassen JA, Kuriva B, Chen L, Harrold LR, Graham DJ et al (2013) Heart failure risk among patients with rheumatoid arthritis starting a TNF antagonist. Ann Rheum Dis 72:1813–1818
Singh JA, Wells GA, Christensen R, Tanjong Ghogomu E, Maxwell L, Macdonald JK et al (2011) Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev 16(2):CD008794
Suzuki A, Tamamura T, Okai T (2014) Five-year administration of tocilizumab to a patient with rheumatoid arthritis complicated by severe chronic heart failure. Jpn J Clin Immunol 37(6):488–492
Kobayashi Y, Kobayashi H, Giles JT, Hirano M, Nakajima Y, Takei M (2015) Association of tocilizumab with changes in measures of regional left ventricular function in rheumatoid arthritis, as assessed by cardiac magnetic resonance imaging. Int J Rheum Dis. doi: 10.1111/1756-185X.12632
Ikonomidis I, Lekakis JP, Nikolaou M, Paraskevaidis I, Andreadou I, Kaplanoglou T et al (2008) Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation 117:2662–2669
Ikonomidis I, Tzortzis S, Lekakis J, Paraskevaidis I, Andreadou I, Nikolaou M et al (2009) Lowering interleukin-1 activity with anakinra improves myocardial deformation in rheumatoid arthritis. Heart 95:1502–1507
Ikonomidis I, Tzortzis S, Andreadou I, Paraskevaidis I, Katseli C, Katsimbri P et al (2014) Increased benefit of Interleukin -1 inhibition on vascular function, myocardial deformation and twisting in patients with coronary artery disease and coexisting rheumatoid arthritis. Circ Cardiovasc Imaging 7:619–628
Fleischamnn RM, Schechtman J, Bennett R, Handel ML, Burmester GR, Tesser J et al (2003) Anakinra, a recombinant human interleukin-1 receptor antagonist, (r-metHuIL-1ra) in patients with rheumatoid arthritis. Arthritis Rheumatism 48:927–934
Abbate A, Van Tassel BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spilmann DW et al (2013) Effects of Interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction (from the Virginia commonwealth University-Anakinra remodeling Trial (2)(VCU-ART2) pilot study). J Cardiol 111:1394–1400
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Lazúrová, I., Tomáš, Ľ. Cardiac Impairment in Rheumatoid Arthritis and Influence of Anti-TNFα Treatment. Clinic Rev Allerg Immunol 52, 323–332 (2017). https://doi.org/10.1007/s12016-016-8566-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-016-8566-3