
Abstract
Several crucial issues remain open in our understanding of primary biliary cirrhosis (PBC), an autoimmune liver disease targeting the small- and medium-sized intrahepatic bile ducts. These issues include the high tissue specificity of the autoimmune injury despite the nontraditional autoantigens found in all mitochondria recognized by PBC-associated autoantibodies, the causes of the commonly observed pruritus, and the disease etiology per se. In all these fields, there has been recent interest secondary to the use of large-scale efforts (such as genome-wide association studies) that were previously considered poorly feasible in a rare disease such as PBC as well as other intuitions. Accordingly, there are now fascinating theories to explain the onset and severity of pruritus due to elevated autotaxin levels, the peculiar apoptotic features of bile duct cells to explain the tissue specificity, and genomic and epigenetic associations contributing to disease susceptibility. We have arbitrarily chosen these four aspects as the most promising in the PBC recent literature and will provide herein a discussion of the recent data and their potential implications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
What is New in the Field of Primary Biliary Cirrhosis
Primary biliary cirrhosis (PBC) is a chronic cholestatic liver disease characterized by high-titer serum anti-mitochondrial autoantibodies (AMA) and an autoimmune-mediated destruction of the small- and medium-sized intrahepatic bile ducts [1]. Over the past few years there has been a growing interest for PBC, as well as represented by numerous research reports in multiple fields ranging from etiopathogenesis to treatment. The aim of the present review article is to briefly illustrate and discuss the most recent developments in terms of clinics (i.e., autotaxin in pruritus), pathogenesis (i.e., the role of apoptosis), and etiology (with genetics and epigenetics) which, albeit not comprehensive of all lines of evidence [2, 3], appear as the most promising issues in this rare but representative autoimmune disease. We recognize that the choice of these issues is arbitrary and does not include all research fields, as numerous other aspects would warrant discussion as hot topics in PBC.
Are the Causes of PBC-Associated Pruritus Finally Solved?
One of the most important clinical features of PBC is represented by pruritus, an undefined symptom that has a prominent impact on patient lifestyle and quality of life, in particular when common therapeutic strategies fail. Similar to other subjective symptoms, pruritus is difficult to evaluate because of its polymorphic clinical manifestations related to cognitive, evaluative, emotional, and sensitive discriminative components. The main causes commonly involved in pruritus generation include skin disorders, systemic disorders (chronic liver diseases or renal failure), neuropathy, psychogenic and mixed origin. Albeit the molecular mechanisms are now understood, the processes of central elaboration of peripheral information remain to be discovered. In chronic liver disease, pruritus is one of the most important symptoms and can be so intense to become the major indication for liver transplantation. Indeed, liver transplantation is the most effective treatment for pruritus even in the absence of liver failure [4] regardless of the disease etiology. Physiology studies on itch mechanisms have led to the identification of specific neuronal pathways, i.e., mediators and receptors involved in signal transmission but also in the perception of stimuli [5–7]. Moreover, new molecular bases of cholestatic diseases itch have recently been hypothesized [8]. In spite of the main cause that lead to cholestasis, the various disorders are all defined by pruritus. Not only PBC but also primary sclerosing cholangitis, intrahepatic cholestasis of pregnancy, and benign recurrent cholestasis or progressive familial intrahepatic cholestasis are, indeed, clinically identified by intense pruritus. All of these liver disorders are marked by impairment of bile production or secretion leading to hepatocyte injury and worsening cholestasis [9]. The treatment options for the pruritus associated with cholestasis remain quite disappointing in terms of efficacy (as in the case of the first-line option, cholestyramine) or tolerability (as for rifampin) while other treatments remain poorly supported by experimental data [10].
Studying the increasing of calcium levels in neuronal cells caused by serum proteins in patients with cholestatic pruritus, lysophosphatidic acid (LPA) has been recently addressed as a potential common key factor [8]. Indeed, in this seminal study the authors demonstrated how LPA is able to enhance neuronal cells activity by increasing intracytoplasmatic calcium levels [8]. Circulating LPA is produced in the blood through cleavage of cholin from lysophosphatidylcholine acid (LPC) by a defined enzyme called autotaxin (ATX) [11, 12] (Fig. 1). This phenomenon is reversed by treatment with a specific LPA receptor blocker, called Ki16425 [8].

The reaction mediated by autotaxin modifies lysophosphatidycholine generating lysophosphatidic acid with the loss of a choline
The history of ATX warrants further discussion as the molecule was firstly identified years ago in the human melanoma cell line A2058 [13] and was defined as a motility factor involved in the generation of cancer metastases [14]. ATX is, indeed, an enzyme overexpressed in several tumors and is involved not only in cell proliferation but primarily in the regulation of motility [15]. It is also essential in processes such as angiogenesis as well as in neuronal development, as demonstrated by the fact that ATX knockout mice manifest lethal vascular or neuronal malformations [16, 17]. ATX acts on LPC with its lysophospholipase D activity generating LPA (Fig. 1) [11, 12]. The recent data report significantly increased ATX levels in serum of patients with cholestatic disorders and pruritus [8]. The same group reported that LPA and ATX serum levels correlate with itch intensity [8]. On the other hand, although not as high as in primarily cholestatic diseases, also patients with chronic hepatitis C are characterized by an increased expression of the enzyme compared to healthy controls [8]. Another suggestive finding by Kremer et al. is that the retention of bile salts does not correlate with pruritus intensity as demonstrated by the use of nasobiliary drainage to treat most severe itch is correlated with a reduction of the ATX activity. Surprisingly, when the symptom becomes evident the enzyme activity is at pre-drainage levels [8] even if it is not cleared by bile flow (ATX and ATX activity are not found in bile of patient affected by cholestatic diseases) [8]. Another correlation between itch and ATX activity is represented by the induction of pruritus in mice that are treated with a subcutaneous injection of LPA, in a dose-dependent manner [8].
Of note, LPA is a phospholipid involved in cell migration and cytoskeletal structural modifications, with a central role in platelet activation but also in cytokine production [15, 18] and in the induction of neuropathic pain [19, 20]. The last target of this bioactive lipid is a family of G protein-coupled receptors that are involved in a great variety of biological processes discovered analyzing ATX effects of melanoma cells. Moreover, LPA has been implicated in the process of neurite remodeling and myelin modifications after a trauma [21]. The LPA intrathecal injection in mice is responsible of the ignition of neuropathic pain [19] and mice with a lack of specific receptors for this lipid are unable to develop histological signs of demyelination or pain [19]. Studies focusing on pain neurotransmission signals have discovered that LPA is responsible of changes in fiber activation by increasing the stimulation of type 3A fibers, but reduces the possibility of activation of type 1C fibers in opposition to substance P [20]. Accordingly, the most recent hypothesis defines LPA as one of the most important molecules involved in the onset of pain and pruritus. LPA could indeed activate specific receptors and pain fibers inducing itch and pain but also stimulate the releasing of other skin-specific mediators [22]. The recent study also reported that pruritus due to chronic liver diseases causing cholestasis is secondary to LPA and ATX along with liver function impairment. This finding is sustained at a physiological level by tissue histology that discovered itch-specific sensory neurons categories [23, 24]. One group of neurons responds only to pruritogenic stimuli as histamine but not to mechanical pain signals [4, 23, 24], while another group is activated independently by histamine even if it is not implicated in cholestatic itching [4, 23, 24]. Nerve fibers, which are activated in pruritus are probably defined in the recent past as nociceptive fibers and even if the identification of stable circuits is far from being completely understood, these findings could explain the wide variability in subjective pruritus degree and localization [25]. Other molecules have been considered to be actors of cholestatic pruritus, some of these are opioid peptides and bile salts. Serum concentrations of these molecules are not correlated with clinical severity of symptom as Kremer et al. demonstrated about LPA [8]. This experimental evidence is strengthened by clinical evidence that antihistamine drugs are ineffective against cholestatic itch [4]. However, even if ATX activity and LPA serum levels are correlated with itching in cholestatic diseases, the molecular pathways remain unknown (Fig. 2). Blood sources of ATX levels are, indeed, poorly defined so an increased enzyme activity could be derived from an increased expression or a reduced degradation as shown in some studies on the role of sinusoidal endothelial cells in ATX clearance [26]. Several research groups demonstrated that ATX blood half-life is very short in healthy individuals but circulating protein level is in a steady state probably due to endothelial and adipocyte cells secretion [27, 28]. Interestingly, ATX is also expressed within the liver [29] and an elevated ATX mRNA expression is observed in patients with chronic hepatitis C [30, 31].

The proposed model for pruritus in chronic cholestatic liver disease. The specific molecular pathways by which cholestasis leads to a rise in ATX blood expression, ATX activity, and LPA levels remain unknown but the discover of this molecule opens new physiological and therapeutical perspectives (ATX autotaxin, LPA lysophosphatidic acid)
The recent finding of ATX and LPA changes opens new research scenarios in pruritus onset and perpetuation; it is possible that these molecules could be involved in development of pruritus regardless of the underlying disease. A study on Hodgkin lymphoma cells infected with Epstein–Barr virus reported high expression levels of ATX [32] that could produce a large amount of LPA involved not only in tumor progression but also in the genesis of pruritogenic stimuli recognized by specific neuronal cells [15]. Patients affected by Hodgkin lymphoma often experience intense pruritus [33] which is associated with poorer prognosis than patients with mild itch and the symptom seems to be negatively related to life expectancy [34]. Finally, another example of a correlation between pruritus and LPA molecular activity is represented by cutaneous wound healing that is typically associated with pruritus. It has been hypothesized that this is secondary to a local overproduction of the bioactive lipid that is strongly related to healing, as LPA promotes and stimulates reepithelialization especially in regenerating tissues [35]. These observations could have, if independently confirmed, important developments in the near future. ATX inhibitors and LPA receptors blockers, which have been developed mainly for cancer therapy with the purpose to reduce metastases [36] could be used to resolve the great problem of PBC-associated pruritus.
Genome-Wide Association Studies
One of the frontiers of genetics in complex diseases is currently represented by the search for specific genomic sites involved in disease pathogenesis through a genome-wide analysis. In fact, over the past decades, researchers have identified over 1,200 gene candidates for specific diseases using fine gene mapping techniques [37, 38]. However, this investigating model has not been successful in identifying the genetic modifiers of the most common diseases lacking pure Mendelian laws of inheritance. Some examples are represented by most of the common age-related diseases such as arterial hypertension [39], type 2 diabetes [40, 41], coronary heart disease [42], and dementia [43], which are presumed to be determined by several genes with a fundamental interaction of several and predisposing environmental agents. For these reason, a wide genetic approach on complex and polygenic diseases seems to be the best way to understand and identify the possible subjects of the pathological conditions that afflict a great part of public health systems budget and impose enormous social costs. Further, few candidate genes have been independently confirmed in more than one population thus reducing the applicability of reported genetic associations. In this scenario, the advent of genome-wide association studies (GWAS) appears as the ideal tool to determine the complex diseases genetic susceptibility due to multiple genes that concur to determine the pathological phenotype. The major hypothesis states that most of genetic predisposition for common diseases is due to nucleotide polymorphisms and copy number variants (CNVs) which represent 0.5% of human DNA [44]. The former are defined by single, double, triple, or more bases substitutions, but the most frequent are covered by single nucleotide polymorphisms (SNPs) while the latter are represented by several repetition of small DNA non-coding sequences. SNPs cover the most widespread proportion of these genomic modifications with approximately ten million SNPs identified in the human genome [45, 46] influencing the majority of genomic sequences. Even if nucleotide substitutions are evenly distributed throughout the genome (estimated in one out of 1,000 base pairs) some studies demonstrated how with the exception of hotspots of high recombination, the human genome is characterized by a block structure with sequences of SNPs that are strongly linked in blocks coined linkage disequilibrium (LD) [47, 48]. CNV are defined as inherited duplications and deletions of 1,000 to 10,000 base lengths of DNA; these sequences are relatively frequent covering approximately 12% of the human genome [49, 50]. CNV mechanism of action is represented by the amplification or suppression of genetic transcription leading to substantial modification in cellular metabolic pathways conducing toward a pathologic state or a disease protective activity. The recent technological improvements justify the recent attention to CNVs in GWAS [51–53]. The results of CNV analysis reveal associations with various phenotypes or disease states like glomerulonephritis [54, 55], subarachnoid hemorrhage [55], body mass index [56], and even cultural dietary preferences [57]. These findings may be due to the effects of CNVs on gene dosages, but also to the possible changes of transcription factor binding sites and modifications of epigenetic mechanisms.
It is important to understand the aims of GWAS, which should be intended as a solid statistical association between a genomic variant and a specific phenotype. The important aspect of this procedure is represented by the absence of major bias, due to the possible influence of personal experience selection of risk factors and/or the choice of candidate genes. The finding of statistically significant differences between the subjects of the study and the controls leads to identify specific chromosomal regions. These identified sequences are related to an impairment control of genetic expression, so they have to be considered like a genetic mark that predispose to disease development, albeit the need for correction for multiple comparisons remain of seminal importance.
The power of GWAS was first illustrated in large studies on type 1 diabetes and Crohn's disease where more than 30 genetic loci have been identified thus far [58, 59]; of note, most of these findings were unexpected and allowed to discover new pathogenetic pathways. From this point of view, GWAS could be considered as an etiological but also pathogenetic generator of hypotheses.
Similar to previous studies on candidate genes, data from GWAS warrant independent replication and this process has failed in the case of Crohn's disease, when some statistically significant findings were not be recapitulated in other populations [60]. These data are ascribed to differences among geographic regions in the genetic susceptibility for the same condition [61, 62] proving how much control populations and environmental influences cannot be overlooked.
In the field of chronic liver diseases, GWAS were introduced relatively late and currently cover a minority (2%) of the published studies, ranging from gallstone [63, 64] to liver autoimmunity [65, 66]. Some of these focus the attention on response to therapy in C hepatitis [67] while others on drug-induced liver injury [68, 69]. The first GWAS on PBC was performed by Hirschfield et al. only in 2009. The most consistent associations are related to the human leukocyte antigen (HLA) class II, interleukin 12s (IL-12a) and interleukin 12 receptor b2 (IL-12RB2) genes [66]. Even if association with HLA class II has been reported by other studies [70], only Hirschfield et al. have demonstrated in a large multicenter study with a relevant statistic significance that this region is involved in PBC pathogenesis. The other two genes identified by the study are strongly involved in susceptibility to PBC as their products have important immunoregulatory functions related to other study findings as several SNPs associated with some inflammatory DNA sequences, i.e., signal transducer and activator of transcription 4 (STAT4), cytotoxic T lymphocyte-associated protein 4 (CTLA4) and interferon regulatory factor 5 (IRF5) [66].These genes are known to be associated with other autoimmune diseases, STAT4 beings related with rheumatoid arthritis, systemic lupus erythematosus [71], and type 1 diabetes [72] while IL-12A gene celiac disease [73]. These findings suggest and sustain the hypothesis of the presence of impaired immune mechanisms leading to overt autoimmunity and PBC. In fact, there is evidence that IL-12A and IL-12RB2 are fundamental in immunity responses against infectious diseases [74], thus the variants associated with PBC could be responsible of an impaired response to infection determining an augmented risk to generate autoimmunity clones [75]. Moreover, this mechanism of PBC autoimmune process is sustained by additional data referring to the development of autoimmune and lymphoproliferative disease in the IL-12RB2 knockout mice [76]. This is not the only example because a case report describing the development of a PBC-like condition in a child with IL-12 deficiency [77] supports this view. Another GWAS has been recently performed in a wide PBC Italian cohort and published as a meta-analysis along with the Canadian data [78]. The researchers comparing the two data set in a included meta-analysis confirmed previous associations and reported additional SNP matching on chromosome 17 (17q12–21 region) [78]. This area is strongly related with inflammatory bowel diseases [59], asthma [79], and type 1 diabetes [58]. Some of these SNPs refer to IKAROS family zinc finger 3 (also known as Aiolos), a transcription factor that prevents apoptosis of IL-2-deprived B cells [80], regulates B cell activation [81] and has been implicated in autoimmunity [82]. The meta-analysis has revealed the involvement of previous unidentified genes as Spi-B transcription factor (Spi-1/PU.1 related) [78], an important mediator of B cell receptor signaling [83], and the association with a locus including IRF5 and transportin 3 (TNPO3) [78]. This result confirms the findings of the combined genome-wide association and replication datasets performed by Hirschfield et al. that has revealed IRF5-TNPO3, 17q12-21 and encoding membrane metallo-endopeptidase like 1 (MMEL1) as new PBC susceptibility loci [84]. Of note, MMEL1 is also associated with rheumatoid arthritis and celiac disease [85–90], two immune-related conditions, but all data require further investigation.
In conclusion, it is possible to consider GWAS as indispensable tools to understand the crucial genomic aspects of common and rare polygenic diseases with the aim to identify diagnostic, prognostic, and therapeutic markers to allow individualized medicine.
Epigenetics
The past years have witnessed a continuous growth in the search for a better understanding of complex diseases, particularly when genomic data appear necessary but insufficient to explain the pathology development. PBC well fits this model as represented by the solid associations reported by GWAS in limited subgroups of patients [78, 84] or the incomplete concordance in monozygotic twins [91]. The field of epigenetics thus appears as the ideal link between genes and environment to determine the onset of complex diseases, particularly considering that new epigenetic therapies are becoming increasingly used.
Epigenetics determine signal or perpetuate gene functions without changing DNA nucleotide sequence while adding or removing chemical functional groups on nucleotides or on specific amino acid residues, generating a real code that can be read by defined proteins. The epigenetic code is determined by different enzymes that ultimately determine gene expression modulation through sterical changes, i.e., DNA methylation and histone acetylation. Epigenetic modifications are crucial for cell type development and differentiation, sexual differentiation in embryo, tissue specialization, gene imprinting, and cell metabolic plasticity. The DNA functional packaging unit is called nucleosome, described as a double wrapping of the double helix around a protein core defined histones, which are highly conserved (Fig. 3). Histone subunits can be modified by functional chemical groups at defined amino acids to modify the packaging of DNA molecules leading to transcriptional accessibility or unavailability of specific nuclear factors or binding proteins. Epigenetic regulatory mechanisms include acetylation or deacetylation of nucleosomal histones lysine residues by histone acetyltransferase (HAT) and histone deacetylase (HDAC) enzymes, respectively [92–95]. HATs promote gene expression by allowing transcription factors to access genetic sequences [96–98] while HDACs removes acetyl groups and is generally associated with gene repression [99–101]. The epigenetic landscape is completed by the discussion of DNA methylation as the most studied mechanism. The addition of a methyl group to cytosine nucleotides is performed by DNA methyltransferases enzymes through a methyl group donor, S-adenosylmethionine. Methylation occurs most commonly at CpG islands, 500-bp DNA regions containing an high density of GC dinucleotides [102–104] with key regulatory functions [105] and an altered CpG island methylation induces changes in genes transcription modifying chromatin structure [106]. Histone methylation also modifies gene expression depending on the position and the number of the modified lysine residue within the histone tails [107] to activate or block transcription [108]. Other epigenetic mechanisms include arginine methylation/demethylation [109] and protein ubiquitination to control the stability and intracellular localization of numerous proteins influencing the status of histone methylation or acetylation [110]. The importance of DNA methylations is represented by the X chromosome inactivation in women which is completely regulated by methylation of specific genetic sequences [111–115].

Nucleosome representation. One hundred and forty-seven base pairs of DNA are double-wrapped around histone core proteins in a 1.7 left-handed superhelical turn. The histone in the center is constituted by eight core proteins folded together in antiparallel pairs (H3 with H4 and H2A with H2B) to constitute tetramers. The assembly of two tetramers forms the octameric core structure (H3/H4-H2A/H2B1) of the nucleosome. [White thick strand (spacefill) and yellow thin one (spheres and sticks) correspond to DNA double helix; red is 5′ and orange 3′ extremities. Blue, histone core proteins]
Specific impairments in the regulation of epigenetic processes in the immune system cells could be responsible for the breakdown of tolerance [116–118] as underlined by the development of autoreactive T cells clones in vitro following treatment with chemical agents inhibiting cellular enzymes responsible of DNA methylation process [119, 120]. In the field of autoimmunity, the epigenetics of rheumatoid arthritis (RA) were largely investigated [121] and it has been proposed that RA-associated joint cartilage destruction derives from a reduced global DNA methylation [122] or a hypomethylation of CpG islands within the LINE-1 promoter [122, 123]. Furthermore, unmethylated CpG islands of the IL-6 promoter in monocytes were described with a local hyperactivation of the inflammation circuit [124] and RA monocytes manifest a change in the methylation status of CpG islands within the promoter of death receptor 3 which is downregulated to induce resistance to apoptosis [125].
We are convinced that PBC recognizes an important role for genome variants, possibly stronger than in other autoimmune disorders [126, 127] since the 63% concordance rate reported in PBC monozygotic (MZ) twins is the highest among autoimmune diseases with the exception of celiac disease [128]. MZ twins represent an ideal model for the study of epigenetics of complex diseases as recent data demonstrated the presence of an epigenetic drift for the development of phenotypic differences between identical twins [129, 130]. Data on DNA methylation in PBC are limited to one study from our group. We reported that MZ twins discordant for PBC are characterized by differential expression of two X-linked genes (PIN4, CLIC2) that are differentially methylated [131]. This report is of particular interest based on previous data pointing towards an X chromosome haploinsufficiency [132] through X chromosome loss in peripheral blood lymphocytes.
Epitopes and Apotopes in PBC
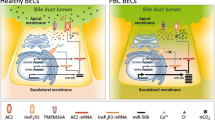
A fascinating line of evidence on the pathogenesis of PBC was produced over the past few years by studies on the role of apoptosis. As previously mentioned, PBC is a chronic cholestatic liver disease where the most important pathogenetic processes is determined by an autoimmune destruction of small- and medium-sized intrahepatic bile ducts [133]. The main serological finding is represented by circulating highly PBC-specific AMA [134], which are autoantibodies directed against the lipoilated domain of the E2 component of the pyruvate dehydrogenase complex (PDC-E2) [135–138] (Fig. 4a) and are present in about 95% of PBC patients. PDC-E2 is a member of the 2-oxo-acid dehydrogenase complex, [139, 140] a family of enzymes that are mainly implicated in oxidative phosphorilation. The common chemical structure, which leads to become immunologically reactive is the E2 subunit (Fig. 4b). This protein motif is formed by an N-terminal domain containing a lysine-lipoyl group, a peripheral subunit binding domain and a C-terminal inner core (Fig. 4c). It has been previously shown that lipoic acid residue attached to AMA epitopes is necessary for autoantibody binding [141, 142]. The map of recognized epitopes has revealed how the autoimmune responses are directed against highly conserved molecular sequences and they include not only the activation of B cells but also CD4+ and CD8+ T cells immunity programs [143]. The understanding of PBC autoantigens and tissue specificity are counterintuitive in fact that a ubiquitous antigen is capable to elicit a bile duct specific immune-mediated injury and the peculiar apoptotic features of intrahepatic biliary epithelial cells (BECs) may recapitulate this contrast. Albeit the biliary epithelium represent only a small proportion of liver cells [144], BECs play an essential role in determining bile composition [145] and in the induction of immune activity against microbes and foreign antigens [146] through the expression of human leukocyte antigen molecules [139] and costimulatory [147] and adhesion molecules [148]. BECs could be implicated in the loss of immune tolerance via an impairment of a correct clearance process of apoptotic cells by specific local phagocytes [149, 150]. In fact, when the uptake of apoptotic residues is ineffective, cells lysates can release intracellular molecules initiating the autoimmune reaction [151–155]. The presence of self-antigens within apoptotic bodies [156] can then be uptaken by local macrophages as antigen-presenting cells (APCs) producing proinflammatory cytokines [157, 158]. Data sustaining the role of BECs in pathogenesis of PBC are related to the presence of AMA-accessible PDC-E2 within BEC apoptotic bodies [159] (Fig. 5). This finding is probably the explanation of the failure of different therapeutical strategies such as immunosuppressive drugs [160] and the disease recurrence following liver transplantation [161]. On the other hand, the cited data describe the efficacy of ursodeoxycholic acid action in cholestatic liver diseases, because the biliary salt acts decreasing biliary apoptotic rate and sustains bile flow improving apoptotic remnants clearance [162, 163] reducing self-antigens.

Structural illustration of the PBC autoantigen. a Pyruvate dehydrogenase complex is a multiple subunits enzyme composed by E1, E2, and E3 main subunits; b the autoimmune reaction is directed against the E2 subunit; c the E2 lipoyl domain represents the major AMA epitope. Blue background images are structural representation; purple, α-helices; yellow, β-sheet; gray, polypeptidic chain. White background images are chemical representation

The proposed role of BECs apoptosis in PBC pathogenesis. First, PDC-E2 maintains its chemical properties tanks to a glutathiolation deficit. This feature in addition to the surface availability is fundamental in interaction with circulating AMAs. The subsequent immune complexes formation determines APCs activation that ignites specific immune systems. CD8+ T cells determines BECs destruction and CD4+ T cells induce AMA production activating autoreactive B cells leading to biliary injuries. Biliary epithelium damage closes the cycle, in fact colangiocytes destruction lead to production of proinflammatory mediators but also the release of other PDC-E2 that sustains the autoreactive immune clone cells. PDC-E2, pyruvate dehydrogenase complex; APCs, antigen presenting cells; BECs, biliary epithelial cells; AMAs, anti-mitochondrial antibodies; TLR, toll-like receptor
Apoptotic cells express some apoptotic signals on their surface before the complete cell destruction occur [150, 164], which are necessary to regulate the clearance of apoptotic bodies. This process is crucial to maintain self-tolerance in the liver microenvironment and to limit the amount of intracellular self antigens which could be disposable to produce new epitopes and to induce autoimmune response [165]. Indeed, apoptotic cells phagocytosis is followed by the secretion and action of anti-inflammatory cytokines in opposition to microbes or necrotic cells which are associated with a production of proinflammatory mediators [166] while under impaired conditions apoptotic bodies and cell fragments can be a source of autoantigens [167] (Fig. 5).
Data from the study of molecular mimicry and xenobiotics [142, 168] well support the apoptosis-based working hypothesis. Indeed, PDC-E2 is found on the cell membrane during apoptosis [169] and xenobiotic-modified PDC-E2 peptides are recognized by PBC sera [170]. In fact, only in BECs PDC-E2 is immunologically active after apoptosis while in other cells types the PDC-E2 autoantibody recognition is abrogated [159]. The responsibility of autoimmune process in recognition of this antigenic epitopes is another time confirmed by the presence of PDC-E2 in apoptotic bodies where it is accessible to AMA recognition and immune cells actions [169]. Data sustain that the interactions between BEC apoptosis epitopes coined “apotopes”, macrophages of PBC patients, and serum AMA lead to a powerful production of cytokines [171]. Moreover, PBC macrophages cultured with BEC apotopes and AMAs produce TNF-alpha inducing apoptosis in BECs [172]. These new findings lead us to understand the real tissue specificity of this pathology, in fact, as mentioned above, the subject of immune responses are BECs.
Taken altogether, the data from BEC apoptosis suggest that different cells maintain the balance between the various local stimuli and the numerous cellular types involved in the microbiliary-environment immune activation and regulation. The presence of specific stimuli with a genetically susceptible background leads to the disequilibrium in immune balance that regulates the activation or suppression of different genetic cellular activities generating the explosion of autoimmune process.
Future Directions
Our brief discussion of four novel developments of old aspects of PBC illustrates the solid effort of researchers worldwide to unravel this mysterious condition. In particular, we now await major advancements based on these findings, particularly in terms of a new treatment for pruritus (based on autotaxin modulation) and the disease (using new monoclonal antibody approaches possibly targeting the IL-12 pathway) as well as new diagnostic tools (based on solid genetic and epigenetic associations).
References
Selmi C, Bowlus CL, Gershwin ME, Coppel RL (2011) Primary biliary cirrhosis. Lancet (in press)
Lan RY, Salunga TL, Tsuneyama K et al (2009) Hepatic IL-17 responses in human and murine primary biliary cirrhosis. J Autoimmun 32:43–51
Padgett KA, Lan RY, Leung PC et al (2009) Primary biliary cirrhosis is associated with altered hepatic microRNA expression. J Autoimmun 32:246–253
European Association for the Study of the Liver (2009) EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol 51:237–267
Paus R, Schmelz M, Biro T, Steinhoff M (2006) Frontiers in pruritus research: scratching the brain for more effective itch therapy. J Clin Invest 116:1174–1186
Sun YG, Chen ZF (2007) A gastrin-releasing peptide receptor mediates the itch sensation in the spinal cord. Nature 448:700–703
Sun YG, Zhao ZQ, Meng XL, Yin J, Liu XY, Chen ZF (2009) Cellular basis of itch sensation. Science 325:1531–1534
Kremer AE, Martens JJ, Kulik W (2010) Lysophosphatidic acid is a potential mediator of cholestatic pruritus. Gastroenterology 139:1008–1018, 1018 e1001
Beuers U (2006) Drug insight: mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat Clin Pract Gastroenterol Hepatol 3:318–328
Kremer AE, Beuers U, Oude-Elferink RP, Pusl T (2008) Pathogenesis and treatment of pruritus in cholestasis. Drugs 68:2163–2182
Umezu-Goto M, Kishi Y, Taira A et al (2002) Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol 158:227–233
Tokumura A, Majima E, Kariya Y et al (2002) Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J Biol Chem 277:39436–39442
Stracke ML, Krutzsch HC, Unsworth EJ et al (1992) Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J Biol Chem 267:2524–2529
Liotta L, Schiffmann E (1988) Tumor autocrine motility factors. Important Adv Oncol 17–30
Mills GB, Moolenaar WH (2003) The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer 3:582–591
Tanaka M, Okudaira S, Kishi Y et al (2006) Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J Biol Chem 281:25822–25830
van Meeteren LA, Ruurs P, Stortelers C et al (2006) Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol 26:5015–5022
van Meeteren LA, Moolenaar WH (2007) Regulation and biological activities of the autotaxin-LPA axis. Prog Lipid Res 46:145–160
Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H (2004) Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med 10:712–718
Ueda H (2006) Molecular mechanisms of neuropathic pain-phenotypic switch and initiation mechanisms. Pharmacol Ther 109:57–77
Savaskan NE, Rocha L, Kotter MR et al (2007) Autotaxin (NPP-2) in the brain: cell type-specific expression and regulation during development and after neurotrauma. Cell Mol Life Sci 64:230–243
Zhao Y, Tong J, He D et al (2009) Role of lysophosphatidic acid receptor LPA2 in the development of allergic airway inflammation in a murine model of asthma. Respir Res 10:114
Schmelz M, Schmidt R, Bickel A, Handwerker HO, Torebjork HE (1997) Specific C-receptors for itch in human skin. J Neurosci 17:8003–8008
Davidson S, Zhang X, Yoon CH, Khasabov SG, Simone DA, Giesler GJ Jr (2007) The itch-producing agents histamine and cowhage activate separate populations of primate spinothalamic tract neurons. J Neurosci 27:10007–10014
Stander S, Schmelz M (2006) Chronic itch and pain—similarities and differences. Eur J Pain 10:473–478
Jansen S, Andries M, Vekemans K, Vanbilloen H, Verbruggen A, Bollen M (2009) Rapid clearance of the circulating metastatic factor autotaxin by the scavenger receptors of liver sinusoidal endothelial cells. Cancer Lett 284:216–221
Ferry G, Tellier E, Try A et al (2003) Autotaxin is released from adipocytes, catalyzes lysophosphatidic acid synthesis, and activates preadipocyte proliferation. Up-regulated expression with adipocyte differentiation and obesity. J Biol Chem 278:18162–18169
Kanda H, Newton R, Klein R, Morita Y, Gunn MD, Rosen SD (2008) Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat Immunol 9:415–423
Giganti A, Rodriguez M, Fould B et al (2008) Murine and human autotaxin alpha, beta, and gamma isoforms: gene organization, tissue distribution, and biochemical characterization. J Biol Chem 283:7776–7789
Cooper AB, Wu J, Lu D, Maluccio MA (2007) Is autotaxin (ENPP2) the link between hepatitis C and hepatocellular cancer? J Gastrointest Surg 11:1628–1634
Watanabe N, Ikeda H, Nakamura K et al (2007) Both plasma lysophosphatidic acid and serum autotaxin levels are increased in chronic hepatitis C. J Clin Gastroenterol 41:616–623
Baumforth KR, Flavell JR, Reynolds GM et al (2005) Induction of autotaxin by the epstein-barr virus promotes the growth and survival of hodgkin lymphoma cells. Blood 106:2138–2146
Rubenstein M, Duvic M (2006) Cutaneous manifestations of Hodgkin's disease. Int J Dermatol 45:251–256
Gobbi PG, Attardo-Parrinello G, Lattanzio G, Rizzo SC, Ascari E (1983) Severe pruritus should be a B-symptom in Hodgkin's disease. Cancer 51:1934–1936
Balazs L, Okolicany J, Ferrebee M, Tolley B, Tigyi G (2001) Topical application of the phospholipid growth factor lysophosphatidic acid promotes wound healing in vivo. Am J Physiol Regul Integr Comp Physiol 280:R466–R472
Peyruchaud O (2009) Novel implications for lysophospholipids, lysophosphatidic acid and sphingosine 1-phosphate, as drug targets in cancer. Anticancer Agents Med Chem 9:381–391
Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2048
Botstein D, Risch N (2003) Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet 33(Suppl):228–237
Wang Y, O'Connell JR, McArdle PF et al (2009) From the cover: whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc Natl Acad Sci USA 106:226–231
Hakonarson H, Grant SF, Bradfield JP et al (2007) A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 448:591–594
Sladek R, Rocheleau G, Rung J et al (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445:881–885
(2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678
Waring SC, Rosenberg RN (2008) Genome-wide association studies in Alzheimer disease. Arch Neurol 65:329–334
Tregouet DA, Konig IR, Erdmann J et al (2009) Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet 41:283–285
Lander ES, Linton LM, Birren B et al (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921
(2003) The International HapMap Project. Nature 426:789–796
Gabriel SB, Schaffner SF, Nguyen H et al (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229
Sebastiani P, Lazarus R, Weiss ST, Kunkel LM, Kohane IS, Ramoni MF (2003) Minimal haplotype tagging. Proc Natl Acad Sci USA 100:9900–9905
Perry GH, Ben-Dor A, Tsalenko A et al (2008) The fine-scale and complex architecture of human copy-number variation. Am J Hum Genet 82:685–695
Redon R, Ishikawa S, Fitch KR et al (2006) Global variation in copy number in the human genome. Nature 444:444–454
McCarroll SA (2008) Extending genome-wide association studies to copy-number variation. Hum Mol Genet 17:R135–R142
McCarroll SA, Kuruvilla FG, Korn JM et al (2008) Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet 40:1166–1174
Korn JM, Kuruvilla FG, McCarroll SA et al (2008) Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet 40:1253–1260
Aitman TJ, Dong R, Vyse TJ et al (2006) Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature 439:851–855
Bae JS, Cheong HS, Kim JO et al (2008) Identification of SNP markers for common CNV regions and association analysis of risk of subarachnoid aneurysmal hemorrhage in Japanese population. Biochem Biophys Res Commun 373:593–596
Willer CJ, Speliotes EK, Loos RJ et al (2009) Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet 41:25–34
Perry GH, Dominy NJ, Claw KG et al (2007) Diet and the evolution of human amylase gene copy number variation. Nat Genet 39:1256–1260
Barrett JC, Clayton DG, Concannon P et al (2009) Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 41:703–707
Barrett JC, Hansoul S, Nicolae DL et al (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet 40:955–962
Mathew CG (2008) New links to the pathogenesis of Crohn disease provided by genome-wide association scans. Nat Rev Genet 9:9–14
Karlsen TH, Hov JR (2010) Genetics of cholestatic liver disease in 2010. Curr Opin Gastroenterol 26:251–258
Gaya DR, Russell RK, Nimmo ER, Satsangi J (2006) New genes in inflammatory bowel disease: lessons for complex diseases? Lancet 367:1271–1284
Buch S, Schafmayer C, Volzke H et al (2007) A genome-wide association scan identifies the hepatic cholesterol transporter ABCG8 as a susceptibility factor for human gallstone disease. Nat Genet 39:995–999
Sanna S, Busonero F, Maschio A et al (2009) Common variants in the SLCO1B3 locus are associated with bilirubin levels and unconjugated hyperbilirubinemia. Hum Mol Genet 18:2711–2718
Karlsen TH, Franke A, Melum E et al (2010) Genome-wide association analysis in primary sclerosing cholangitis. Gastroenterology 138:1102–1111
Hirschfield GM, Liu X, Xu C et al (2009) Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med 360:2544–2555
Ge D, Fellay J, Thompson AJ et al (2009) Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461:399–401
Yuan X, Waterworth D, Perry JR et al (2008) Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet 83:520–528
Daly AK, Donaldson PT, Bhatnagar P et al (2009) HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet 41:816–819
Invernizzi P, Selmi C, Poli F et al (2008) Human leukocyte antigen polymorphisms in Italian primary biliary cirrhosis: a multicenter study of 664 patients and 1992 healthy controls. Hepatology 48:1906–1912
Remmers EF, Plenge RM, Lee AT et al (2007) STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 357:977–986
Fung EY, Smyth DJ, Howson JM et al (2009) Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun 10:188–191
Hunt KA, Zhernakova A, Turner G et al (2008) Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet 40:395–402
Filipe-Santos O, Bustamante J, Chapgier A et al (2006) Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol 18:347–361
Selmi C, Gershwin ME (2009) The role of environmental factors in primary biliary cirrhosis. Trends Immunol 30:415–420
Airoldi I, Di Carlo E, Cocco C et al (2005) Lack of Il12rb2 signaling predisposes to spontaneous autoimmunity and malignancy. Blood 106:3846–3853
Pulickal AS, Hambleton S, Callaghan MJ et al (2008) Biliary cirrhosis in a child with inherited interleukin-12 deficiency. J Trop Pediatr 54:269–271
Liu X, Invernizzi P, Lu Y et al (2010) Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet 42:658–660
Bouzigon E, Corda E, Aschard H et al (2008) Effect of 17q21 variants and smoking exposure in early-onset asthma. N Engl J Med 359:1985–1994
Romero F, Martinez AC, Camonis J, Rebollo A (1999) Aiolos transcription factor controls cell death in T cells by regulating Bcl-2 expression and its cellular localization. EMBO J 18:3419–3430
Wang JH, Avitahl N, Cariappa A et al (1998) Aiolos regulates B cell activation and maturation to effector state. Immunity 9:543–553
Sun J, Matthias G, Mihatsch MJ, Georgopoulos K, Matthias P (2003) Lack of the transcriptional coactivator OBF-1 prevents the development of systemic lupus erythematosus-like phenotypes in Aiolos mutant mice. J Immunol 170:1699–1706
Garrett-Sinha LA, Su GH, Rao S et al (1999) PU.1 and Spi-B are required for normal B cell receptor-mediated signal transduction. Immunity 10:399–408
Hirschfield GM, Liu X, Han Y et al (2010) Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cirrhosis. Nat Genet 42:655–657
Raychaudhuri S, Remmers EF, Lee AT et al (2008) Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat Genet 40:1216–1223
Coenen MJ, Trynka G, Heskamp S et al (2009) Common and different genetic background for rheumatoid arthritis and coeliac disease. Hum Mol Genet 18:4195–4203
Cutolo M, Montagna P, Brizzolara R et al (2009) Sex hormones modulate the effects of Leflunomide on cytokine production by cultures of differentiated monocyte/macrophages and synovial macrophages from rheumatoid arthritis patients. J Autoimmun 32:254–260
Kochi Y, Suzuki A, Yamada R, Yamamoto K (2009) Genetics of rheumatoid arthritis: underlying evidence of ethnic differences. J Autoimmun 32:158–162
Kushida T, Ueda Y, Umeda M et al (2009) Allogeneic intra-bone marrow transplantation prevents rheumatoid arthritis in SKG/Jcl mice. J Autoimmun 32:216–222
Rojas-Villarraga A, Diaz FJ, Calvo-Paramo E et al (2009) Familial disease, the HLA-DRB1 shared epitope and anti-CCP antibodies influence time at appearance of substantial joint damage in rheumatoid arthritis. J Autoimmun 32:64–69
Selmi C, Mayo MJ, Bach N et al (2004) Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology 127:485–492
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403:41–45
Turner BM (2000) Histone acetylation and an epigenetic code. Bioessays 22:836–845
Yoshida M, Matsuyama A, Komatsu Y, Nishino N (2003) From discovery to the coming generation of histone deacetylase inhibitors. Curr Med Chem 10:2351–2358
Yang XJ (2004) The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res 32:959–976
Gregory PD, Wagner K, Horz W (2001) Histone acetylation and chromatin remodeling. Exp Cell Res 265:195–202
Kalkhoven E (2004) CBP and p300: HATs for different occasions. Biochem Pharmacol 68:1145–1155
Roth SY, Denu JM, Allis CD (2001) Histone acetyltransferases. Annu Rev Biochem 70:81–120
de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370:737–749
Michan S, Sinclair D (2007) Sirtuins in mammals: insights into their biological function. Biochem J 404:1–13
Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF (2003) Histone deacetylases: unique players in shaping the epigenetic histone code. Ann NY Acad Sci 983:84–100
Gardiner-Garden M, Frommer M (1987) CpG islands in vertebrate genomes. J Mol Biol 196:261–282
Illingworth RS, Bird AP (2009) CpG islands–‘a rough guide’. FEBS Lett 583:1713–1720
Hewagama A, Richardson B (2009) The genetics and epigenetics of autoimmune diseases. J Autoimmun 33:3–11
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Baylin SB, Herman JG (2000) DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet 16:168–174
Bannister AJ, Kouzarides T (2005) Reversing histone methylation. Nature 436:1103–1106
Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705
Wysocka J, Allis CD, Coonrod S (2006) Histone arginine methylation and its dynamic regulation. Front Biosci 11:344–355
Osley MA, Fleming AB, Kao CF (2006) Histone ubiquitylation and the regulation of transcription. Results Probl Cell Differ 41:47–75
Migeon BR (2007) Why females are mosaics, X-chromosome inactivation, and sex differences in disease. Gend Med 4:97–105
Sun BK, Tsao H (2008) X-chromosome inactivation and skin disease. J Invest Dermatol 128:2753–2759
Erwin JA, Lee JT (2008) New twists in X-chromosome inactivation. Curr Opin Cell Biol 20:349–355
Chow J, Heard E (2009) X inactivation and the complexities of silencing a sex chromosome. Curr Opin Cell Biol 21:359–366
Dementyeva EV, Shevchenko AI, Zakian SM (2009) X-chromosome upregulation and inactivation: two sides of the dosage compensation mechanism in mammals. Bioessays 31:21–28
Ezhkova E, Pasolli HA, Parker JS et al (2009) Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 136:1122–1135
Dupont C, Armant DR, Brenner CA (2009) Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med 27:351–357
Egger G, Liang G, Aparicio A, Jones PA (2004) Epigenetics in human disease and prospects for epigenetic therapy. Nature 429:457–463
Zhou Y, Lu Q (2008) DNA methylation in T cells from idiopathic lupus and drug-induced lupus patients. Autoimmun Rev 7:376–383
Yung RL, Richardson BC (1994) Drug-induced lupus. Rheum Dis Clin North Am 20:61–86
Maciejewska-Rodrigues H, Karouzakis E, Strietholt S et al (2010) Epigenetics and rheumatoid arthritis: the role of SENP1 in the regulation of MMP-1 expression. J Autoimmun 35:15–22
Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M (2009) DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 60:3613–3622
Neidhart M, Rethage J, Kuchen S et al (2000) Retrotransposable L1 elements expressed in rheumatoid arthritis synovial tissue: association with genomic DNA hypomethylation and influence on gene expression. Arthritis Rheum 43:2634–2647
Nile CJ, Read RC, Akil M, Duff GW, Wilson AG (2008) Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum 58:2686–2693
Takami N, Osawa K, Miura Y et al (2006) Hypermethylated promoter region of DR3, the death receptor 3 gene, in rheumatoid arthritis synovial cells. Arthritis Rheum 54:779–787
Liu X, Invernizzi P, Lu Y et al (2010) Genome-wide meta-analyses identifies three loci associated with primary biliary cirrhosis. Nat Genet 42(8):658–660
Invernizzi P, Selmi C, Mackay IR, Podda M, Gershwin ME (2005) From bases to basis: linking genetics to causation in primary biliary cirrhosis. Clin Gastroenterol Hepatol 3:401–410
Selmi C, Invernizzi P, Miozzo M, Podda M, Gershwin ME (2004) Primary biliary cirrhosis: does X mark the spot? Autoimmun Rev 3:493–499
Fraga MF, Ballestar E, Paz MF et al (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 102:10604–10609
Jarvinen P, Aho K (1994) Twin studies in rheumatic diseases. Semin Arthritis Rheum 24:19–28
Mitchell MM, Lleo A, Zammataro L et al (2011) Epigenetic investigation of variably X chromosome inactivated genes in monozygotic female twins discordant for primary biliary cirrhosis. Epigenetics 6(1)
Invernizzi P, Miozzo M, Battezzati PM et al (2004) Frequency of monosomy X in women with primary biliary cirrhosis. Lancet 363:533–535
Kaplan MM, Gershwin ME (2005) Primary biliary cirrhosis. N Engl J Med 353:1261–1273
Oertelt S, Rieger R, Selmi C et al (2007) A sensitive bead assay for antimitochondrial antibodies: chipping away at AMA-negative primary biliary cirrhosis. Hepatology 45:659–665
Lleo A, Invernizzi P, Mackay IR, Prince H, Zhong RQ, Gershwin ME (2008) Etiopathogenesis of primary biliary cirrhosis. World J Gastroenterol 14:3328–3337
Moteki S, Leung PS, Dickson ER et al (1996) Epitope mapping and reactivity of autoantibodies to the E2 component of 2-oxoglutarate dehydrogenase complex in primary biliary cirrhosis using recombinant 2-oxoglutarate dehydrogenase complex. Hepatology 23:436–444
Leung PS, Chuang DT, Wynn RM et al (1995) Autoantibodies to BCOADC-E2 in patients with primary biliary cirrhosis recognize a conformational epitope. Hepatology 22:505–513
Gershwin ME, Mackay IR, Sturgess A, Coppel RL (1987) Identification and specificity of a cDNA encoding the 70 kd mitochondrial antigen recognized in primary biliary cirrhosis. J Immunol 138:3525–3531
Ichiki Y, Selmi C, Shimoda S, Ishibashi H, Gordon SC, Gershwin ME (2005) Mitochondrial antigens as targets of cellular and humoral auto-immunity in primary biliary cirrhosis. Clin Rev Allergy Immunol 28:83–91
Agmon-Levin N, Shapira Y, Selmi C et al (2010) A comprehensive evaluation of serum autoantibodies in primary biliary cirrhosis. J Autoimmun 34:55–58
Bruggraber SF, Leung PS, Amano K et al (2003) Autoreactivity to lipoate and a conjugated form of lipoate in primary biliary cirrhosis. Gastroenterology 125:1705–1713
Rieger R, Leung PS, Jeddeloh MR et al (2006) Identification of 2-nonynoic acid, a cosmetic component, as a potential trigger of primary biliary cirrhosis. J Autoimmun 27:7–16
Gershwin ME, Mackay IR (2008) The causes of primary biliary cirrhosis: convenient and inconvenient truths. Hepatology 47:737–745
Alpini G, Lenzi R, Sarkozi L, Tavoloni N (1988) Biliary physiology in rats with bile ductular cell hyperplasia.Evidence for a secretory function of proliferated bile ductules. J Clin Invest 81:569–578
Alvaro D, Cho WK, Mennone A, Boyer JL (1993) Effect of secretion on intracellular pH regulation in isolated rat bile duct epithelial cells. J Clin Invest 92:1314–1325
Harada K, Isse K, Nakanuma Y (2006) Interferon gamma accelerates NF-kappaB activation of biliary epithelial cells induced by Toll-like receptor and ligand interaction. J Clin Pathol 59:184–190
Saidman SL, Duquesnoy RJ, Zeevi A, Fung JJ, Starzl TE, Demetris AJ (1991) Recognition of major histocompatibility complex antigens on cultured human biliary epithelial cells by alloreactive lymphocytes. Hepatology 13:239–246
Fava G, Glaser S, Francis H, Alpini G (2005) The immunophysiology of biliary epithelium. Semin Liver Dis 25:251–264
Savill J, Dransfield I, Gregory C, Haslett C (2002) A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol 2:965–975
Ravichandran KS, Lorenz U (2007) Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol 7:964–974
Torok NJ (2007) Apoptotic cell death takes its toll. Hepatology 46:1323–1325
Perniok A, Wedekind F, Herrmann M, Specker C, Schneider M (1998) High levels of circulating early apoptic peripheral blood mononuclear cells in systemic lupus erythematosus. Lupus 7:113–118
Salunga TL, Cui ZG, Shimoda S et al (2007) Oxidative stress-induced apoptosis of bile duct cells in primary biliary cirrhosis. J Autoimmun 29:78–86
Allina J, Hu B, Sullivan DM et al (2006) T cell targeting and phagocytosis of apoptotic biliary epithelial cells in primary biliary cirrhosis. J Autoimmun 27:232–241
Lleo A, Invernizzi P, Selmi C et al (2007) Autophagy: highlighting a novel player in the autoimmunity scenario. J Autoimmun 29:61–68
Schiller M, Bekeredjian-Ding I, Heyder P, Blank N, Ho AD, Lorenz HM (2008) Autoantigens are translocated into small apoptotic bodies during early stages of apoptosis. Cell Death Differ 15:183–191
Mandron M, Martin H, Bonjean B, Lule J, Tartour E, Davrinche C (2008) Dendritic cell-induced apoptosis of human cytomegalovirus-infected fibroblasts promotes cross-presentation of pp 65 to CD8+ T cells. J Gen Virol 89:78–86
Nagata S, Hanayama R, Kawane K (2010) Autoimmunity and the clearance of dead cells. Cell 140:619–630
Odin JA, Huebert RC, Casciola-Rosen L, LaRusso NF, Rosen A (2001) Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J Clin Invest 108:223–232
Combes B, Emerson SS, Flye NL et al (2005) Methotrexate (MTX) plus ursodeoxycholic acid (UDCA) in the treatment of primary biliary cirrhosis. Hepatology 42:1184–1193
Van de Water J, Gerson LB, Ferrell LD (1996) Immunohistochemical evidence of disease recurrence after liver transplantation for primary biliary cirrhosis. Hepatology 24:1079–1084
Chamulitrat W, Burhenne J, Rehlen T, Pathil A, Stremmel W (2009) Bile salt-phospholipid conjugate ursodeoxycholyl lysophosphatidylethanolamide as a hepatoprotective agent. Hepatology 50:143–154
Amaral JD, Viana RJ, Ramalho RM, Steer CJ, Rodrigues CM (2009) Bile acids: regulation of apoptosis by ursodeoxycholic acid. J Lipid Res 50:1721–1734
Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RA, Henson PM (2000) A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 405:85–90
Henson PM (2005) Dampening inflammation. Nat Immunol 6:1179–1181
Huynh ML, Fadok VA, Henson PM (2002) Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest 109:41–50
Casciola-Rosen LA, Anhalt G, Rosen A (1994) Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med 179:1317–1330
Amano K, Leung PS, Rieger R et al (2005) Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J Immunol 174:5874–5883
Lleo A, Selmi C, Invernizzi P et al (2009) Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology 49:871–879
Long SA, Quan C, Van de Water J (2001) Immunoreactivity of organic mimeotopes of the E2 component of pyruvate dehydrogenase: connecting xenobiotics with primary biliary cirrhosis. J Immunol 167:2956–2963
Lleo A, Bowlus CL, Yang GX et al (2010) Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology 52:987–998
Takeda K, Kojima Y, Ikejima K et al (2008) Death receptor 5 mediated-apoptosis contributes to cholestatic liver disease. Proc Natl Acad Sci USA 105:10895–10900
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Folci, M., Meda, F., Gershwin, M.E. et al. Cutting-Edge Issues in Primary Biliary Cirrhosis. Clinic Rev Allerg Immunol 42, 342–354 (2012). https://doi.org/10.1007/s12016-011-8253-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-011-8253-3