
Abstract
Serine protease inhibitors (serpins) are evolutionary old, structurally conserved molecules which encompass nearly all branches of life. More than 1,000 serpins were characterized to date which are subdivided into 16 subgroups (A–P) according to their common ancestry; among them, 37 are found in humans. Serpins were termed after their capability to inhibit serine proteases, but mounting evidence suggests that they may achieve a greater deal of functions, ranging from embryological growth to synaptic plasticity, development of both myeloid and lymphoid immune cells, and modulation of apoptosis. Serpins are mainly extracellular molecules, although some of them (namely, ov-serpins or clade B serpins) mostly act inside the cells, being either ubiquitously or tissue-specifically expressed. Among newly characterized serpin functions, regulation of cellular proliferation through apoptosis modulation and proteasome disturbance seems to play a major role. Accordingly, several serpins were found to be hyperexpressed in tumor cells. Indeed, apoptosis dysregulation is likely to be a cornerstone in both tumorigenesis and autoimmunity, since uncontrolled cellular viability results in tumor proliferation, while inefficient disposal of apoptotic debris may favor the rescue of autoreactive immune cells. Such a process was widely documented in systemic lupus erythematosus (SLE). Interestingly, alterations in the expression of some serpins, e.g., the ov-serpin SERPINB3, are being unraveled in patients affected with SLE and other autoimmune disorders, suggesting that a failure in serpin function might affect immune homeostasis and self-tolerance, thereby contributing to autoimmunity. Here, we provide an overview of serpin origin, function, and dysfunction, focusing on human serpins and ov-serpins, with a hub on SERPINB3.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
General Features of the Serpin Superfamily
Serine protease inhibitors (serpins) are a superfamily of functionally distinct but structurally conserved proteins named after their capability to inhibit serine proteases [1, 2], although some of them can bind cysteine proteases as well (so-called cross-class serpins), while others do not possess any binding activity, carrying out other cellular functions [1]. Serpins are the greatest group of peptidase inhibitors identified to date [3].
Both inhibitory and non-inhibitory serpins may achieve a number of biological tasks beyond or irrespective of proteinase inhibition, including hormone transport (SERPINA6 or corticosteroid-binding globulin, SERPINA7 or thyroxin-binding globulin), blood pressure regulation and renal development (SERPINA8 or angiotensinogen), B cell development (SERPINA9 or centerin), neurological development (SERPINI1 or neuroserpin), and still others [4]. However, the roles of many serpins remain elusive.
Nomenclature and Structure of the Serpins
Globally, more than 1,000 serpins have been identified to date across all the living kingdoms, encompassing viruses as well as simple organisms, humans, and plants [2, 5, 6], which can be subdivided into 16 subgroups (clades) from A to P, according to their phylogenetic relationship. Actually, an additional group exists comprising “orphan” serpins that have not been located in any other clade yet [1]. Sorting of serpins into different clades is based on the conservation in their amino acidic sequence that may underlie a common kinship [7]. Among the 16 clades, P and K comprise plant and insect serpins, while viral serpins fall into the N and O clades. The remaining 12 subgroups contain animal serpins, 3 clades (J, L, and M) being species-based (nematodes, trematodes, and horseshoe crab) and 9 (A–I) comprising high-animal serpins (including human serpins) which segregate according to function rather than species [7]. The nomenclature by which serpins are termed is SERPINXY, with X being the clade and Y being the number within the clade [5], and newly discovered serpins that are further added proceed sequentially in this way.
Although serpins are very ancient molecules, they are prevalent among eukaryotes, suggesting that they may have developed after prokaryotes/eukaryotes separation or that simpler organisms might have lost them during evolution [8]. Moreover, since serpins are more widely found in metazoans, they could have scattered to other branches of life (i.e., plants) by lateral gene transfer [8], or plant serpins might have evolved as a separate evolutionary unit, since no orthology links them to animal serpins [7].
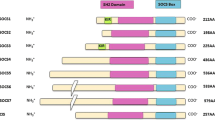
Serpins are encoded by genes mapping on different chromosomes (human serpin genes map on 10 chromosomes, see succeeding paragraphs), so that one clade may have its members split throughout the genome; however, serpin-encoding genes are often clustered and, within each cluster, all serpins belong to the same clade [1]. Notably, all serpins share a conserved tertiary structure, made of 3 beta-sheets, 8 or 9 alpha-helices, and a reactive center loop (RCL) which is about 17 amino acids long and is tethered between the A and C beta-sheets [1, 2, 5] (Fig. 1). Despite their common folding, the homology in their primary structure accounts for <25 % [1].

All serpins share a conserved tertiary structure made of 3 beta-sheets, 8 or 9 alpha-helices, and an RCL which is about 17 amino acids long and is tethered between the A and C beta-sheets
The RCL is essential to serpin specificity and function [1] since residues within the RCL match with amino acids of the protease active site, thus defining which proteases will be recognized. The most critical residue for RCL specificity is called P1, which is flanked by recognition residues named P4–P4′ [9, 10]. P1–P1′ is called the scissile bond [2] and consists of some residues extending from the aminoacyl-terminal (P1) to the carboxyl-terminal (P1′) [5, 10].
Serpins usually act at an extracellular level [1] and exist in two alternative conformations, switching from the native metastable stressed form to a relaxed stable form when binding the protease (i.e., stressed-to-relaxed transition), thus reaching a firmer conformation during inhibition [2, 11]. Indeed, during activation, the RCL inserts itself into the center of the beta-sheet A, forming an extra strand [1, 11] and leading to a hyperstable state [2]. Only serpins displaying an effective inhibitory behavior are able to incorporate the RCL into the beta-sheet A [2]. Incorporation of subsequent residues causes the thermodynamic stability of the RCL insertion to increase, thus rendering the process favorable and prone to self-perpetuation [1, 12].
At steady conditions, native serpins are restrained from reaching their stable form by disadvantageous energetic interactions (so-called unfavorable interactions, e.g., overpacking of side chains, presence of hydrophobic pockets, polar–nonpolar interactions or burial of polar groups) accomplished by critical amino acids in the molecule [11], so that a dynamic balance between stability and metastability is provided overall. Nevertheless, such interactions may be overcome, resulting in spontaneous conversion of the native form into a more stable one, i.e., the latent form, by which the RCL inserts into the beta-sheet A irrespective of the interaction with a protease, thus resulting in burial of the serpin binding site and premature loss of function [4, 10, 12].
Serpin Functions
Inhibitory serpins drive the inhibition of several serine proteases, thereby modulating their function. Serine proteases are enzymes with a lytic behavior that carry out several tasks, e.g., bacterial killing and inflammation: neutrophil elastase, granzymes; coagulation: thrombin, factor XI; fibrinolysis: plasmin, tissue plasminogen activator; complement activation: C1q; and others. Tissue damage due to protease hyperactivation is usually avoided because protease activity is tightly regulated by different mechanisms, including serpin-mediated inhibition [9].
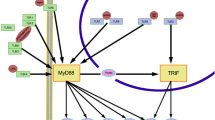
Serpins may proceed along two alternative pathways to interact with proteases, termed the inhibitory pathway and the substrate pathway [6], which are not mutually exclusive and result in protease inhibition through a suicide mechanism, meaning that the serpin undergoes irreversible structural changes in order to inhibit the protease, thereby losing its function [2, 13] (Fig. 2). Along the inhibitory pathway, the RCL binds the protease and is then cleaved at the P1–P1′ bond, subsequently slipping in the beta-sheet A and shuttling the protease from one extremity of the molecule to the other. As a consequence, the protease is crushed against the bottom of the serpin and undergoes a dramatic distortion, thus completely mislaying its lytic capability [2, 9]. The energy required for protease distortion is released during the stressed-to-relaxed transition [4] and it is estimated to be as great as −32 kcal/mol [14]. During this process, a covalent complex is formed that irreversibly links the serpin to the protease (Fig. 2).

Serpins interact with proteases through two alternative pathways, termed the inhibitory pathway and the substrate pathway, which are not mutually exclusive and result in protease inhibition through a suicide mechanism. In addition, SERPINB3 and SERPINB4 proceed along a different pathway which has not been elucidated yet, resulting in a decrease of protease function. SI refers to the amount of serpins required to inhibit a single molecule of protease: the effective inhibition occurs when SI approaches 1; conversely, the substrate pathway is favored when SI is >1. SI stoichiometry of inhibition
According to the substrate pathway, serpin serves as a real substrate for the protease that is not structurally modified but has its function transiently hindered by the binding with the RCL [13]. Some serpins, namely, SERPINB3 and SERPINB4, proceed along a different pathway which has not been elucidated yet, resulting in alteration of both the serpin and the protease, thereby blunting protease function [13] (Fig. 2). Whether the inhibitory pathway or the substrate pathway predominates is determined by two kinetic parameters, namely, the association constant (K ass) and the stoichiometry of inhibition (SI) [10, 13], i.e., the rate of RCL insertion (K ass) and the amount of serpins required to inhibit a single molecule of protease (SI); the greater the SI, the rarer the effective inhibition. In fact, relevant interactions take place when SI approaches 1; conversely, the substrate pathway is favored when the RCL resembles the protease target too closely [10]. Additionally, cofactors that enhance serpin activity may skew toward the inhibitory pathway rather than the substrate pathway (Fig. 2).
Cofactors are a compelling device for serpins to have their functions regulated and to be activated specifically when and where they are needed. In fact, many serpin cofactors are glycosaminoglycans whose expression varies across different tissues in the body according to the ongoing biological processes, e.g., heparin availability increases after endothelial damage and coagulation initiation or vitronectin–plasminogen activator inhibitor (PAI-1, SERPINE1) complexes at wounded sites may control both fibrinolysis and wound healing [1]. Cofactors may serve as bridging molecules that bring the serpin and the protease together, thus favoring their encounter and inhibition, or they can bind the serpin only, causing it to undergo a conformational change that enables interaction with the protease [1]. Such mechanisms are not mutually exclusive and may occur contemporaneously, as is the case of heparin, which is a well-characterized serpin cofactor [1, 2, 5].
Cofactors may also guarantee that serpin metastability and responsiveness are maintained, preventing serpins from undergoing latency before they have bound their target protease; this is likely the case with PAI-1 binding to vitronectin, by which circulating PAI-1 is preserved, leading to a modulation of plasminogen activation [1, 12]. Furthermore, the cleavage of serpins, either inhibitory or not, elsewhere from the scissile bond by nontarget proteinases (especially metalloproteinases) may effectively hinder serpin function [1], decreasing its binding affinity for the substrate. Notably, SERPINA6 (corticosteroid-binding globulin) undergoes such a nonspecific cleavage by neutrophil elastase, thereby releasing the glucocorticoids at the site of inflammation [1].
Beside cofactors and nonspecific cleavage, other mechanisms still have to be addressed that may regulate serpin activation and function. Hence, inhibitory serpins modulate the activity of a wide array of proteases mainly through the establishment of a tight binding with their substrate, after a profound conformational change has occurred. Such a conformational inhibition is quite different from the standard reversible lock-and-key mechanism which is widespread among living organisms and is carried out by all nonserpin protease inhibitors [14]. The mechanisms diverge in that serpins change their conformation dramatically, inhibit their substrate irreversibly, and need P1 residue to interact with protease-catalytic serine to function properly [15]. Serpin binding hence ensures inhibition to be at the same time tough (since a covalent complex is formed) and very finely tuned, according to what is needed in a given tissue at a given moment [1, 2].
Human Serpins
Origin and Genomic Organization
In high animals, including humans, nine serpin clades (A–I) are found, the largest being clade A and B (alpha-1-antitrypsin (α1AT)-like serpins and ov-serpins, with 13 members each) [5]. In humans, 37 serpins have been described so far, among which at least 26 show an inhibitory attitude, while the remainder carry out other notable functions, e.g., hormone transport (corticosteroid-binding globulin, thyroxin-binding globulin) or tumor suppression (maspin) [1, 5]. The majority of human serpins act at an extracellular level, with the exception of ov-serpins which are intracellular molecules (reviewed in the next paragraph) [1]. The first human serpins to be identified were antithrombin and α1AT which were unexpectedly found to share a common tertiary structure with chicken ovalbumin, suggesting that they could derive from a common ancestor [16]. Afterwards, antithrombin and α1AT were located in clade C and clade A, respectively, and they can somewhat be seen as the founding elements of their series. Moreover, α1AT is sometimes referred to as the prototype serpin, since other serpins share with it as much as 30 % of similarity in their primary sequence [1, 9, 17].
Genes encoding human serpins map on 10 different chromosomes and 25 out of 37 are clustered on chromosomes 6, 14, and 18, with a smaller group on chromosome 3 [1]. Notably, clusters on chromosome 6 and 18 build up the serpin clade B (ov-serpins), and all but two of the serpins belonging to clade A are encoded by the gene cluster on chromosome 14 [1]. Serpin gene clustering might be explained either by chromosomal duplications (including large chromatin fragment duplications as well as multiple intrachromosomal duplications) or conversely by splitting of an ancestral locus [1, 18], which might particularly fit the origin of the ov-serpins (see further). Genes clustered on chromosome 6 show high homology with each other, as do the serpins they encode, meaning that they have similar amino acidic sequences [17]. On the other hand, genes mapping on chromosome 18 are not so similar to each other and rather seem to be related to distant counterparts on chromosome 6 [19], suggesting that they might have arisen as paralogues (genes derived from duplication of a common ancestor). Hence, gene analysis may inform about serpin evolutionary roots, while protein structure may account for their function and intragroup relationships [8], so a comparison between the two may increase classification reliability. Tables 1, 2, and 3 summarize the general features of human serpins known to date [20–34].
Serpin Perturbations and Homeostatic Failure
Although non-inhibitory serpins exist, the master function of serpin appears to be the inhibition of several proteases through an irreversible conformational-based mechanism (see previous section), which provides high inhibitory efficiency, but on the other hand, renders serpins susceptible to even subtle changes in their sequence. Indeed, since protease binding requires specific interactions between matching residues, mutations of critical amino acids may impair serpin affinity and effectiveness [2], by either slowing or abnormally promoting RCL insertion in the beta-sheet A. For instance, the Cambridge I and II variants of antithrombin result in increased risk of venous thrombosis since they hamper antithrombin conformational transition and thrombin flipping through the serpin molecule; conversely, the Rouen VI, Wibble, and Wobble variants of antithrombin cause the RCL to be incorporated too efficiently in the beta-sheet, irrespective of peptidase binding, thus reaching the latent state and again increasing the risk of thrombosis [14]. Moreover, substitution of specific residues may change serpin specificity with harmful effects, e.g., the Pittsburgh variant of α1AT (Met to Arg in P1), recognizing thrombin in spite of neutrophil elastase and leading to fatal bleeding [1, 5, 10].
Besides point mutations causing functional failure, abnormal serpin folding may occur as well, leading to latency of the serpin molecules (see previous paragraph) as well as to anomalous insertion of the RCL of one serpin into the beta-sheet of another, thereby chaining the serpins and causing them to polymerize [4]. Unlike latency, polymerization occurs only if serpins display any polymerogenic mutations in their sequence, with the exception of PAI-2 (SERPINB2) which tends to polymerize spontaneously and reversibly under physiological conditions [22]. Recently, a new challenging mechanism has been proposed, postulating that serpin polymerization occurs during the folding process [35, 36], and not after it is accomplished; however, the precise mechanisms are not fully understood.
Transition in serpin conformation is unlikely to occur as a sudden one-step mechanism, rather encompassing diverse intermediates with different thermodynamic stabilities [1, 35, 37] among which a dynamic balance may be established [37]. Moreover, the presence of intermediates along the pathway toward polymerization may slow the folding rate since noncovalent interactions take place inside the alpha-helixes [37], which preserve native metastable conformation. However, since both polymers and latent forms are far more stable than the native shape [13, 35], the folding transition might accelerate once it has started. It has to be highlighted that serpin polymerization generates ordered polymers and lateral associations may occur [35, 36], causing insoluble serpin aggregates to form and precipitate either in or out of the cells, thereby resulting in cellular toxicity.
The mechanisms by which precipitated serpin polymers may harm the cells concern both the loss of serpin function with uncontrolled protease activity (e.g., unbalanced elastase activation and emphysema, C1-inhibitor (INH) deficiency and angioedema, antithrombin deficiency and thrombosis [38]) and the accumulation of serpin chains with subsequent endoplasmic reticulum (ER) overload, resulting in abnormal activation of nuclear factor-kappa B (NF-κB) proinflammatory signaling (e.g., liver cirrhosis due to α1AT accumulation or neuronal tangles of mutated neuroserpin) [39, 40]. Moreover, accumulation of polymers outside the cells may result in increased inflammation, since some of the polymers (e.g., α1AT polymer) may recruit and trap neutrophils from circulation, thereafter favoring their local degranulation [38]. Therefore, whatever the mechanism and although further evidence is needed, polymerization of neighbor serpins results in cytotoxic accumulation of polymers inside the ER, a pathological condition known as serpinopathy [1, 41, 42].
Two major serpinopathies have been reported to date, i.e., liver cirrhosis due to the accumulation of mutated α1AT (Z null allele Glu342Lys) and familial encephalopathy with neuroserpin inclusion bodies dementia due to the accumulation of mutated neuroserpin [38, 42, 43]. Moreover, other serpinopathies are likely to affect other districts in the body, e.g., overexpression of megsin in glomerular and tubular cells may account for renal damage and increased proteinuria both in rats and humans, being involved in different types of glomerulopathies [44].
Human Ov-serpins and SERPINB3
Genes and Cellular Localization
Serpins falling into clade B were originally termed ov-serpins since chicken ovalbumin represented the archetypal member of that group [8]. Initially, five molecules, i.e., chicken ovalbumin, chicken gene Y, PAI-2, squamous cell carcinoma antigen (SCCA), and elastase inhibitor, were classified as being ov-serpins according to some common features [45], and to date, 13 clade B serpins (SERPINB1–SERPINB13) have been found in humans (Table 2).
Human ov-serpins are intracellular molecules that carry out several functions, including protease inhibition, tumor suppression, regulation of apoptosis and inflammation, regulation of angiogenesis, and others [8, 13], and map on two different loci at 6p25 (three genes: SERPINB1, SERPINB6, and SERPINB9) and 18q21 (10 genes: SERPINB2, SERPINB3, SERPINB4, SERPINB5, SERPINB7, SERPINB8, SERPINB10, SERPINB11, SERPINB12, and SERPINB13) [8, 46]. Mammals appear to be the only class to have clade B genes split into two separate loci, whereas fish, amphibians, and birds all display a single ov-serpin locus [18, 46]. Therefore, it was suggested that the two mammalian loci resulted from an early chromosomal breakage, and this was strikingly supported by the finding that human clade B genes from both loci have several orthologues on the chicken single clade B locus, and moreover, the chicken locus is flanked by upstream and downstream genes that have corresponding human orthologues in the same positions [18]. Gene duplications rather than splitting may likewise occur in living beings. In this regard, a recent comparison between the human 6p25 and the mouse chromosome 13 has shown a broadened serpin repertoire in the mouse genome, with 15 expanded members [47] which probably arose through subsequent duplications under selective evolutionary pressure [8].
All human ov-serpins are found in the cytoplasm or associated with cytoplasmic organelles and usually require an ATP-independent active process to be driven to the nucleus [8]. Since they lack the N-terminal signal peptide required in the secretory pathway [1, 8, 13], human ov-serpins are retained in the cell where they probably play a cytoprotective role owing to their protease inhibitory attitude [10, 12, 48], as well as to their antiapoptotic capability [49]. Nevertheless, modest secretion of some ov-serpins (e.g., SERPINB2, SERPINB3, SERPINB5, SERPINB7) has been reported [12, 50, 51], but in the majority of cases, extracellular distribution might be due to passive loss or cellular lysis (e.g., ripping of tumor cell and SCCA1 release in plasma) [8]. Compelling evidence was drawn only for SERPINB5 (which may be found in secretory vesicles at the cell surface) [52] and SERPINB2 [53], although the exporting mechanisms remain elusive [12, 54].
Ov-serpin Functions with a Focus on Immunity and Cell Death
All human ov-serpins, except SERPINB5 (maspin) and SERPINB11 (epipin), display an inhibitory phenotype, mainly targeting trypsin-like or chymotrypsin-like serine proteases; in fact, ov-serpins can inhibit a great deal of molecules, including caspases, subtilisins, pepsin, allergens, and papain-like cysteine proteases [13]. SERPINB4 and SERPINB9 are cross-class serpins, while SERPINB3 and SERPINB13 can only inhibit cysteine proteases [1, 13]. The mechanisms of cysteine protease inhibition probably overlap those of serine protease inhibition, but it has to be mentioned that SERPINB3 and SERPINB13 are intrinsically predisposed to inhibit papain-like cysteine proteases owing to their structural conformation [55]. Although ov-serpin functions are not fully characterized (Table 2), one of their major tasks appears to be cellular protection against cell own cytotoxic molecules that are released during cellular activation (e.g., granzyme B) or may otherwise leak in the cytoplasm, e.g., by lysosome loss (cathepsins) [10, 12, 56]. Ov-serpins are expressed by a wide variety of cells and tissues, e.g., skin, placenta (PAI-2) [36], endothelial cells (SERPINB9, SERPINB6) [6, 57], platelets (SERPINB6) [6], and noteworthy, in immune-competent cells, such as monocytes (SERPINB2), dendritic cells (DC; SERPINB9), or lymphocytes (SERPINB9) [20, 58], where they may rescue cells from unwanted apoptosis and sustain along their development. Several ov-serpins are thought to help myeloid cell maturation in physiological conditions, and variations in their levels of expression were seen to correlate with the cell maturation state [59].
It should be noted that ov-serpins target molecules that are seldom targeted by extracellular serpins as well (e.g., neutrophil elastase, cathepsin G, and proteinase 3 are targets of both SERPINB6 and SERPINA1) [20, 57], consistent with the fact that such proteinases play a dual role, acting both at the intracellular and extracellular levels (i.e., inside phagocytic vesicles to dispose ingested material and as inflammatory mediators released by granules). Accordingly, ov-serpins exert a cytoprotective effect, whereas serpins belonging to other clades are more likely to protect the surrounding cells and tissues. However, ov-serpins may safeguard neighboring cells from cytolytic death as well. In this regard, SERPINB9 is thought to be paramount in cytotoxic lymphocyte (CTL) protection from endogenous granzyme B which is inactivated in the cytoplasm [10] as well as in shielding activated DC which are exposed to proteolytic peptides, including granzymes [58]. This may in turn preserve DC contribution to the maintenance of cytotoxic responses, since DC are required in CTL activation or the response would prematurely fade. Clustering of death receptors is another mechanism by which CTL may kill target cells, and although SERPINB9 was said not to interact with caspases [60], conflicting observations have been reported [10, 57], suggesting that SERPINB9 may actually interact with caspase 8 and caspase 10 and thereby modulate Fas-mediated and TNFα-mediated cytotoxicity, albeit in a cell-specific fashion [57]. Besides the cytotoxic pathways to cell death, serpins may interfere with the intrinsic apoptotic pathway as well [6, 22, 61]. Some ov-serpins were found to be involved in tumor genesis or progression, particularly SERPINB2 and SERPINB5 expressions were reported to hamper tumor spreading and metastasis and thereby ameliorate the prognosis; conversely, SERPINB3 was reported to correlate with a poor prognosis in diverse epithelial or endodermal cancers [62], since it might interfere with canonical apoptosis and, therefore, rescue cancer cells from death.
SERPINB3
SERPINB3 was originally named SCCA since it was found to be highly expressed in some squamous epithelial cancers, such as uterine cervix carcinoma, esophagus carcinoma, and head and neck carcinomas [13]; more recently, increased SERPINB3 expression was reported in liver carcinoma as well [63]. Originally, SERPINB3 was reported to inhibit apoptosis in cancer cells, thus favoring their spreading and worsening the prognosis [64]. The mechanisms by which SERPINB3 may hamper apoptosis are not clear; however, it was recently hypothesized to interfere with mitochondrial release of cytochrome c [61] or it might be responsible for resistance to anticancer drugs as well as to TNFα-induced apoptosis through inhibition of caspase 3 or upstream proteins [64] (Fig. 3). Previous findings also demonstrated that TNFα could elicit SERPINB3 expression in tumor cells [65], thus establishing a prosurvival loop for cancer cells.

Mechanisms involved in tumor spreading and autoimmunity by SERPINB3. HBV hepatitis B virus, TNF tumor necrosis factor, CytC cytochrome c
In normal conditions, SERPINB3 and its close homologue SERPINB4 are widely coexpressed in a variety of epithelia, including tongue, tonsils, uterus, cervix, vagina, and upper airways, as well as in thymus or Hassall’s corpuscles [13]; moreover, SERPINB3 was recently reported to be expressed on CD27+ B cells [66]. Despite such a broadened expression, however, the physiological functions of SERPINB3 remain elusive. Actually, SERPINB3 looks intriguingly involved in the regulation of apoptosis and cell death, and a great deal of functions may be attributed to this serpin encompassing cell death and immunity [67]. As a protease inhibitor, SERPINB3 is able to inhibit cysteine proteases (cathepsins, papain) and it may be found either outside or inside the cells (mainly in the cytoplasm), although whether its secretion plays a physiological role remains to be addressed [13, 68]. SERPINB3 antiprotease activity may also be involved in the dampening of anticancer drug-induced apoptosis; however, SERPINB3 was shown to trigger a decreased phosphorylation of the proapoptotic p38 mitogen-activated protein kinase and to halt ultraviolet-induced apoptosis [67] in a way which does not seem to be related to its antiprotease activity.
Although SERPINB3 antiapoptotic capability is likely to explain at least in part how SERPINB3 may prolong cellular survival, recent evidence suggests that SERPINB3 may avoid cell death triggered by intracellular damage (e.g., by lysosome loss of lytic enzymes) but may actually favor ER stress-induced apoptosis [62], thus modulating cell survival in both directions. Lysosome damage, induced by various types of stress, including hypotonic stress, hypoxia, heat shock, or DNA alkylation, leads to intracellular release of cathepsins, which are targeted by SERPINB3, and indeed, cell death owing to aberrant hydrolase release is avoided [62]. However, SERPINB3 hyperexpression (as seen in some tumor cells) causes inhibition of the proteasome function (Fig. 3), leading to accumulation and aggregation of polyubiquitinated proteins including caspase 8 which is pushed to activation, thus initiating an apoptotic cascade. Such a process begins with an aberrant ER stress, meaning that an abortive unfolded protein response (UPR) is carried out which loses the capability to dispose the unfolded material because of proteasome inhibition, thus skewing the pathway toward a caspase 8-driven cell death [62]. However, cells may survive until proteasome function is not completely overcome; therefore, tumor cells may escape lysosome stress (e.g., by alkylating agents) without undergoing ER stress-induced apoptosis, thereby resulting in anticancer drug resistance. Accordingly, SERPINB3 expression was seen to correlate with a poor prognosis in breast cancer patients [69].
Not only tumor cells but also viruses may exploit the serpin repertoire to induce cell survival and, therefore, escape killing by immune cells. In this regard, SERPINB3 expression was shown to be induced in human cells infected with Toxoplasma gondii [70], thus preserving parasite viability. Moreover, SERPINB3 was reported to serve as a surface binding receptor for human hepatitis B virus, not only in hepatocytes but also in peripheral blood mononuclear cells [71, 72]. Finally, serum concentrations of SERPINB3 were found to be elevated not only in patients with squamous carcinomas but also in some of those affected with systemic sclerosis (especially if lung fibrosis or diffuse skin involvement occurred) and psoriasis [73, 74]. In psoriatic patients, autoreactive IgG may be produced, which target SERPINB3 [75]. These findings, together with the knowledge that apoptosis dysregulation is likely to play a role in the induction of aberrant immune responses, raise the question whether SERPINB3 might be involved in the development of autoimmunity [67].
Serpins in Autoimmunity and SLE
Failure in serpin function was shown to associate with dysregulation in cell survival (detailed in the previous section) as well as with some autoimmune traits, meaning that people carrying serpin dysfunction often display an altered immune response. For instance, hereditary C1-INH-deficient patients are prone to develop autoantibodies (especially antinuclear antibodies) and immunoregulatory disorders [76, 77], and patients affected with autoimmune diseases (e.g., systemic lupus erythematosus [SLE]) may develop anti-C1-INH antibodies and may acquire C1-INH deficiency [78, 79], displaying severe clinical features [80]. On the other hand, it has recently been observed that autoantibodies against SERPINB13 may delay diabetes onset in nonobese diabetic mice and that children who experience early diabetes lack effective anti-clade B serpin activity [81], thus suggesting that there are multiple ways by which clade B serpins are implied in immune homeostasis, although many of them remain elusive.
Of interest, serpins may be double-faced in modulating immunity. Administration of α1-antichymotrypsin (α1ACT, SERPINA3) was shown to ameliorate disease and delay autoimmunity in a mouse model of arthritis, the treated mice displaying lower levels of anticollagen autoantibodies and of B cell activating factor, suggesting that α1ACT may somehow influence B cell function [82]. On the other hand, α1ACT is found in amyloid plaques in Alzheimer’s disease and is thought to accelerate disease onset and severity [20, 83].
Abnormal accumulation of serpins inside the ER (i.e., serpinopathies) may also fuel aberrant autoimmune responses. Classically, abnormal accumulation of misfolded proteins within the ER leads to ER stress and initiation of salvage pathways, i.e., the UPR, which can prevent the suffering cells from being flooded by an excessive protein load [84]. Misfolded polypeptides that cannot be properly refolded are delivered to the proteasome in the cytoplasm and undergo ER-associated degradation (ERAD). If all these measures fail, the UPR leads to the activation of apoptosis and cellular death [85–87]. Notably, serpin polymers do not evoke an effective UPR on their own, being rather associated with endoplasmic overload response (EOR) or autophagy hyperactivation [43, 88, 89], most probably because of the polymers’ ordered structure [43].
UPR and EOR have some common features; most notably, they both culminate in the activation of NF-κB and subsequent expression of several genes mostly involved in antiapoptosis, inflammation, and cellular survival and proliferation [90], and their activating stimuli are sometimes overlapping.
With regard to polymer degradation, both proteasome and autophagy are involved, meaning ERAD is somehow activated despite the scarce UPR response; it could be that different signaling pathways are initiated, but they still remain elusive [91]. In fact, autophagy and ERAD seem to carry out different tasks, perhaps playing complementary roles, since autophagy might be responsible for a bulk degradation of both mutants and wild-type proteins depending on their abnormal aggregation, whereas ERAD seems to selectively target mutant monomers, thus shaping the pool of proteins tagged for degradation [91, 92]. The sorting of abnormal proteins along the proteasome or the autophagy pathway may, therefore, depend on their conformation. However, it has to be pointed out that autophagy plays a major role in α1AT polymers degradation, while it may not be very effective in the removal of neuroserpin polymers [93], suggesting that further devices are exploited to target serpin aggregates.
Both autophagy and clearance of misfolded proteins ensure a kind of cellular homeostasis, thus their alteration may account for aberrant exposition of autoantigens or modified self-antigens that are not properly removed, recalling what is likely to happen in dysregulated apoptosis [94–101]. In this regard, autophagy has gained increased importance as an antigen-presenting mechanism, since it may both enhance major histocompatibility complex (MHC) I presentation of endogenous antigens and enable MHC II molecules to be loaded with both nuclear and cytoplasmic intracellular antigens, thus favoring abnormal presentation and recognition of autoantigens by T CD4+ lymphocytes, eventually triggering an autoimmune response [102, 103]. Indeed, MHC II molecules are usually charged with extracellular peptides coming from lysosomal degradation, but autophagosomes may fuse with MHC class II containing compartments and aberrantly deliver intracellular antigens to MHC II pockets [102].
Autophagy is also involved in central lymphocyte selection and maintenance of peripheral immune homeostasis [104, 105]; accordingly, autophagy perturbations have been described in several autoimmune conditions, including SLE [106]. Although ov-serpins mainly act at the intracellular level, membrane-bound expression of SERPINB3 was recently demonstrated on peripheral blood mononuclear cells, especially on CD27+ (antigen-exposed) B cells [66]. Interestingly, in the same study, SERPINB3 was found to be absent on SLE B CD27+ B lymphocytes, consistent with its expression being suppressed by high levels of type I interferon, which is a typical finding in SLE [66]. Thus, a link between SLE underlying abnormalities and lack of SERPINB3 on lupus B lymphocytes seems conceivable.
Since SERPINB3 displays an antiapoptotic behavior, alterations in its expression might contribute to the apoptotic dysregulation seen in SLE, thereby increasing the autoantigen burden. Furthermore, SERPINB3 expression and CD27 positivity were found to be directly related, suggesting that this serpin might also be implied in normal B cell activation. It has to be noted that the peripheral B cell repertoire and particularly CD27+ B cell number is heterogeneously altered in SLE [107–109]. Interestingly, administration of an α1AT fragment (termed UBE) to lupus-prone mice was found to be associated with reduced double-negative lymphocytes and B220+ cells in lymph nodes and spleen, decreased interleukin-17 secretion, lower serum anti-DNA antibodies, and a better prognosis [110]. Of interest, UBE peptide production was induced in mice after the administration of the histone fragment H2A.
In summary, serpins seem to play a relevant role in maintaining immune homeostasis, and impairment in serpin function may contribute to the development of autoimmune disorders. Further analyses are needed to clearly unravel their mechanism of action and exploit serpin therapeutic potential.
Take-Home Messages
-
1.
Serpins are a superfamily of functionally distinct but structurally conserved proteins named after their capability to inhibit serine proteases, although some of them can bind cysteine proteases as well.
-
2.
More than 1,000 serpins have been identified to date across all the living kingdoms, encompassing viruses as well as simple organisms, humans, and plants. They can be subdivided into 16 subgroups (clades) from A to P, according to their phylogenetic relationship.
-
3.
Human ov-serpins (clades A–I) are intracellular molecules which carry out several functions, including protease inhibition, tumor suppression, regulation of apoptosis and inflammation, regulation of angiogenesis, and others.
-
4.
Apoptosis dysregulation is a cornerstone in both tumorigenesis and autoimmunity, since uncontrolled cellular viability results in tumor proliferation, while inefficient disposal of apoptotic debris may favor the rescue of autoreactive immune cells.
-
5.
SERPINB3 is physiologically expressed on the surface of CD27+ B lymphocytes, but its expression is not detectable in SLE patients. These results may suggest a role for SERPINB3 in B cell defects typically found in autoimmune disorders.
References
Gettings PG (2002) Serpin structure, mechanism, and function. Chem Rev 102:4751–4804
Huntington JA (2011) Serpin structure, function and dysfunction. J Thromb Haemost 9(Suppl 1):26–34
Rawlings ND, Tolle DP, Barrett AJ (2004) Evolutionary families of peptidase inhibitors. Biochem J 378:705–716
Silverman GA, Bird PI, Carrell RW, Church FC, Coughlin PB, Gettings PG et al (2001) The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem 276:33293–33296
Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W et al (2006) An overview of the serpin superfamily. Genome Biol 7:216
Mangan MS, Kaiserman D, Bird PI (2008) The role of serpins in vertebrate immunity. Tissue Antigens 72:1–10
Irving JA, Pike RN, Lesk AM, Whisstock JC (2000) Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res 10:1845–1864
Silverman GA, Whisstock JC, Askew DJ, Pak SC, Luke CJ, Cataltepe S et al (2004) Human clade B serpins (ov-serpins) belong to a cohort of evolutionarily dispersed intracellular proteinase inhibitor clades that protect cells from promiscuous proteolysis. Cell Mol Life Sci 61:301–325
Bots M, Medema JP (2008) Serpins in T cell immunity. J Leukoc Biol 84:1238–1247
Kaiserman D, Bird PI (2010) Control of granzymes by serpins. Cell Death Differ 17:586–595
Khan MS, Singh P, Azhar A, Naseem A, Rashid Q, Kabir MA et al (2011) Serpin inhibition mechanism: a delicate balance between native metastable state and polymerization. J Amino Acids 2011:606797
Pike RN, Bottomley SP, Irving JA, Bird PI, Whisstock JC (2002) Serpins: finely balanced conformational traps. IUBMB Life 54:1–7
Izuhara K, Ohta S, Kanaji S, Shiraishi H, Arima K (2008) Recent progress in understanding the diversity of the human ov-serpin/clade B serpin family. Cell Mol Life Sci 65:2541–2553
Huntington JA (2006) Shape-shifting serpins—advantages of a mobile mechanism. Trends Biochem Sci 31:427–435
Olson ST, Bock PE, Kvassman J, Shore JD, Lawrence DA, Ginsburg et al (1995) Role of the catalytic serine in the interactions of serine proteinases with protein inhibitors of the serpin family. Contribution of a covalent interaction to the binding energy of serpin–proteinase complexes. J Biol Chem 270:30007–30017
Hunt LT, Dayhoff MO (1980) A surprising new protein superfamily containing ovalbumin, antithrombin-III, and alpha 1-proteinase inhibitor. Biochem Biophys Res Commun 95:864–871
Van Gent D, Sharp P, Morgan K, Kalsheker N (2003) Serpins: structure, function and molecular evolution. Int J Biochem Cell Biol 35:1536–1547
Benarafa C, Remold-O’Donnell E (2005) The ovalbumin serpins revisited: perspective from the chicken genome of clade B serpin evolution in vertebrates. Proc Natl Acad Sci USA 102:11367–11372
Scott FL, Eyre HJ, Lioumi M, Ragoussis J, Irving JA, Sutherland GA et al (1999) Human ovalbumin serpin evolution: phylogenic analysis, gene organization, and identification of new PI8-related genes suggest that two interchromosomal and several intrachromosomal duplications generated the gene clusters at 18q21–q23 and 6p25. Genomics 62:490–499
Janciauskiene S (2001) Conformational properties of serine proteinase inhibitors (serpins) confer multiple pathophysiological roles. Biochim Biophys Acta 1535:221–235
Remold-O’Donnell. Involvement of SerpinB1 in generation of NETs (neutrophil extracellular traps). The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Medcalf RL, Stasinopoulos SJ (2005) The undecided serpin. The ins and outs of plasminogen activator inhibitor type 2. FEBS J 272:4858–4867
Inagi R, Nangaku M, Miyata T, Kurokawa K (2003) Mesangial cell-predominant functional gene, megsin. Clin Exp Nephrol 7:87–92
Wang J, Yang L, Li J, Rezaie R. The cardioprotective activity of antithrombin through interaction with vascular HSPGs. The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Tollefsen DM. Heparin cofactor II modulates the response to arterial injury. The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Andreasen PA. PAI-1 in cancer. The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Selbonne S, Azibani F, Iatmanen S, Boulaftali Y, Jandrot-Perrus M, Bouton MC, et al. In vitro and in vivo anti-angiogenic properties of the tissue serpin protease nexin-1. The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract)
Bouton MC. Emerging role of PN-1 in thrombosis and vascular biology. The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Fitzgerald DP, Subramanian P, Deshpande M, Graves C, Gordon I, Qian Y, et al. PEDF: opposing effects of metastatic cells and neurons in the brain. The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Becerra SP, Deshpande MM, Locatelli-Hoops S, Moghaddam-Taaheri S, Guerrier S, Balko N, et al. Identification of pigment epithelium-derived factor (PEDF) protein versions with distinct activities on tumor cells lines. The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Yepes M. Tissue-type plasminogen activator and neuroserpin in the central nervous system. The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Lee TW, Montgomery J, Christie DL, Birch NP. Neuroserpin and neural development: modulation of growth and morphological characteristics. The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Valiente M, Massague J. Neuroserpin mediates brain metastasis. The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Higgins WJ, Grehan G, Alhayek S, Worrall DM. Characterisation of human serpin I2; a protective role against prematurely activated pancreatic zymogens? The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Huntington JA, Sendall TJ, Yamasaki M (2009) New insight into serpin polymerization and aggregation. Prion 3:12–14
Yamasaki M, Li W, Johnson DJ, Huntington JA (2008) Crystal structure of a stable dimer reveals the molecular basis of serpin polymerization. Nature 455:1255–1258
Whisstock JC, Bottomley SP (2006) Molecular gymnastics: serpin structure, folding and misfolding. Curr Opin Struct Biol 16:761–768
Gooptu B, Lomas DA (2008) Polymers and inflammation: disease mechanisms of the serpinopathies. J Exp Med 205:1529–1534
Gooptu B, Lomas DA (2009) Conformational pathology of the serpins: themes, variations, and therapeutic strategies. Annu Rev Biochem 78:147–176
Devies MJ, Lomas DA (2008) The molecular aetiology of the serpinopathies. Int J Biochem Cell Biol 40:1273–1286
Lomas DA, Mahadeva R (2002) Alpha1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest 110:1585–1590
Miyata T, Inagi R, Sugiyama S, Usuda N (2005) Serpinopathy and endoplasmic reticulum stress. Med Mol Morphol 38:73–78
Roussel BD, Irving JA, Ekeowa UI, Belorgey D, Haq I, Ordóñez A et al (2011) Unravelling the twists and turns of the serpinopathies. FEBS J 278:3859–3867
Inagi R, Nangaku M, Usuda N, Shimizu A, Onogi H, Izuhara Y et al (2005) Novel serpinopathy in rat kidney and pancreas induced by overexpression of megsin. J Am Soc Nephrol 16:1339–1349
Remold-O’Donnell E (1993) The ovalbumin family of serpin proteins. FEBS Lett 315:105–108
Kaiserman D, Bird PI (2005) Analysis of vertebrate genomes suggests a new model for clade B serpin evolution. BMC Genomics 6:167
Kaiserman D, Knaggs S, Scarff KL, Gillard A, Mirza G, Cadman M et al (2002) Comparison of human chromosome 6p25 with mouse chromosome 13 reveals a greatly expanded ov-serpin gene repertoire in the mouse. Genomics 79:349–362
Hirst CE, Buzza MS, Bird CH, Warren HS, Cameron PU, Zhang M et al (2003) The intracellular granzyme B inhibitor, proteinase inhibitor 9, is up-regulated during accessory cell maturation and effector cell degranulation, and its overexpression enhances CTL potency. J Immunol 170:805–815
Zhang YQ, Li P, Hou M, Wang X, Fan J, Tan L et al (2003) Identification of interaction between PAI-2 and IRF-3. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 35:661-5.
Palmiter RD, Gagnon J, Walsh KA (1978) Ovalbumin: a secreted protein without a transient hydrophobic leader sequence. Proc Natl Acad Sci U S A 75:94–98
Belin D (1993) Biology and facultative secretion of plasminogen activator inhibitor-2. Thromb Haemost 70:144–147
Pemberton PA, Tipton AR, Pavloff N, Smith J, Erickson JR, Mouchabeck ZM et al (1997) Maspin is an intracellular serpin that partitions into secretory vesicles and is present at the cell surface. J Histochem Cytochem 45:1697–1706
Von Heijne G, Liljeström P, Mikus P, Andersson H, Ny T (1991) The efficiency of the uncleaved secretion signal in the plasminogen activator inhibitor type 2 protein can be enhanced by point mutations that increase its hydrophobicity. J Biol Chem 266:15240–15243
Ritchie H, Booth NA (1998) Secretion of plasminogen activator inhibitor 2 by human peripheral blood monocytes occurs via an endoplasmic reticulum–Golgi-independent pathway. Exp Cell Res 242:439–450
Irving JA, Pike RN, Dai W, Brömme D, Worrall DM, Silverman GA et al (2002) Evidence that serpin architecture intrinsically supports papain-like cysteine protease inhibition: engineering alpha(1)-antitrypsin to inhibit cathepsin proteases. Biochemistry 41:4998–5004
Bird PI (1999) Regulation of pro-apoptotic leucocyte granule serine proteinases by intracellular serpins. Immunol Cell Biol 77:47–57
Kummer JA, Micheau O, Schneider P, Bovenschen N, Broekhuizen R, Quadir R et al (2007) Ectopic expression of the serine protease inhibitor PI9 modulates death receptor-mediated apoptosis. Cell Death Differ 14:1486–1496
Medema JP, Schuurhuis DH, Rea D, van Tongeren J, de Jong J, Bres SA et al (2001) Expression of the serpin serine protease inhibitor 6 protects dendritic cells from cytotoxic T lymphocyte-induced apoptosis: differential modulation by T helper type 1 and type 2 cells. J Exp Med 194:657–667
Missen MA, Haylock D, Whitty G, Medcalf RL, Coughlin PB (2006) Stage specific gene expression of serpins and their cognate proteases during myeloid differentiation. Br J Haematol 135:715–724
Bots M, Van Bostelen L, Rademaker MT, Offringa R, Medema JP (2006) Serpins prevent granzyme-induced death in a species-specific manner. Immunol Cell Biol 84:79–86
Ciscato F, Sciacovelli M, Ruvoletto MG, Quarta S, Turato C, Gatta A, et al. SERPINB3 inhibits the mitochondrial permeability transition pore. The 6th International Symposium of the Chemistry and Biology of Serpins, Chapel Hill, NC, USA, 23–26 October 2011 (abstract).
Ullman E, Pan JA, Zong WX (2011) Squamous cell carcinoma antigen 1 promotes caspase-8-mediated apoptosis in response to endoplasmic reticulum stress while inhibiting necrosis induced by lysosomal injury. Mol Cell Biol 31:2902–2919
Pontisso P, Calabrese F, Benvegnù L, Lise M, Belluco C, Ruvoletto MG et al (2004) Overexpression of squamous cell carcinoma antigen variants in hepatocellular carcinoma. Br J Cancer 90:833–837
Suminami Y, Nagashima S, Vujanovic NL, Hirabayashi K, Kato H, Whiteside TL (2000) Inhibition of apoptosis in human tumour cells by the tumour-associated serpin, SCC antigen-1. Br J Cancer 82:981–989
Numa F, Takeda O, Nakata M, Nawata S, Tsunaga N, Hirabayashi K et al (1996) Tumor necrosis factor-alpha stimulates the production of squamous cell carcinoma antigen in normal squamous cells. Tumour Biol 17:97–101
Vidalino L, Doria A, Quarta SM, Crescenzi M, Ruvoletto M, Frezzato F et al (2012) SERPINB3 expression on B-cell surface in autoimmune diseases and hepatitis C virus-related chronic liver infection. Exp Biol Med (Maywood) 237:793–803
Vidalino L, Doria A, Quarta S, Zen M, Gatta A, Pontisso P (2009) SERPINB3, apoptosis and autoimmunity. Autoimmun Rev 9:108–112
Uemura Y, Pak SC, Luke C, Cataltepe S, Tsu C, Schick C et al (2000) Circulating serpin tumor markers SCCA1 and SCCA2 are not actively secreted but reside in the cytosol of squamous carcinoma cells. Int J Cancer 89:368–377
Catanzaro JM, Guerriero JL, Liu J, Ullman E, Sheshadri N, Chen JJ et al (2011) Elevated expression of squamous cell carcinoma antigen (SCCA) is associated with human breast carcinoma. PLoS One 6:e19096
Song KJ, Ahn HJ, Nam HW (2012) Anti-apoptotic effects of SERPIN B3 and B4 via STAT6 activation in macrophages after infection with Toxoplasma gondii. Korean J Parasitol 50:1–6
Pontisso P, Morsica G, Ruvoletto MG, Zambello R, Colletta C, Chemello L et al (1991) Hepatitis B virus binds to peripheral blood mononuclear cells via the pre S1 protein. J Hepatol 12:203–206
Pontisso P, Vidalino L, Quarta S, Gatta A (2008) Biological and clinical implications of HBV infection in peripheral blood mononuclear cells. Autoimmun Rev 8:13–17
Hamanaka S, Ujihara M, Numa F, Kato H (1997) Serum level of squamous cell carcinoma antigen as a new indicator of disease activity in patients with psoriasis. Arch Dermatol 133:393–395
Giannelli G, Iannone F, Fransvea E, Chialà A, Lapadula G, Antonaci S (2007) Squamous cellular carcinoma immunocomplexed is increased in scleroderma patients with lung fibrosis. Clin Exp Rheumatol 25:794–795
El-Rachkidy RG, Young HS, Griffiths CE, Camp RD (2008) Humoral autoimmune responses to the squamous cell carcinoma antigen protein family in psoriasis. J Invest Dermatol 128:2219–2224
Khan S, Tarzi MD, Doré PC, Sewell WA, Longhurst HJ (2007) Secondary systemic lupus erythematosus: an analysis of 4 cases of uncontrolled hereditary angioedema. Clin Immunol 123:14–17
Brickman CM, Tsokos GC, Chused TM, Balow JE, Lawley TJ, Santaella M et al (1986) Immunoregulatory disorders associated with hereditary angioedema. II. Serologic and cellular abnormalities. J Allergy Clin Immunol 77:758–767
Cacoub P, Frémeaux-Bacchi V, De Lacroix I, Guillien F, Kahn MF, Kazatchkine MD et al (2001) A new type of acquired C1 inhibitor deficiency associated with systemic lupus erythematosus. Arthritis Rheum 44:1836–1840
Nettis E, Colanardi MC, Loria MP, Vacca A (2005) Acquired C1-inhibitor deficiency in a patient with systemic lupus erythematosus: a case report and review of the literature. Eur J Clin Invest 35:781–784
Lahiri M, Lim AY (2007) Angioedema and systemic lupus erythematosus—a complementary association? Ann Acad Med Singapore 36:142–145
Czyzyk J, Henegariu O, Preston-Hurlburt P, Baldzizhar R, Fedorchuk C, Esplugues E et al (2012) Enhanced anti-serpin antibody activity inhibits autoimmune inflammation in type 1 diabetes. J Immunol 188:6319–6327
Grimstein C, Choi YK, Wasserfall CH, Satoh M, Atkinson MA, Brantly ML et al (2011) Alpha-1 antitrypsin protein and gene therapies decrease autoimmunity and delay arthritis development in mouse model. J Transl Med 9:21
Sardi F, Fassina L, Venturini L, Inguscio M, Guerriero F, Rolfo E et al (2011) Alzheimer’s disease, autoimmunity and inflammation. The good, the bad and the ugly. Autoimmun Rev 11:149–153
Costa CZ, da Rosa SE, de Camargo MM (2011) The unfolded protein response: how protein folding became a restrictive aspect for innate immunity and B lymphocytes. Scand J Immunol 73:436–448
Rasheva VI, Domingos PM (2009) Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis 14:996–1007
Inagi R (2009) Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron Exp Nephrol 112:e1–e9
Vitadello M, Doria A, Tarricone E, Ghirardello A, Gorza L (2010) Myofiber stress-response in myositis: parallel investigations on patients and experimental animal models of muscle regeneration and systemic inflammation. Arthritis Res Ther 12(2):R52
Ekeowa UI, Freeke J, Miranda E, Gooptu B, Bush MF, Pérez J et al (2010) Defining the mechanism of polymerization in the serpinopathies. Proc Natl Acad Sci U S A 107:17146–17151
Lawless MV, Greene CM, Mulgrew A, Taggart CC, O’Neill SJ, McElvaney NG (2004) Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J Immunol 172:5722–5760
Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13:1211–1233
Kroeger H, Miranda E, MacLeod I, Pérez J, Crowther DC, Marciniak SJ et al (2009) Endoplasmic reticulum-associated degradation (ERAD) and autophagy cooperate to degrade polymerogenic mutant serpins. J Biol Chem 284:22793–22802
Ishida Y, Nagata K (2009) Autophagy eliminates a specific species of misfolded procollagen and plays a protective role in cell survival against ER stress. Autophagy 5:1217–1219
Davies MJ, Miranda E, Roussel BD, Kaufman RJ, Marciniak SJ, Lomas DA (2009) Neuroserpin polymers activate NF-kappaB by a calcium signaling pathway that is independent of the unfolded protein response. J Biol Chem 284:18202–18209
Singh RP, Waldron RT, Hahn BH (2012) Genes, tolerance and systemic autoimmunity. Autoimmun Rev 11:664–669
Varin MM, Guerrier T, Devauchelle-Pensec V, Jamin C, Youinou P, Pers JO (2012) In Sjögren’s syndrome, B lymphocytes induce epithelial cells of salivary glands into apoptosis through protein kinase C delta activation. Autoimmun Rev 11:252–258
Iaccarino L, Ghirardello A, Canova M, Zen M, Bettio S, Nalotto L et al (2011) Anti-annexins autoantibodies: their role as biomarkers of autoimmune diseases. Autoimmun Rev 10:553–558
Comi C, Fleetwood T, Dianzani U (2012) The role of T cell apoptosis in nervous system autoimmunity. Autoimmun Rev 12:150–156
Zen M, Canova M, Campana C, Bettio S, Nalotto L, Rampudda M et al (2011) The kaleidoscope of glucorticoid effects on immune system. Autoimmun Rev 10:305–310
Racanelli V, Prete M, Musaraj G, Dammacco F, Perosa F (2011) Autoantibodies to intracellular antigens: generation and pathogenetic role. Autoimmun Rev 10:503–508
Costenbader KH, Gay S, Alarcón-Riquelme ME, Iaccarino L, Doria A (2012) Genes, epigenetic regulation and environmental factors: which is the most relevant in developing autoimmune diseases? Autoimmun Rev 11:604–609
Rekvig OP, Putterman C, Casu C, Gao HX, Ghirardello A, Mortensen ES et al (2012) Autoantibodies in lupus: culprits or passive bystanders? Autoimmun Rev 11:596–603
Münz C (2012) Antigen processing for MHC Class II presentation via autophagy. Front Immunol 3:9
Münz C (2010) Antigen processing via autophagy—not only for MHC class II presentation anymore? Curr Opin Immunol 22:89–93
Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L (2008) Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 455:396–400
Deretic V (2009) Multiple regulatory and effector roles of autophagy in immunity. Curr Opin Immunol 21:53–62
Pierdominici R, Vomero M, Barbati C, Colasanti T, Maselli A, Vacirca D et al (2012) Role of autophagy in immunity and autoimmunity, with a special focus on systemic lupus erythematosus. FASEB J 26:1400–1412
Rodríguez-Bayona B, Ramos-Amaya A, Pérez-Venegas JJ, Rodríguez C, Brieva JA (2010) Decreased frequency and activated phenotype of blood CD27 IgD IgM B lymphocytes is a permanent abnormality in systemic lupus erythematosus patients. Arthritis Res Ther 12:R108
Hostmann A, Jacobi AM, Mei H, Hiepe F, Dörner T (2008) Peripheral B cell abnormalities and disease activity in systemic lupus erythematosus. Lupus 17:1064–1069
Korganow AS, Knapp AM, Nehme-Schuster H, Soulas-Sprauel P, Poindron V, Pasquali JL et al (2010) Peripheral B cell abnormalities in patients with systemic lupus erythematosus in quiescent phase: decreased memory B cells and membrane CD19 expression. J Autoimm 34:426–434
Shapira E, Proscura E, Brodsky B, Wormser U (2011) Novel peptides as potential treatment of systemic lupus erythematosus. Lupus 20:463–472
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gatto, M., Iaccarino, L., Ghirardello, A. et al. Serpins, Immunity and Autoimmunity: Old Molecules, New Functions. Clinic Rev Allerg Immunol 45, 267–280 (2013). https://doi.org/10.1007/s12016-013-8353-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-013-8353-3