
Abstract
Excessive Th2 cell signaling and IgE production play key roles in the pathogenesis of atopic dermatitis (AD). Yet, recent information suggests that the inflammation in AD instead is initiated by inherited insults to the barrier, including a strong association between mutations in FILAGGRIN and SPINK5 in Netherton syndrome, the latter of which provides an important clue that AD is provoked by excess serine protease activity. But acquired stressors to the barrier may also be required to initiate inflammation in AD, and in addition, microbial colonization by Staphylococcus aureus both amplifies inflammation, but also further stresses the barrier in AD. Therapeutic implications of these insights are as follows: While current therapy has been largely directed toward ameliorating Th2-mediated inflammation and/or pruritus, these therapies are fraught with short-term and potential long-term risks. In contrast, “barrier repair” therapy, with a ceramide-dominant triple-lipid mixture of stratum corneum lipids, is more logical, of proven efficacy, and it provides a far-improved safety profile.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Both a defective epidermal permeability barrier [1–4] and a propensity to develop secondary infections [5] are well-recognized features of atopic dermatitis (AD). While it has been previously assumed that these abnormalities reflect Th2-driven immunologic abnormalities (the historical “inside-to-outside” view of AD pathogenesis), we and others have long proposed that the barrier abnormality is not merely a secondary phenomenon but rather the “driver” of disease activity in AD (“outside-to-inside” view of disease pathogenesis) [6–8] because it was well-known that (1) the extent of the permeability barrier abnormality parallels severity of disease phenotype in AD [1, 2, 4]; (2) clinically uninvolved skin sites, as well as skin cleared of inflammation for ≤5 years, continue to display barrier abnormalities [2]; (3) emollient therapy comprises effective ancillary therapy for AD [9]; and finally, (4) as will be discussed further below, specific lipid replacement therapy, which targets the prominent lipid abnormalities that account for the barrier abnormality [6, 9], not only corrects the barrier abnormality but also ameliorates inflammation in AD.
In this review, we will first provide relevant background information about normal epidermal barrier structure and function. Second, we will update recent information about how inherited defects in either filaggrin (FLG) and/or the serine protease inhibitor, LEKTI 1, contribute to the development of AD. Third, we will explain the multiple mechanisms whereby a primary barrier abnormality in AD can lead to inflammation. Fourth, we will explore how certain acquired stressors, such as a reduced external humidity, high pH soaps, and surfactants, psychological stress, as well as secondary Staphylococcus aureus infections, initiate or further aggravate AD. Finally and most importantly, we will compare various therapeutic paradigms for AD, highlighting the risks and benefits of glucocorticoids (GC), immunomodulators, and corrective lipid replacement therapy.
Barrier Functions of Normal Skin
The outer epidermis generates a set of protective and sensory functions (Table 1), in part attributable largely to its anucleate, but metabolically active, differentiation end-product, the stratum corneum (SC) [10]. These defensive functions include (1) the permeability barrier, which both retards transcutaneous evaporative water loss in a potentially desiccating external environment; (2) an antimicrobial barrier, which simultaneously encourages colonization by non-pathogenic “normal” flora, while resisting growth and invasion by microbial pathogens [11]; (3) multiple other, key protective functions, such as antioxidant and defense against ultraviolet irradiations; and finally, (4) recently appreciated, biosensory functions clearly place the epidermis as the distal outpost of the nervous system [12], as well as performing important regulatory and signaling functions within the epidermis itself.
The SC comprises a multilayered tissue composed of vertically stacked arrays of anucleate corneocytes, embedded in a hydrophobic extracellular matrix filled with multilayers of planar lamellar bilayers, enriched in a family of at least 10 ceramides (Cer), cholesterol, and both essential and non-essential free fatty acids (FFA). These lipids are delivered to the SC interstices as their precursors (i.e., glucocylceramides, sphingomyelin, cholesterol sulfate, and phospholipids) through secretion of the epidermal lamellar body (LB) contents (Fig. 1). However, this epidermis-unique organelle delivers not only lipid precursors but also lipid hydrolases (β-glucocerebrosidase, acidic sphingomylinase, secretory phospholipase A2, and steroid sulfate) that generate Cer, cholesterol, and FFA, which then self-organize into lamellar membranes. In addition, lamellar body-derived proteases/anti-proteases orchestrate the orderly digestion of epidermis-unique, junctional structures, corneodesmosomes, allowing invisible shedding of corneocytes from the skin surface [13, 14]. Pertinent to distal innate immunity, certain antimicrobial peptides (i.e., the carboxy terminal fragment of human cathelicidin (hCAP)18 (LL-37) and human β-defensin 2) also are delivered to the SC intercellular domains through secretion of LB contents [15, 16].

Lamellar body secretion delivers key components of both permeability and antimicrobial barriers (modified from Elias [11])
Inherited Barrier Abnormalities in Atopic Dermatitis
Relationship of Ichthyosis Vulgaris to Atopic Dermatitis:
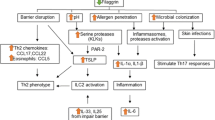
Preliminary studies in genotyped AD subjects and in FLG-deficient mouse models [17, 18] suggest that the phenotype in both ichthyosis vulgaris (IV) and AD is linked to an underlying abnormality in permeability barrier homeostasis [19]. Moreover, as in AD, the pH of SC is elevated in IV [19, 20], and the increase in pH, in turn, could activate serine proteases (kallikreins), with a host of negative downstream consequences, including (a) PAR2-mediated blockade of lamellar body secretion [21, 22], (b) possible downstream alterations in keratin filament organization that could impede lamellar body secretion (see below), and (c) both Th1- and KLK5-activated Th2 inflammation [23] (Figs. 2 and 3). According to this view, while the primary phenotype in IV is one of scaling, it also represents the forme fruste of AD, displaying clinical inflammation only when affected skin is either exposed to sustained antigen ingress and/or to additional acquired stressors to the barrier (e.g., high pH surfactants, exposure to a reduced external humidity, or sustained psychological stress).

Relationship of ichthyosis vulgaris and Netherton syndrome to atopic dermatitis

Mechanisms whereby filaggrin (FLG) deficiency in IV could predispose to the development of atopic dermatitis
The strongest evidence that a primary structural abnormality of SC underlies the pathogenesis of AD derives from the recent link between loss-of-function mutations in the gene encoding, filament aggregating protein (FLG), and AD [24, 25]. Up to 60% of Europeans with AD reveal single- or double-allele mutations in FLG on chromosome 1q21. FLG is the main component of F-type keratohyalin granules, responsible for the designation of the stratum granulosum. Decreased FLG expression results in a paucity of keratohyalin granules, a hallmark of IV [26] and reduced FLG is also common in AD [3, 27, 28]. Accordingly, IV is associated with concomitant AD, allergic rhinitis, and/or asthma in approximately two thirds of patients [3].
FLG deficiency has been ascribed to both nonsense and frame shift mutations. Although more than 20 different mutations have been reported, six of them account for the majority of European cases [29, 30]. Most FLG mutations result in truncation of pro-FLG, explaining reduced-to-absent FLG expression in the epidermis of IV/AD. While heterozygous patients show residual FLG with a milder phenotype, IV patients with homozygous or compound heterozygous mutations lack FLG and exhibit generalized scaling, as well as an increased propensity to develop severe and persistent AD (op. cit.).
The initial product of FLG translation is pro-FLG, a large, histidine-rich, highly cationic phosphoprotein, consisting of ten-to-12 FLG repeats, enriched in hydrophobic amino acids [31–33]. Pro-FLG contains an amino-terminal sequence, including a calcium-binding A domain; the B domain is a putative S100-like, calcium-binding domain. In contrast to the cytoplasmatic localization of C-terminal FLG monomers, the N terminus of pro-FLG appears to tether to the nucleus via its nuclear localization sequence. In normals, pro-FLG is dephosphorylated and proteolytically processed to FLG monomers during cornification. Processed FLG peptides then induce aggregation of keratins within the corneocyte cytosol and attach to the cornified envelope (CE), a unique structure that replaces the plasma membrane as granular cells transform into corneocytes [34, 35]. The CE provides a relatively inflexible, mechanically resistant barrier (Table 1). However, as the water content of the SC drops in the mid-to-outer SC, FLG detaches from the CE, to the C-terminal portion of FLG is proteolyzed into its constituent amino acids, followed by their deimination into polycarboxylic acids (= “natural moisturizing factors”) [36, 37], such as pyrrolidine carboxylic acid and trans-urocanic acid (t-UCA) [38] (Fig. 4). These metabolites, in turn, act as osmolytes, drawing water into corneocytes, thereby accounting in large part for corneocyte hydration.

Filaggrin proteolytic pathway impacts multiple SC functions: potential implications for pathogenesis of AD (modified from Elias et al. [70])
While it is widely hypothesized that FLG deficiency provokes a permeability barrier abnormality [25], until very recently, the cellular basis for such an abnormality is unknown. Indeed, abnormal permeability barrier function was noted in IV patients, lacking AD, in the era prior to genetic FLG analysis [39–41]. But Hubiche et al. failed to find defective barrier function in IV [42], thereby challenging the prevailing hypothesis that FLG deficiency causes an impaired barrier to transcutaneous water loss. Our recent studies confirm that both single- and double-allele IV patients display a barrier abnormality [43]. Yet, how loss of FLG (an intracellular protein) provokes a permeability barrier abnormality (almost always an extracellular defect) is only now becoming clear (Figs. 2 and 3). One possibility is that loss of this quantitatively important protein could alter corneocyte shape, perhaps inducing flattening that could disrupt extracellular lamellar bilayer organization. Alternatively, our recent results suggest that decreased FLG cornified envelope integrity, contributing to poor SC integrity. Moreover, our very recent studies suggest yet another mechanism, i.e., that unprocessed pro-FLG interferes with loading of lamellar body contents, as well as secretion [44]. But the most immediate result of FLG deficiency in AD likely is decreased SC hydration, which leads to a steeper water gradient across the SC, thereby “driving” increased transcutaneous water loss. Thus, decreased SC hydration, leading to increased water loss, is likely an important cause of barrier dysfunction in FLG-deficient AD.
Yet, none of these mechanisms alone suffices to explain enhanced antigen penetration in AD, which could best explained by yet another consequence of FLG deficiency, i.e., decreased downstream production of acidic metabolites resulting from FLG proteolysis (Figs. 3 and 4). Indeed, t-UCA, in particular, is a purported, endogenous acidifier of the SC [45]. Thus, decreased generation of FLG products could result in an initial increase in the SC pH, sufficient activate multiple SP in SC, which all exhibit neutral-to-alkaline pH optima [14]. Such a pH-induced increase in SP activity, if prolonged, could precipitate multiple downstream structural and functional alterations, including Th2 inflammation in AD (see below).
Relationship of Netherton Syndrome and Atopic Dermatitis:
Netherton syndrome (NS) always presents with concurrent, severe AD, including elevated IgE levels, mucosal atopy, anaphylactic food reactions, and a characteristic hairshaft abnormality, trichorrhexis invaginata (“bamboo hair”). Because of the severity of AD, an exploration of pathogenic mechanisms in NS could provide further insights into AD pathogenesis (Fig. 5). NS is associated with loss-of-function mutations in SPINK5, which encodes the serine protease inhibitor, lymphoepithelial kazal-type inhibitor 1 (LEKTI) [46]. Diminished LEKTI 1 activity in NS likely leads to cutaneous inflammation by multiple mechanisms. First, because serine proteases activate IL-1α/β in corneocytes [47], initiating the “cytokine cascade” [48], and second, an acquired deficiency of LEKTI 1, due to proteolytic consumption of the inhibitor, could also contribute to the pathogenesis of AD [49] (Fig. 2). Third, KLK5 can activate Th2 cytokines, independent of allergen exposure [23]. Moreover, in some population studies, polymorphisms in the SPINK5 gene (missense variants) also are associated with AD and mucosal atopy [50]. Yet several control studies have been found with an increased frequency of single nucleotide polymorphisms (Glu420Lys) in SPINK5 [50], recent studies cast doubts upon this association. Likewise, a British case–control study describing putative, gain-of-function polymorphisms (AACCAACC vs. AACC) in the 3′ region of KLK7, which encodes the SP, SC chymotryptic enzyme (KLK7) [51], is now disputed. Furthermore, in a recent genetic study involving 2,500 AD cases and 10,000 controls, there was no evidence for an epistatic (additive) interaction between SPINK5/KLK7 polymorphisms and filaggrin mutations [52]. Yet, transgenic mice that express human KLK7 display a severe AD-like dermatosis. Moreover, in experimental animals, a net increase in SP activity, achieved by a variety of means, has been shown to compromise barrier function through accelerated degradation of both corneodesmosomes (accounting for flawed SC integrity) and lipid-processing enzymes [53] (Fig. 3), resulting in a failure to generate Cer, a characteristic lipid abnormality in AD [54, 55].

Pathogenesis of Netherton syndrome
Residual LEKTI expression in NS correlates inversely with the extent of SP activation within the outer epidermis [56], resulting in a gene dose-dependent permeability barrier defect [56], due to unrestricted, SP-dependent degradation of lipid-processing enzymes. Moreover, excess SP activity leads to dramatic thinning of SC, due to degradation of corneodesmosome-constituent proteins [56].
In addition to KLK5 upregulation of Th2 inflammation via signaling of the plasminogen activator type 2 receptor (PAR2), elevated SP activity likely provokes the barrier abnormality via PAR2 because SP binding to PAR2 downregulates LB secretion [57], entombing some of these organelles in nascent corneocytes [58]. Failure of LB secretion accounts, in turn, for the global decrease in SC lipids in AD [3, 59], which correlates with a decrease in extracellular lamellar bilayers in AD [4]. Thus, increased SP activity provides a mechanistic basis for the immunologic and lipid abnormalities, as well as the further decline in Cer levels that occur in AD (Fig. 3).
Consequences of a Flawed Antimicrobial Barrier in Atopic Dermatitis
The antimicrobial barrier also is compromised in AD, commonly leading to colonization of lesional and non-lesional skin by S. aureus [5], initiating yet another series of vicious cycle in AD (Fig. 6). Although colonization is most often not apparent, impetiginization, widespread folliculitis, or less frequently cutaneous abscesses or cellulitis are well-recognized complications in AD. Moreover, superantigen producing S. aureus strains colonize AD more commonly in steroid-resistant patients [60] and further exacerbate disease in AD through augmentation of IgE production, as well as through stimulation of IgE, specifically directed toward epidermal structural proteins (review in [19]). Over time, non-toxigenic strains of S. aureus that colonize AD can be replaced by enterotoxin-generating strains [61], which in turn could aggravate AD by at least three mechanisms (Fig. 6): (1) toxigenic strains are more likely to produce clinical infections than are non-toxigenic strains [61], (2) some toxins stimulate pruritus [62] and production of specific IgE [5, 63], and (3) some toxins serve as “superantigens” that stimulate T and B cell proliferation, as well as immunoglobulin class-switching to allergen-specific or “superallergens” that stimulate IgE production [5]. Activated T cells produce IL-31, which also induces pruritus [64]. Finally, clinical infections, particularly folliculitis, are notoriously pruritic, even in non-atopics, eliciting an “itch-scratch” vicious cycle that creates additional portals of entry for pathogens. It is self-evident that excoriations create further defects in the permeability barrier, representing yet another potentially important vicious cycle in AD pathogenesis. Finally, patients with atopic dermatitis are also susceptible to widespread cutaneous viral infections, including molluscum contagiosum, Herpes simplex (Kaposi’s varicelliform eruption), and life-threatening Vaccinia infections. Widespread dermatophytosis (tinea corporis) and Malassezia infections also occur in AD and the latter can also stimulate specific IgE production. Together, these observations point to loss of a competent antimicrobial barrier in AD. While failure of both permeability and antimicrobial function is well-recognized in AD, only recently has it become clear that these two functions share common structural and biochemical features [11], and both are co-regulated and interdependent [65] (Table 2). Thus, failure of the permeability barrier in itself favors secondary infection, and conversely, pathogen colonization/infection further aggravates the permeability barrier abnormality.

Secondary infections further aggravate barrier abnormality in AD (modified from Elias et al. [70])
Increased colonization with S. aureus [66] occurs both as a result of the barrier abnormality (a structurally competent, lipid-replete, acidic SC itself comprises a formidable barrier to pathogen colonization [11], and it can further aggravate barrier function in AD by several mechanisms; Fig. 6). The antimicrobial barrier is intimately linked to the permeability barrier [65], and as with water egress, pathogen ingress occurs via the extracellular domains [67]. Moreover, an impaired permeability barrier alone predisposes to pathogen colonization, not only because of the increase in surface pH but also because levels of FFA and the Cer metabolite, sphingosine, which exhibit potent in vitro antimicrobial activity [67, 68], decline in AD [11]. Surface proteins on S. aureus can downregulate epidermal FFA production, thereby aggravating both permeability and antimicrobial function in parallel, a strategy that could also facilitate microbial invasion. In addition, at least one member of epidermal antimicrobial peptides, the hCAP product, LL-37, is downregulated in a Th2-dependent fashion in AD [66, 69], and LL-37 is required for normal permeability barrier function [65]. Notably, LL-37 also displays robust activity against S. aureus and is required not only for normal epidermal permeability barrier function but also for the integrity of extracutaneous epithelia. Thus, it is likely that decreased LL-37 amplifies the barrier defect in AD.
Exogenous and Endogenous Stressors Further Aggravate Barrier Function in AD
That FLG mutations alone do not suffice is shown in IV, where the same single- or double-allele FLG mutations reduce FLG content, but inflammation (i.e., AD) does not inevitably occur. We and others have repeatedly proposed that certain stressors aggravate the barrier abnormality [70–72]. One or more of these stressors likely provoke an incremental increase in pH of the SC, leading to a further amplification of SP activity (Fig. 3). For example, pH-dependent increases in SP activity likely accounts for the precipitation of AD following the use of neutral-to-alkaline soaps [73].
Prolonged exposure to a reduced environmental humidity, as occurs in radiant-heated homes in temperate climates during the winter, is also a well-known risk factor for AD. Under these conditions, transcutaneous water loss would accelerate across a defective SC, aggravating the underlying hydration and permeability barrier abnormalities and amplifying cytokine signaling of inflammation. Because FLG proteolysis is regulated by changes in external humidity [38], sustained reductions in environmental relative humidity could further deplete residual FLG in single-allele FLG-deficient patients. Finally, sustained psychological stress aggravates permeability barrier function in humans [74], and PS is both a well-known precipitant of AD and cause of resistance to therapy. In experimental animals, PS induces an increase in endogenous GC, which in turn alter permeability barrier homeostasis, SC integrity, and epidermal antimicrobial defense [70, 71]. The putative mechanism for the negative effects of PS is GC-mediated inhibition of synthesis of the three key epidermal lipids that mediate barrier function, i.e., Cer, cholesterol, and FFA. Accordingly, a topical mixture of these three lipids largely normalizes all of these functions, even in the face of ongoing PS or GC therapy [75, 76], and should comprise particularly effective therapy for AD patients with unusual levels of stress.
Basis for Inflammation in AD
As noted above, one important downstream determinant of cutaneous inflammation in AD results from increased SP activity, which activates the primary cytokines, IL-1α and IL-1β [47], from their 33-kDa pro-forms that are stored in large quantities in the cytosol of corneocytes. The putative pH-induced increase in SP activity would generate 17-kDa active forms of these cytokines [47], the first step in the cytokine cascade that we have long proposed is an important contributor to inflammation in AD [6, 7]. Sustained antigen ingress through a defective barrier leading to a Th2-dominant infiltrate then would be a second cause of inflammation in AD [70] (Fig. 3). Certain antigens, such as cat dander, mites, and cockroach antigens, are preferentially associated with AD and are frequent triggers of AD, particularly in FLG-deficient patients [77]. Mites themselves activate SP activity with further damage to the barrier [78]. Yet, the damaged barrier in AD is due to lipid depletion, which also explains the preferential penetration of water-soluble haptens, such as nickel, in AD [79]. Accordingly, correction of the barrier abnormality alone should ameliorate both the cytokine cascade and allergen-induced inflammation in AD.
Despite accumulating evidence in support of a barrier-initiated pathogenesis of AD, recent studies suggest further mechanisms whereby Th2-generated cytokines could also further aggravate AD [70]. Exogenous application of the Th2 cytokines, IL-4 or IL-13, impede permeability barrier homeostasis in experimental animals [80]. The basis for these negative effects of IL-4 includes (1) inhibition of Cer synthesis [81], providing yet another mechanism accounting for decreased Cer in AD; (2) inhibition of keratinocyte differentiation-linked proteins, most notably loricrin and FLG [27]; and (3) decreased desmoglein 3 expression, which would further compromise SC integrity (cited in [80, 81]). In a further “vicious cycle,” serum IgE from AD patients auto-reacts against a variety of keratinocyte antigens [82]. Together, these observations provide acquired mechanisms that could further compromise barrier function in AD [27]. Thus, primary inherited barrier abnormalities in AD ultimately stimulate downstream paracrine mechanisms that further compromise permeability barrier function, completing a potential “outside–inside–outside” pathogenic loop in AD (Figs. 3 and 4).
Therapeutic Implications
The evidence cited above creates a powerful rationale that measures aimed at barrier repair should prevent and/or ameliorate the inflammatory disease component in AD and could break the vicious cycle of inflammation-induced barrier impairment [27]. Yet, anti-inflammatory steroids and immunomodulators remain the mainstay of therapy in AD. We provide below some reasons for concern with over-reliance on these agents.
Immunosuppressive Therapy—Efficacy and Concerns:
Topical macrolactin immunosuppressants (IS), deployed in the 1980s as systemic therapy to prevent organ transplant rejection, display modest anti-inflammatory activity, comparable to low-to-mid-potency steroids. But IS continue to be prescribed as an alternate to topical steroids because of their presumed safety. Yet, despite many protestations from the dermatologic community that topical IS are safe, concerns continue to be raised. The result of initial concerns was the still-controversial “Black Box” warning, which has dampened enthusiasm for prescribing IS among many practitioners. Summarized below are the FDA’s concerns that lead to the imposition of the “Black Box” warning.
Off-Label Prescribing:
The direct-to-consumer and physician-to-provider promotional activities that accompanied the marketing of Elidel® and Protopic® lead to widespread off-label prescribing of these agents for children with AD under the age of 2 years [83]. This market-driven effort prompted an FDA Advisory Group, convened in 2005, to reconsider the risk/benefit profile of these agents and to re-establish their appropriate role in the treatment of AD [83, 84]. Even critics of the FDA’s imposition of black box warnings on Elidel® and Protopic® acknowledge the strong causal link between systemic (internal) use of these agents and the development of both lymphatic cancers (lymphomas) and UV-induced skin cancers [85]. The current controversy centers on whether long-term, topical use of these immunosuppressive drugs also presents a risk for the development of lymphoma or other cancers. In contrast, the advisory group noted that in contrast, long-term use of topical steroids poses no increased risk of either lymphoma or sun-induced skin cancer [86].
Misleading Blood Level Data:
Although the average blood concentrations of patients using topical Elidel® and Protopic® are much lower than blood levels of these drugs when administered to organ transplant recipients, some patients, particularly small children with severe AD, demonstrate high blood levels that occasionally persist with prolonged use [87] (Fig. 7 [from FDA Advisory Group]). Some of these patients may have previously undiagnosed NS, where toxic blood levels can occur following topical applications of tacrolimus [87, 88], due to a highly defective barrier [89]. Thus, when tacrolimus or pimecrolimus are employed as either maintenance or preventive therapy, rather than for short-term use, as currently approved, physicians should consider checking blood levels periodically in order to identify patients at risk for unusually high blood levels.

Tacrolimus blood levels in adults and children (6–12 year) following application of 0.1% ointment
Tissue vs. Blood Levels:
The FDA Advisory Group also noted that blood levels do not necessarily reflect tissue levels of these drugs. Unfortunately, neither Novartis nor Astellas provided information about drug levels in either skin or regional lymph nodes, though it was available from their preclinical studies. The stated reason was that this information was not requested by the FDA. Despite very low blood levels, it is possible that absorbed drugs could move through the lymphatics to regional lymph nodes, where lymphomas occur not infrequently in animals following topical use of these agents.
Tumors at or Near Sites of Topical Applications:
Some of the cancers reported to the FDA have developed either directly at sites of prolonged Elidel® or Protopic® applications (most commonly squamous cell carcinomas or lymphomas in draining, regional lymph nodes; Table 3). Importantly, some of the reported cases of “recalcitrant” eczema proved not to be eczema, but malignant or premalignant disorders, such as Bowen disease, parapsoriasis en plaque, or Paget disease of the nipple. Therefore, it would be advisable to biopsy and diagnose “recalcitrant” eczematous lesions prior to initiating therapy with topical immunomodulators.
Relation of Length of Therapy/Total Absorbed Dosage to Tumor Development:
Some of the tumors reported to the FDA and others that the author has since become aware of have been linked to both the total applied dose and duration of Elidel® or Protopic® therapy. Thus, the more prolonged IS drug use and the higher the drug concentration (e.g., 0.3% vs. 0.1% tacrolimus) could increase the likelihood of developing cancer. One must also question the wisdom, if not the ethics, of clinical trials that are investigating the utility of topical IS for the long-term treatment of seborrheic dermatitis, or the prevention of atopic dermatitis [90, 91], i.e., the “atopic march” [92], since it will take several more years before the issue of long-term safety is resolved.
UV-Induced Skin Cancers:
Long-term use of systemic IS, including cyclosporin and tacrolimus, is associated with an increased risk of both melanoma and non-melanoma skin cancer [93]. Yet, even the current package inserts for Protopic® and Elidel® are vague in dealing with this potential concern, despite several reports of multiple lentigines [94], melanoma, squamous cell carcinomas, and basal cell carcinoma at sites of prolonged applications to sun-exposed skin (Table 3). Although the package inserts advise caution in the use of these agents in sun-exposed skin, they provide no specific recommendations to either prescribing physicians or patients about the use of protective clothing, sun-avoidance, or sunscreens. As a result, off-label prescribing for peri-oral/peri-orbital eczema and seborrheic dermatitis is now common, fueled by reports of efficacy in rosacea and seborrheic dermatitis [95, 96]. While the lengthy latency period for the development of non-melanoma skin cancers suggests that the short-term use of these agents should not present a problem even in sun-exposed skin, either melanoma and/or non-melanoma skin cancers could arise with long-term, topical therapy of dermatoses in sun-exposed areas. Yet, if these agents are applied to small, premalignant lesions, such as actinic keratoses or Bowen disease, they could transform into cancers, even after only short-term therapy.
Infectious Complications:
The risk of serious and life-threatening infections, due to prolonged immunosuppression, has been down-played in recent review articles [91, 97, 98]. Yet, both topical pimecrolimus and tacrolimus compromise permeability barrier function in normal skin [99], and defects in permeability barrier function could predict reduced antimicrobial defense [100]. Thus, compromised barrier function could provide an explanation for the not infrequent reports of viral infections, including eczema herpeticum and molluscum contagiosum [101–103], in earlier trials with topical tacrolimus [91, 104, 105]. Furthermore, the overall incidence of respiratory/gastrointestinal and cutaneous streptococcal/staphylococcal infection reportedly is increased by about 2-fold in patients receiving topical IS (op. cit.). To circumvent this potential pitfall, the package inserts rather misleadingly lump together non-life-threatening infections with more serious infections, under broad, banal-sounding categories, such as “skin, GI and respiratory infections.” This somewhat disingenuous marketing approach could mask the actual incidence of more serious infectious complications in these patients.
Rationale for Barrier Repair Therapy
Even though anti-inflammatory therapies effectively reduce disease severity by suppressing immune function in AD, they do not address the primary underlying barrier abnormality that “drives” disease pathogenesis [70].Footnote 1 As noted above, recent studies have shown that topical immunomodulators compromise barrier function in normal skin [99], as do topical glucocorticoids [76, 106]. Thus, as AD improves, continued topical applications of steroids and IS likely will increasingly compromise barrier function. In contrast, studies in both AD animal models and in patients show that barrier repair interventions reduce the inflammatory component of disease by promoting normal epidermal function [70, 72, 107, 108].
The rationale for barrier repair therapy goes beyond trapping moisture and preventing dry skin because of the numerous other protective functions of the outermost skin layer (SC), including its antimicrobial function (Table 1). The “pro-inflammatory” functions of the SC begin with the large pool of pre-formed IL-1α and IL-1β, which are stored in large quantities in corneocytes. These molecules are released into the lower epidermis and dermis when barrier function is perturbed, helping to normalize function in normal skin. Since barrier is always abnormal in AD, these repair mechanisms are unsuccessful in AD. Hence, the cytokine cascade is sustained, resulting in the downstream recruitment of additional, pro-inflammatory molecules (the “outside-to-inside” concept of AD pathogenesis). In addition to the ongoing cytokine cascade, repeated access of haptens across a defective barrier ultimately stimulates the characteristic TH2-cytokine response [70, 71], and serine protease activation, an invariable feature of AD [109], alone can initiate Th2 inflammation [23]. Along with exotoxins released from colonizing S. aureus, these four barrier-initiated mechanisms account for downstream inflammation in AD. Together, these data provide a strong scientific rationale for barrier repair therapy in AD.
Now that AD is increasingly ascribed to a primary defect in cutaneous barrier function [110], primary therapy logically should be directed at correcting barrier function. Although emollient moisturizers decrease steroid use through moisturization [111], they consist of non-physiologic lipids, such as petrolatum and lanolin, which actually impede rather than correcting the underlying defect in barrier function [112]. Yet, effective, disease-specific formulations that correct the underlying barrier abnormality in AD have not been available until recently. The permeability barrier defect in AD is characterized by a global reduction in the contents of all three key lipids (i.e., cholesterol, free fatty acids, and ceramides), with a further reduction in ceramide content [54, 55]. Thus, correction of the barrier abnormality in AD requires topical applications not only of sufficient quantities of all three key lipids that mediate barrier function but also provision of the lipids in a ceramide-dominant proportion that corrects the underlying lipid biochemical abnormality in AD [112]. Under these conditions, restoration of normal skin barrier function alone then can downregulate inflammation in deeper skin layers.
Several recent studies show that targeted correction of the lipid biochemical abnormality in AD, with a ceramide-dominant form of barrier repair therapy (EpiCeram® emulsion), comprises effective therapy for AD [107]. Specifically, in separate, multicenter, blinded studies, EpiCeram® demonstrated comparable efficacy to both fluticasone cream (Cutivate®) [113] (Fig. 8) and desonide cream for moderate-to-severe pediatric AD.Footnote 2 Moreover, in a small, double-blinded study, EpiCeram® proved comparable to Elidel® for mild-to-moderate AD [114]. Since EpiCeram® contains only ingredients that are present (individually) in currently available moisturizers, it displays none of the safety concerns of either topical steroids or IS, and it therefore could assume a central place in the treatment of AD [107]. Notably, several other currently available products that make “barrier repair” claims incorporate either incomplete mixtures of the three key lipids, incorrectly formulated mixtures, incorrect types of the three lipids, or, in some cases, insufficient quantities of the three lipids [107]. Not surprisingly, these products fail to improve barrier repair in animal models of AD (op. cit.).

Percent changes in mean SCORAD scores—EpiCeram® vs. Cutivate (Fluticasone) cream 0.05% in moderate-to-severe AD
Since some patients might develop “rebound flares” while being withdrawn from topical steroids, it is advisable to employ a combination of EpiCeram® with a short-term course of a low-potency steroid (e.g., desonide), which should both yield faster initial results, which should allow withdrawal from steroids with less risk of rebound flares. After 2–4 weeks, however, EpiCeram® alone should suffice as stand-alone therapy. Whether barrier repair therapy will also reduce secondary colonization by pathogenic S. aureus and whether it will prevent emergence of mucosal atopy (“atopic march”) remain to be determined.
Barrier Repair Therapeutics—Possible Future Options
Together, the converging pathogenic features described above create a strong rationale for the deployment of specific strategies that restore barrier function in AD. Based upon the mechanisms described above, these approaches could range from a prolonged reduction in the pH of SC alone (hyperacidification [notably, EpiCeram® is formatted at pH 5]), applications of serine protease inhibitors, topical PAR2 antagonists, topical activators of peroxisomal proliferator activator or liver X receptors, recently shown to be effective in animal models of AD [115, 116].
In summary, we have reviewed here emerging evidence that the inflammation in AD results from inherited to acquired insults to the barrier and the therapeutic implications of this new paradigm. Moreover, these preliminary, recent studies suggest that pathogenesis-based therapy is effective and could comprise a new paradigm for the therapy of AD. Yet, an important question remains: will restoration of permeability barrier function alone simultaneously improve antimicrobial defense in AD? Since recent studies have shown that these two key functions are both regulated in parallel and interdependent [65], there is reason to be optimistic on this score, as well. A final consequence of the defective epidermal barrier in AD could be that it would allow epicutaneous delivery of antigens that induce asthma and allergic rhinitis. Thus, the “atopic march,” i.e., the tendency for AD to precede the later development of mucosal atopy, can be explained by cutaneous penetration of aeroallergens of all types. FLG deficiency is associated with mucosal atopy, independent of AD [117], though FLG is not expressed in either bronchial or other non-keratinizing mucosal epithelia [118]. An implication of this observation is that again barrier repair therapy could block development of the “atopic march.”
Notes
Reductions in inflammation alone can, however, reduce TEWL due to the “vicious cycle” that is operative in AD (Elias et al. [70]).
Data on file with Promius Pharma, Bridgewater, NJ, USA.
Abbreviations
- AD:
-
Atopic dermatitis
- AMP:
-
Antimicrobial peptides
- Cer:
-
Ceramides
- FLG:
-
Filaggrin
- FFA:
-
Free fatty acids
- hBD:
-
Human β-defensins
- hCAP:
-
Human cathelicidin
- IV:
-
Ichthyosis vulgaris
- LB:
-
Lamellar bodies
- LEKTI:
-
Lymphoepithelial Kazal-type inhibitor
- NS:
-
Netherton syndrome
- PAR2:
-
Plasminogen activator type 2 receptor
- SP:
-
Serine protease
- SC:
-
Stratum corneum
- t-UCA:
-
Trans-urocanic acid
References
Sugarman JL, Fluhr JW, Fowler AJ et al (2003) The objective severity assessment of atopic dermatitis score: an objective measure using permeability barrier function and stratum corneum hydration with computer-assisted estimates for extent of disease. Arch Dermatol 139:1417–1422
Seidenari S, Giusti G (1995) Objective assessment of the skin of children affected by atopic dermatitis: a study of pH, capacitance and TEWL in eczematous and clinically uninvolved skin. Acta Derm Venereol 75:429–433
Proksch E, Folster-Holst R, Jensen JM (2006) Skin barrier function, epidermal proliferation and differentiation in eczema. J Dermatol Sci 43:159–169
Chamlin SL, Kao J, Frieden IJ et al (2002) Ceramide-dominant barrier repair lipids alleviate childhood atopic dermatitis: changes in barrier function provide a sensitive indicator of disease activity. J Am Acad Dermatol 47:198–208
Baker BS (2006) The role of microorganisms in atopic dermatitis. Clin Exp Immunol 144:1–9
Elias PM, Wood LC, Feingold KR (1999) Epidermal pathogenesis of inflammatory dermatoses. Am J Contact Dermat 10:119–126
Elias PM, Feingold KR (2001) Does the tail wag the dog? Role of the barrier in the pathogenesis of inflammatory dermatoses and therapeutic implications. Arch Dermatol 137:1079–1081
Taieb A (1999) Hypothesis: from epidermal barrier dysfunction to atopic disorders. Contact Dermat 41:177–180
Grimalt R, Mengeaud V, Cambazard F (2007) The steroid-sparing effect of an emollient therapy in infants with atopic dermatitis: a randomized controlled study. Dermatology 214:61–67
Elias PM (2005) Stratum corneum defensive functions: an integrated view. J Invest Dermatol 125:183–200
Elias PM (2007) The skin barrier as an innate immune element. Sem Immunopath 29:3–14
Denda M, Nakatani M, Ikeyama K, Tsutsumi M, Denda S (2007) Epidermal keratinocytes as the forefront of the sensory system. Exp Dermatol 16:157–161
Caubet C, Jonca N, Brattsand M et al (2004) Degradation of corneodesmosome proteins by two serine proteases of the kallikrein family, SCTE/KLK5/hK5 and SCCE/KLK7/hK7. J Invest Dermatol 122:1235–1244
Brattsand M, Stefansson K, Lundh C, Haasum Y, Egelrud T (2005) A proteolytic cascade of kallikreins in the stratum corneum. J Invest Dermatol 124:198–203
Braff MH, Di Nardo A, Gallo RL (2005) Keratinocytes store the antimicrobial peptide cathelicidin in lamellar bodies. J Invest Dermatol 124:394–400
Oren A, Ganz T, Liu L, Meerloo T (2003) In human epidermis, beta-defensin 2 is packaged in lamellar bodies. Exp Mol Pathol 74:180–182
Fallon PG, Sasaki T, Sandilands A et al (2009) A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet 41:602–608
Scharschmidt TC, Man MQ, Hatano Y et al (2009) Filaggrin deficiency confers a paracellular barrier abnormality that reduces inflammatory thresholds to irritants and haptens. J Allergy Clin Immunol 124:496–506, 506.e1–6
Schmuth M, Crumrine D, Presland RB et al (2005) Basis for the epidermal functional abnormalities in granular layer-absent (AGL) vs. -present (PGL) ichthyosis vulgaris. J Invest Dermatol 124:A72
Ohman H, Vahlquist A (1994) In vivo studies concerning a pH gradient in human stratum corneum and upper epidermis. Acta Derm Venereol 74:375–379
Hachem JP, Man MQ, Crumrine D et al (2005) Sustained serine proteases activity by prolonged increase in pH leads to degradation of lipid processing enzymes and profound alterations of barrier function and stratum corneum integrity. J Invest Dermatol 125:510–520
Demerjian M, Hachem JP, Tschachler E et al (2008) Acute modulations in permeability barrier function regulate epidermal cornification: role of caspase-14 and the protease-activated receptor type 2. Am J Pathol 172:86–97
Briot A, Deraison C, Lacroix M et al (2009) Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med 206:1135–1147
Irvine AD, McLean WH (2006) Breaking the (un)sound barrier: filaggrin is a major gene for atopic dermatitis. J Invest Dermatol 126:1200–1202
Hudson TJ (2006) Skin barrier function and allergic risk. Nat Genet 38:399–400
Fleckman P, Brumbaugh S (2002) Absence of the granular layer and keratohyalin define a morphologically distinct subset of individuals with ichthyosis vulgaris. Exp Dermatol 11:327–336
Howell MD, Kim BE, Gao P et al (2007) Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol 120:150–155
Bieber T (2008) Atopic dermatitis. N Engl J Med 358:1483–1494
O'Regan GM, Sandilands A, McLean WH, Irvine AD (2008) Filaggrin in atopic dermatitis. J Allergy Clin Immunol 122:689–693
Sandilands A, Smith FJ, Irvine AD, McLean WH (2007) Filaggrin’s fuller figure: a glimpse into the genetic architecture of atopic dermatitis. J Invest Dermatol 127:1282–1284
Lynley AM, Dale BA (1983) The characterization of human epidermal filaggrin. A histidine-rich, keratin filament-aggregating protein. Biochim Biophys Acta 744:28–35
Harding CR, Scott IR (1983) Histidine-rich proteins (filaggrins): structural and functional heterogeneity during epidermal differentiation. J Mol Biol 170:651–673
Fleckman P, Dale BA, Holbrook KA (1985) Profilaggrin, a high-molecular-weight precursor of filaggrin in human epidermis and cultured keratinocytes. J Invest Dermatol 85:507–512
Takahashi M, Tezuka T, Katunuma N (1996) Filaggrin linker segment peptide and cystatin alpha are parts of a complex of the cornified envelope of epidermis. Arch Biochem Biophys 329:123–126
Steinert PM, Marekov LN (1995) The proteins elafin, filaggrin, keratin intermediate filaments, loricrin, and small proline-rich proteins 1 and 2 are isodipeptide cross-linked components of the human epidermal cornified cell envelope. J Biol Chem 270:17702–17711
Scott IR, Harding CR, Barrett JG (1982) Histidine-rich protein of the keratohyalin granules. Source of the free amino acids, urocanic acid and pyrrolidone carboxylic acid in the stratum corneum. Biochim Biophys Acta 719:110–117
Rawlings AV, Scott IR, Harding CR, Bowser PA (1994) Stratum corneum moisturization at the molecular level. J Invest Dermatol 103:731–741
Scott IR, Harding CR (1986) Filaggrin breakdown to water binding compounds during development of the rat stratum corneum is controlled by the water activity of the environment. Dev Biol 115:84–92
Abe T, Ohkido M, Yamamoto K (1978) Studies on skin surface barrier functions:—skin surface lipids and transepidermal water loss in atopic skin during childhood. J Dermatol 5:223–229
Werner Y, Lindberg M (1985) Transepidermal water loss in dry and clinically normal skin in patients with atopic dermatitis. Acta Derm Venereol 65:102–105
Fartasch M, Diepgen TL (1992) The barrier function in atopic dry skin. Disturbance of membrane-coating granule exocytosis and formation of epidermal lipids? Acta Derm Venereol Suppl (Stockh) 176:26–31
Hubiche T, Ged C, Benard A et al (2007) Analysis of SPINK 5, KLK 7 and FLG genotypes in a French atopic dermatitis cohort. Acta Derm Venereol 87:499–505
Elias P, Williams M, Crumrine D, Schmuth M (2010) Disorders of cornification (the ichthyoses): key clinical features, biochemical genetics, cellular pathogenesis, and diagnostic ultrastructure. In: Current problems in dermatology. Karger, Basel, p 110
Scharschmidt T, Hatano Y, Man M et al (2008) Mice with deficiency in epidermal filaggrin display heightened susceptibility to hapten-induced atopic dermatitis. J Instr Dev 128:S92
Krien P, Kermici M (2000) Evidence for the existence of a self-regulated enzymatic process within human stratum corneum—an unexpected role for urocanic acid. J Invest Dermatol 115:414–420
Sprecher E, Chavanas S, DiGiovanna JJ et al (2001) The spectrum of pathogenic mutations in SPINK5 in 19 families with Netherton syndrome: implications for mutation detection and first case of prenatal diagnosis. J Invest Dermatol 117:179–187
Nylander-Lundqvist E, Back O, Egelrud T (1996) IL-1 beta activation in human epidermis. J Immunol 157:1699–1704
Elias PM (1996) Stratum corneum architecture, metabolic activity and interactivity with subjacent cell layers. Exp Dermatol 5:191–201
Roelandt T, Thys B, Heughebaert C et al (2009) LEKTI-1 in sickness and in health. Int J Cosmet Sci 31:247–254
Walley AJ, Chavanas S, Moffatt MF et al (2001) Gene polymorphism in Netherton and common atopic disease. Nat Genet 29:175–178
Vasilopoulos Y, Cork MJ, Murphy R et al (2004) Genetic association between an AACC insertion in the 3′UTR of the stratum corneum chymotryptic enzyme gene and atopic dermatitis. J Invest Dermatol 123:62–66
Weidinger S, Baurecht H, Wagenpfeil S et al (2008) Analysis of the individual and aggregate genetic contributions of previously identified serine peptidase inhibitor Kazal type 5 (SPINK5), kallikrein-related peptidase 7 (KLK7), and filaggrin (FLG) polymorphisms to eczema risk. J Allergy Clin Immunol 122(560–8):e4
Hachem JP, Crumrine D, Fluhr J et al (2003) pH directly regulates epidermal permeability barrier homeostasis, and stratum corneum integrity/cohesion. J Invest Dermatol 121:345–353
Di Nardo A, Wertz P, Giannetti A, Seidenari S (1998) Ceramide and cholesterol composition of the skin of patients with atopic dermatitis. Acta Derm Venereol 78:27–30
Imokawa G, Abe A, Jin K et al (1991) Decreased level of ceramides in stratum corneum of atopic dermatitis: an etiologic factor in atopic dry skin? J Invest Dermatol 96:523–526
Hachem JP, Wagberg F, Schmuth M et al (2006) Serine protease activity and residual LEKTI expression determine phenotype in Netherton syndrome. J Invest Dermatol 126:1609–1621
Hachem JP, Houben E, Crumrine D et al (2006) Serine protease signaling of epidermal permeability barrier homeostasis. J Invest Dermatol 126:2074–2086
Man MQ, Barish GD, Schmuth M et al (2008) Deficiency of PPARbeta/delta in the epidermis results in defective cutaneous permeability barrier homeostasis and increased inflammation. J Invest Dermatol 128:370–377
Sator PG, Schmidt JB, Honigsmann H (2003) Comparison of epidermal hydration and skin surface lipids in healthy individuals and in patients with atopic dermatitis. J Am Acad Dermatol 48:352–358
Schlievert PM, Case LC, Strandberg KL, Abrams BB, Leung DY (2008) Superantigen profile of Staphylococcus aureus isolates from patients with steroid-resistant atopic dermatitis. Clin Infect Dis 46:1562–1567
Lomholt H, Andersen KE, Kilian M (2005) Staphylococcus aureus clonal dynamics and virulence factors in children with atopic dermatitis. J Invest Dermatol 125:977–982
Wehner J, Neuber K (2001) Staphylococcus aureus enterotoxins induce histamine and leukotriene release in patients with atopic eczema. Br J Dermatol 145:302–305
Leung DY, Harbeck R, Bina P et al (1993) Presence of IgE antibodies to staphylococcal exotoxins on the skin of patients with atopic dermatitis. Evidence for a new group of allergens. J Clin Invest 92:1374–1380
Sonkoly E, Muller A, Lauerma AI et al (2006) IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol 117:411–417
Aberg KM, Man MQ, Gallo RL et al (2008) Co-regulation and interdependence of the mammalian epidermal permeability and antimicrobial barriers. J Invest Dermatol 128:917–925
Ong PY, Ohtake T, Brandt C et al (2002) Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 347:1151–1160
Miller SJ, Aly R, Shinefeld HR, Elias PM (1988) In vitro and in vivo antistaphylococcal activity of human stratum corneum lipids. Arch Dermatol 124:209–215
Bibel DJ, Aly R, Shinefield HR (1992) Antimicrobial activity of sphingosines. J Invest Dermatol 98:269–273
Nomura I, Goleva E, Howell MD et al (2003) Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J Immunol 171:3262–3269
Elias PM, Hatano Y, Williams ML (2008) Basis for the barrier abnormality in atopic dermatitis: outside–inside–outside pathogenic mechanisms. J Allergy Clin Immunol 121:1337–1343
Elias PM, Steinhoff M (2008) “Outside-to-inside” (and now back to “outside”) pathogenic mechanisms in atopic dermatitis. J Invest Dermatol 128:1067–1070
Cork MJ, Danby SG, Vasilopoulos Y et al (2009) Epidermal barrier dysfunction in atopic dermatitis. J Invest Dermatol 129:1892–1908
Cork MJ, Robinson DA, Vasilopoulos Y et al (2006) New perspectives on epidermal barrier dysfunction in atopic dermatitis: gene–environment interactions. J Allergy Clin Immunol 118:3–21, quiz 22–3
Garg A, Chren MM, Sands LP et al (2001) Psychological stress perturbs epidermal permeability barrier homeostasis: implications for the pathogenesis of stress-associated skin disorders. Arch Dermatol 137:53–59
Choi EH, Man MQ, Xu P et al (2007) Stratum corneum acidification is impaired in moderately aged human and murine skin. J Invest Dermatol 127:2847–2856
Choi EH, Demerjian M, Crumrine D et al (2006) Glucocorticoid blockade reverses psychological stress-induced abnormalities in epidermal structure and function. Am J Physiol Regul Integr Comp Physiol 291:R1657–R1662
Bisgaard H, Simpson A, Palmer CN et al (2008) Gene–environment interaction in the onset of eczema in infancy: filaggrin loss-of-function mutations enhanced by neonatal cat exposure. PLoS Med 5:e131
Jeong SK, Kim HJ, Youm JK et al (2008) Mite and cockroach allergens activate protease-activated receptor 2 and delay epidermal permeability barrier recovery. J Invest Dermatol 128:1930–1939
Novak N, Baurecht H, Schafer T et al (2008) Loss-of-function mutations in the filaggrin gene and allergic contact sensitization to nickel. J Invest Dermatol 128:1430–1435
Kurahashi R, Hatano Y, Katagiri K (2008) IL-4 suppresses the recovery of cutaneous permeability barrier functions in vivo. J Invest Dermatol 128:1329–1331
Hatano Y, Terashi H, Arakawa S, Katagiri K (2005) Interleukin-4 suppresses the enhancement of ceramide synthesis and cutaneous permeability barrier functions induced by tumor necrosis factor-alpha and interferon-gamma in human epidermis. J Invest Dermatol 124:786–792
Altrichter S, Kriehuber E, Moser J et al (2008) Serum IgE autoantibodies target keratinocytes in patients with atopic dermatitis. J Invest Dermatol 128:2232–2239
Temeck J (2005) Protopic and Elidel presentation for FDA regulatory briefing on January 14, 2005
Stern RS (2006) Topical calcineurin inhibitors labeling: putting the “box” in perspective. Arch Dermatol 142:1233–1235
Fleischer AB Jr (2006) Black box warning for topical calcineurin inhibitors and the death of common sense. Dermatol Online J 12:2
Arellano FM, Wentworth CE, Arana A, Fernandez C, Paul CF (2007) Risk of lymphoma following exposure to calcineurin inhibitors and topical steroids in patients with atopic dermatitis. J Invest Dermatol 127:808–816
Kameda G, Kramm C, Stege H et al (2003) Unexpected high serum levels of tacrolimus after a single topical application in an infant. J Pediatr 143:280
Allen A, Siegfried E, Silverman R et al (2001) Significant absorption of topical tacrolimus in 3 patients with Netherton syndrome. Arch Dermatol 137:747–750
Moskowitz DG, Fowler AJ, Heyman MB et al (2004) Pathophysiologic basis for growth failure in children with ichthyosis: an evaluation of cutaneous ultrastructure, epidermal permeability barrier function, and energy expenditure. J Pediatr 145:82–92
Paller AS, Eichenfield LF, Kirsner RS et al (2008) Three times weekly tacrolimus ointment reduces relapse in stabilized atopic dermatitis: a new paradigm for use. Pediatrics 122:e1210–e1218
Wahn U, Bos JD, Goodfield M et al (2002) Efficacy and safety of pimecrolimus cream in the long-term management of atopic dermatitis in children. Pediatrics 110:e2
Ehrchen J, Sunderkotter C, Luger T, Steinhoff M (2008) Calcineurin inhibitors for the treatment of atopic dermatitis. Expert Opin Pharmacother 9:3009–3023
Jain AB, Yee LD, Nalesnik MA et al (1998) Comparative incidence of de novo nonlymphoid malignancies after liver transplantation under tacrolimus using surveillance epidemiologic end result data. Transplantation 66:1193–1200
Hickey JR, Robson A, Barker JN, Smith CH (2005) Does topical tacrolimus induce lentigines in children with atopic dermatitis? A report of three cases. Br J Dermatol 152:152–154
Cook BA, Warshaw EM (2009) Role of topical calcineurin inhibitors in the treatment of seborrheic dermatitis: a review of pathophysiology, safety, and efficacy. Am J Clin Dermatol 10:103–118
Luger T, Paul C (2007) Potential new indications of topical calcineurin inhibitors. Dermatology 215(Suppl 1):45–54
Schachner LA, Lamerson C, Sheehan MP et al (2005) Tacrolimus ointment 0.03% is safe and effective for the treatment of mild to moderate atopic dermatitis in pediatric patients: results from a randomized, double-blind, vehicle-controlled study. Pediatrics 116:e334–e342
Paller A, Eichenfield LF, Leung DY, Stewart D, Appell M (2001) A 12-week study of tacrolimus ointment for the treatment of atopic dermatitis in pediatric patients. J Am Acad Dermatol 44:S47–S57
Kim M, Jung M, Hong SP et al (2009) Topical calcineurin inhibitors compromise stratum corneum integrity, epidermal permeability and antimicrobial barrier function. Exp Dermatol 19:501–510
Rodriguez-Martin M, Hupe M, Man M, Naya J, Elias P (2009) Topical imiquimod and calcipotriol stimulate antimicrobial peptide expression in mouse epidermis. J Invest Dermatol 129:S72
Goksugur N, Ozbostanci B, Goksugur SB (2007) Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol 24:E63–E65
Papp KA, Werfel T, Folster-Holst R et al (2005) Long-term control of atopic dermatitis with pimecrolimus cream 1% in infants and young children: a two-year study. J Am Acad Dermatol 52:240–246
Segura S, Romero D, Carrera C, Iranzo P, Estrach T (2005) Eczema herpeticum during treatment of atopic dermatitis with 1% pimecrolimus cream. Acta Derm Venereol 85:524–525
Berger TG, Duvic M, Van Voorhees AS, VanBeek MJ, Frieden IJ (2006) The use of topical calcineurin inhibitors in dermatology: safety concerns. Report of the American Academy of Dermatology Association Task Force. J Am Acad Dermatol 54:818–823
Langley RG, Luger TA, Cork MJ, Schneider D, Paul C (2007) An update on the safety and tolerability of pimecrolimus cream 1%: evidence from clinical trials to post-marketing surveillance. Dermatology 215(Suppl 1):27–44
Kao JS, Fluhr JW, Man MQ et al (2003) Short-term glucocorticoid treatment compromises both permeability barrier homeostasis and stratum corneum integrity: inhibition of epidermal lipid synthesis accounts for functional abnormalities. J Invest Dermatol 120:456–464
Elias PM (2008) Barrier-repair therapy for atopic dermatitis: corrective lipid biochemical therapy. Exp Rev Dermatol 3:441–452
Ong PY (2009) Emerging drugs for atopic dermatitis. Expert Opin Emerg Drugs 14:165–179
Voegeli R, Rawlings AV, Breternitz M et al (2009) Increased stratum corneum serine protease activity in acute eczematous atopic skin. Br J Dermatol 161:70–77
Elias PM, Schmuth M (2009) Abnormal skin barrier in the etiopathogenesis of atopic dermatitis. Curr Opin Allergy Clin Immunol 9:437–446
Cork MJ, Britton J, Butler L et al (2003) Comparison of parent knowledge, therapy utilization and severity of atopic eczema before and after explanation and demonstration of topical therapies by a specialist dermatology nurse. Br J Dermatol 149:582–589
Elias PM (2006) Epilogue: fixing the barrier—theory and rational deployment. In: Elias PM, Feingold KR (eds) Skin barrier. Taylor & Francis, New York, pp 591–600
Sugarman J, Parish L (2009) Efficacy of a lipid-based, barrier repair formulation in moderate-to-severe pediatric atopic dermatitis. J Drugs Dermatol 8:1106–1111
Simpson E, Berry T, Tofte S, Hanifin JM, Eichenfield L (2008) EpiCeram for the treatment of mild-to-moderate atopic dermatitis—a pilot study. IID 2008 Kyoto, Japan
Hatano Y, Man MQ, Uchida Y et al (2010) Murine atopic dermatitis responds to peroxisome proliferator-activated receptors alpha and beta/delta (but not gamma) and liver X receptor activators. J Allergy Clin Immunol 125(160–9):e1–e5
Hatano Y, Man MQ, Uchida Y et al (2009) Maintenance of an acidic stratum corneum prevents emergence of murine atopic dermatitis. J Invest Dermatol 129:1824–1835
Galli SJ, Tsai M, Piliponsky AM (2008) The development of allergic inflammation. Nature 454:445–454
De Benedetto A, Qualia CM, Baroody FM, Beck LA (2008) Filaggrin expression in oral, nasal, and esophageal mucosa. J Invest Dermatol 128:1594–1597
Acknowledgments
This work was supported by NIH grants R01-AR019098 and R01-AI059311, DOD grant W81XWH-05-2-0094, and the Medical Research Service, Department of Veterans Affairs. Dr. Matthias Schmuth, Chairman, Department of Dermatology, Innsbruck Medical University, Innsbruck, Austria provided multiple insights that substantially improved the content of this manuscript.
Conflict of Interest Statement
Dr. Elias is a co-inventor of the optimal ratio, triple-lipid therapy for atopic dermatitis. He also is a consultant for Promius Pharma, LLC and Pediapharm, Inc., which market EpiCeram® in the USA and Canada, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Elias, P.M., Wakefield, J.S. Therapeutic Implications of a Barrier-Based Pathogenesis of Atopic Dermatitis. Clinic Rev Allerg Immunol 41, 282–295 (2011). https://doi.org/10.1007/s12016-010-8231-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-010-8231-1