
Abstract
Osteoarthritis (OA) remains incurable in humans or horses and mesenchymal stromal/stem cells (MSCs) represent an attractive solution for producing a neocartilage substitute. However, the best MSC source still needs to be identified. This study compared the chondrogenic potential of equine MSCs derived from bone marrow (BM) and umbilical cord blood (UCB), at their undifferentiated status to check if one cell source is better proned, and after chondrogenic-induced differentiation. Chondrogenesis was induced by culture in collagen scaffold with BMP-2 + TGF-ß1 in hypoxia or normoxia. MSCs chondrogenic potential was evaluated using the mRNA and corresponding protein levels for osteogenic, hypertrophic and chondrogenic markers. MSCs characterization demonstrated that BM- and UCB-MSCs differ in proliferation and tripotencies. At undifferentiated status, they also showed differences in their expression of osteogenic, chondrogenic and hypertrophic markers. Upon chondrogenesis induction, both MSCs sources exhibited increased chondrogenic expression and produce an extracellular matrix (ECM) of better quality in hypoxia, although collagen I remained expressed. UCB-MSCs produced higher amounts of collagen II, particularly its IIB isoform, than BM-MSCs, but also collagen I and Htra1, regardless of the oxygen condition. Finally, immunohistochemistry revealed that the BM-MSCs synthesized an ECM of higher quality, regarding the more homogenous distribution of type IIB collagen, compared to UCB-MSCs. Considering collagen I as the major undesirable component in the neo-synthesis of in vitro cartilage, we recommend using BM-MSCs for horse cartilage engineering.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA) is a common joint disease characterized by the degradation of articular cartilage [1]. Cartilage has a poor intrinsic capacity for self-repair, which renders any degradation almost irreversible [2]. Studying the role and the physiology of articular cartilage can help understand the severity of OA and the pain it causes. Articular cartilage is avascular, non-innervated tissue that covers bone extremities to ensure joint mobility. When cartilage degradation occurs, the bone extremities come into direct contact with each other, causing great pain due to the innervation of bone tissue. OA is a multifactorial age-related disease. In light of the expected growth of the elderly population in the near future, finding a cure for OA is a significant goal and a therapeutic challenge [3].
Current pharmacological treatments can attenuate OA-associated pain, inflammation or decelerate OA outcomes, but do not cure OA. Surgical options, with prosthetic joint replacement, is the definitive solution for the most severe OA cases, but does not provide a long-term solution because the prosthetic implants deteriorate over time. Discovered nearly 25 years ago, autologous chondrocyte implantation (ACI) appears to be a very promising solution to treat cartilage defects [4]. Nevertheless, one step in the strategy involves chondrocyte expansion as a monolayer culture known to induce chondrocyte dedifferentiation [5]. Since the beginning of cartilage tissue engineering with the Brittberg procedure, cartilage-cell-based therapies have been developed [6, 7]. New ACI generations have emerged with the use of 3D cultures, chondrogenic growth factors, hypoxic microenvironments and also new cell types, such as mesenchymal stem cells (MSCs). MSCs have increased in their appeal for cartilage therapy because they have high proliferative potential and, above all, are able to differentiate into mesoderm-derived cell types, including chondrocytes [8]. There are different types of MSCs according to their tissue source, either neonatal tissue or adult tissue. The MSCs properties and differentiation abilities vary with their source [9]. Today, bone marrow (BM) is the most common source of MSCs used for cartilage repair, but has some limitations, namely the synthesis of a fibrocartilage subject to degradation [10], a differentiation ability that decreases with age [11] and a painful harvesting procedure (in humans). On the contrary, harvesting MSCs from umbilical cord blood (UCB) is painless and non-invasive. According to the literature, UCB-MSCs seem to have several advantages over BM-MSCs, and thus, may be a better cell type for cartilage tissue engineering [12, 13].
In this study we compared the chondrogenic differentiation abilities of equine BM-MSCs and equine UCB-MSCs. The equine model provides access to samples from young animals in a minimally-invasive manner, compared to humans. The chondrogenic differentiation protocol has already been developed and optimized for each MSC type [14, 15]. Moreover, UCB-MSCs were chosen because they differentiated into chondrocytes and express an abundant, dense and a hyaline-like cartilage matrix. By contrast, we demonstrated that despite their expression of cartilage markers, umbilical cord matrix MSCs failed to express a relevant cartilage matrix after chondrogenic induction [16]. Our chondrogenic differentiation protocol employed hypoxic or normoxic 3D cultures, using type I/III collagen sponges as biomaterials, and chondrogenic factors (BMP-2, TGF-β1) for 28 days.
Material and Methods
Cell Isolation and Culture
UCB and BM were collected, MSCs were isolated and characterized as previously described [14, 15]. Briefly, equine UCB was collected by venipuncture of the umbilical veins and equine BM was collected from sternal puncture in a cohort of horses with ages and genders which were as follows: 5 mares (two were 3 years old, three 4 years old) and 2 geldings (3 years old).
Then, MSCs were isolated by density gradient centrifugation with Ficoll-Paque PREMIUM (GE healthcare Bio-sciences; Chicago, ILL, USA) followed with a seed of the interphase in a plastic flask filled with an isolation medium (low glucose-Dulbecco’s modified Eagle Medium (LG-DMEM; Invitrogen; Carlsbad, CA, USA) containing 30% fetal calf serum (FCS, Invitrogen Life Technologies; Carlsbad, CA, USA), 10−7 M dexamethasone (Sigma-Aldrich; Saint Louis, MO, USA)) to select cells which were able to adhere and form colonies, the MSCs. Cells forming colonies were detached using trypsin/EDTA (Invitrogen; Carlsbad, CA, USA) and then reseeded at 5000 cells/cm2 (passage one, P1). Cell expansion was performed in LG-DMEM medium containing 20% FCS. All the media used in this study were supplemented with antimicrobial agents (100 IU/ml of penicillin, 100 μg/ml of erythromycin, and 0.25 mg/ml of fungizone). The absence of bacterial or viral contamination in isolated MSCs was carried out by an external laboratory according to their internal protocols (LABÉO, Saint-Contest, France). Characterization of MSCs was carried out by assessing their proliferative capacities, differentiation in adipocytes, chondrocytes and osteoblasts and the presence of cluster of differentiation (CD) characteristics of MSCs.
mRNA of equine articular chondrocytes (eAC) were prepared from healthy metacarpal joints, as previously described [14, 15]. Briefly, cartilage samples were digested in order to obtain pelleted chondrocytes on which we performed RNA extraction. Equine articular cartilage proteins were also obtained as previously described [14, 15]. Cartilage slices were crushed in liquid nitrogen, and then RIPA buffer was added to extract the proteins.
All the methods and procedures were carried out in accordance with relevant guidelines and regulations. The ComEth Anses/ENVA/UPEC Ethical Committee approved the protocol (Permit number: 10/03/15–12; 10/06/14–8).
MSCs Characterization
Population Doubling Level
125,000 MSCs were seeded in 25 cm2 flasks, trypsinized and counted every week. The equation [log10(number of harvested cells) - log10(number of seeded cells)]/log10(2) was used to determine the population doubling level at every passage.
Immunophenotyping
At P3 to P4, MSCs were trypsinized, then suspended in PBS (106 cells/ml) and incubated 25 min in the dark with the following mouse anti-human antibodies: CD29-Allophycocyanin (APC) (BioLegend; San Diego, CA, USA), CD44-Phycoerythrin (PE) (IOTest; Brea, CA, USA), CD45-Pacific Blue (PB) (AbD Serotec; Kidlington, UK), CD73-APC (Abcam; Cambridge, UK), CD90-Fluorescein isothiocyanate (FITC) (InvestCare; London, UK), CD105-FITC (Abcam; Cambridge, UK) and type II MHC-RPE (AbD Serotec; Kidlington, UK). Subsequently, after a washing step, cells were resuspended in 500 μl of PBS. A Beckman Coulter Gallios flow cytometer (Beckman Coulter; Brea, CA, USA) (Federative Research Structure ICORE platform, University of Caen Normandy, France) and FlowJo Software (TreeStar; Ashland, OR, USA) were used to perform the analysis. We used respective mouse isotype antibodies as controls. The antibodies cross reactivity with equine cells, and specificity, were verified using an equine blood sample (Supplemental Fig. 2).
Multilineage Capacity
The capacity of equine MSCs to differentiate in osteogenic, adipogenic and chondrogenic lineages was determined at P4. For this purpose, cells were seeded in 6-well plates. At 50% of the confluence, osteogenic differentiation was induced and then assessed by Alizarin red S staining. Cells were incubated (10 min; room temperature) with 4% paraformaldehyde, washed with PBS (pH 4.1) and stained (20 min; room temperature) with Alizarin red S.
At 80% of the confluence, adipogenic treatments were added. Oil red O staining was used to reveal intracellular accumulation of red lipid droplets. Cells were fixed, washed and stained (10 min; room temperature) with 0.3% Oil red O in isopropanol.
Chondrogenic differentiation was induced when cells were at 90% of confluence. The chondrogenic differentiation was assessed by staining the sulphated proteoglycans with an Alcian blue solution 1% in chloridric acid 1 M (pH 1).
The composition of the three media used for the evaluation of tripotency were described in our previous studies [14, 15].
As negative controls for chondrogenic, osteogenic and adipogenic differentiations, cells were cultured in LG-DMEM supplemented only with 10% FCS.
Chondrogenic Differentiation in 3D
We used collagen sponges as scaffold for cell culture, as previously described [14, 15]. MSCs were grown for 28 days in this 3D scaffold to induce chondrogenesis according to the protocols previously established [14, 15]. Briefly, at P4 the cells were seeded onto the collagen sponges (5 × 105 cells/sponge) and then the cell constructs were incubated with ICM in the presence or absence of 50 ng/ml BMP-2 and 10 ng/ml TGF-β1, in hypoxia (3% O2) or normoxia (21% O2). Sponges were harvested for RNA and protein analyses on days 7, 14, 21 and 28. MSC monolayers before induction were used as controls (day zero, D0).
RNA Isolation and RT-qPCR
Cells were extracted with the Trizol Reagent according to manufacturer’s instruction. Then, one μg of RNA was reverse transcribed into cDNA using reverse transcriptase (MMLV, Thermo Fisher Scientific; Waltham, MA, USA) and oligo dT (Eurogentec; Liège, Belgium). RT-qPCR was performed on a CFX96 touch Real Time PCR Detection System (Bio-Rad; Hercules, CA, USA) using iTaq Universal SYBR Green Supermix (Bio-Rad; Hercules, CA, USA). The Table 1 lists the sequences of the primers we used. The 2-ΔΔCT method was used to calculate the relative gene expression. Each sample was normalized to β-actin.
Protein Extraction and Western-Blots
After treatment, sponges containing cells or cells in monolayers (D0) were washed with PBS, crushed and extraction of the total proteins was performed using the RIPA-lysis buffer. A cocktail of several protease inhibitors was systematically used. We determined the protein concentration according to the Bradford procedure (Bio-Rad; Hercules, CA, USA). Then, 10 μg of the proteins extracts were separated in 7.5% polyacrylamide gels (TGX Stain Free Fast Cast Acrylamide Kit 7.5%, Bio-Rad) and transferred to a PolyVinylidene Difluoride Membrane (Trans-Blot Turbo RTA Midi PVDF Transfer Kit, Bio-Rad). Unspecific binding sites of the membranes were blocked with 10% non-fat milk powder in Tris-buffered saline with 0.1% Tween (TBST, Sigma Aldrich; Saint Louis, MO, USA) for 1 h. Then, membranes were incubated overnight at 4 °C with rabbit anti-human type I collagen (Novotec; Bron, France), rabbit anti-human type II collagen (Novotec; Bron, France), mouse anti-human type X collagen (Sigma-Aldrich; Saint Louis, MO, USA), rabbit anti-human GAPDH (Santa Cruz Biotechnology; Dallas, TX, USA), mouse anti-human Runx2 (Santa Cruz Biotechnology; Dallas, TX, USA), mouse anti-human PCNA (Santa Cruz Biotechnology; Dallas, TX, USA), mouse anti-human Sox9 (Santa Cruz Biotechnology; Dallas, TX, USA), rabbit anti-human HtrA1 (Merck Millipore; Billerica, MA, USA) or rabbit anti-human β-tubulin (Santa Cruz Biotechnology; Dallas, TX, USA). The generation of the anti-human IIB collagen was made by Covalab, through the strategy described previously by Aubert-Foucher et al. [17]. This antibody reveals the proα1(II)B and pNα1(II)B type II collagen isoforms. The next day, PVDF membranes were washed (TBST), incubated one hour with HRP-conjugated goat anti-rabbit or anti-mouse IgG antibody (Jackson Immunoresearch; West Grove, PA, USA). The chemiluminescence (Clarity Western ECL Substrate, Bio-Rad) was visualized on the ChemiDoc MP Imaging System (Bio-Rad; Hercules, CA, USA). To support the main conclusions, the quantification of the blots was performed, and shown in the supplementary data section, using the Image Lab software (Biorad; Hercules, CA, USA). The quantification of the proteins of interest was normalized by the quantification of the corresponding house-keeping protein.
Immunohistochemistry
Biological substitutes, incubated 16 h with 4% paraformaldehyde, were embedded in paraffin and sectioned in 4 μm-thick slices. Sections were deparaffinized using toluene, rehydrated and then immunostained. Immunostaining began by unmasking the antigenic epitopes with 0.5% hyaluronidase in PBS-BSA buffer (3%), washing with PBS and permeabilization. Following several washes with PBS, sections were then treated with a peroxidase blocking solution (Dako Real, Agilent; Santa Clara, CA, USA) and subsequently with PBS-BSA (5%) for 30 min. Then, the sections were treated with specific antibodies against type I (1:1000 dilution) and type IIB (1:500 dilution) collagens and aggrecan (1:500 dilution). The antibodies used are the ones used in western-blots. Section were rinsed, then incubated with a HRP-conjugated secondary antibody (undiluted, Agilent; Santa Clara, CA, USA), and signals were detected using the DAB substrate (Dako, Agilent; Santa Clara, CA, USA). Hematoxylin (Dako, Agilent; Santa Clara, CA, USA) was used as the counter-stain. An Aperio ScanScope slide scanner was used to digitalize histological slides (Leica Biosystems; Wetzlar, Germany).
Statistical Analyses
All experiments were repeated at least four times with cells derived from different horses. We used Graphpad Prism 6 (Graphpad Software Inc.; San Diego, CA, USA) to perform the Mann-Whitney U-test. A P value of ≤0.05 was considered to be significant.
Results
MSCs Characterization
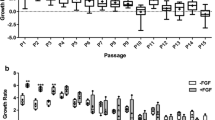
To characterize BM- and UCB-MSCs, we assessed their multilineage capacity, population doubling level and performed immuno-phenotyping (Fig. 1). BM- and UCB-MSCs were able to differentiate into chondrocytes, osteoblasts and adipocytes, nevertheless they showed differences in their multilineage capacity (Fig. 1a). Both types of MSC showed similar staining after their differentiation into osteoblasts. On the contrary, chondrogenic treatment led to a stronger staining in UCB-MSCs. Furthermore, although adipogenic treatment led to a modification of UCB-MSCs morphology, these MSCs showed no or extremely weak Oil Red O staining. Both types of MSC had strong proliferative capacities: their respective cumulative population doubling levels increased from passage 1 (P1) to P15 (Fig. 1b). The proliferation rate was high during the first eight passages and then slowed down. At each passage, the cumulative population doubling level of BM-MSCs tended to be higher than its UCB-MSCs counterpart. Finally, both BM- and UCB-MSCs possessed most MSC cluster of differentiation (CD) characteristics and neither expressed type II MHC or CD45 (PBMC were used as a positive control in this latter case) (Fig. 1c; supplemental Figs. 1 and 2). Surprisingly, our results indicate that both types of MSC were negative for CD105. CD73 was expressed in one MSC sub-population, but this sub-population was significantly larger in UCB-MSCs than in BM-MSCs.

Characterization of MSCs. Ability of bone marrow (BM)- and umbilical cord blood (UCB)- mesenchymal stem cells (MSCs) to differentiate into osteoblasts, adipocytes and chondrocytes (a). The cells were amplified at P4 and cultured to 50% confluence with media specific to osteoblastic (n = 4), adipocyte (n = 4) or chondrogenic (n = 4) differentiation (treated panels) or amplification medium (control panels) for 14 days. After fixing with paraformaldehyde, cells were stained with Alizarin red, Oil Red O and Alcian blue to reveal the presence of calcium deposits, lipid droplets and sulfated proteoglycans, respectively. Proliferative capacity of equine MSCs isolated from BM and UCB (b). The MSCs derived from BM and UCB were isolated and seeded at 5000 cells/cm2. A passage with trypsin/EDTA was performed every 7 days, followed by counting and re-seeding at the same cell density (n = 4). The significance of the results between BM- and UCB-MSCs was evaluated using a Mann-Whitney test (*p < 0.05, **p < 0.01, ***p < 0.001). Immuno-phenotyping of the isolated and amplified MSCs (C). In passage 4 (P4), the cells were trypsinized and suspended in PBS (10 millions cells/ml). Then, cells were incubated 30 min in the dark with the specific antibodies, or relevant anti-IgG isotype controls. After a washing step, the analysis was performed. 10,000 events were collected for each run. The graph represents the mean of the values obtained for each run of four independent experiments. All the original images of cell rate based on cell surface markers obtained in flow cytometry are presented in supplemental Fig. 1 (n = 4 for BM-MSCs and UCB-MSCs). CD: cluster of differentiation; MHC: major histocompatibility complex; BM: bone marrow; UCB: umbilical cord blood. AU: arbitrary units
As a conclusion, all the results obtained in Fig. 1 are summarized in supplemental Table 1.
Comparison of BM- and UCB-MSCs at their Basal Undifferentiated State
Because MSCs may exhibit differences involved in their chondrogenic abilities, we compared both types of MSC at their basal state in order to know if a cell source is may be proned for chondrogenesis. Interestingly, both types of MSC expressed chondrogenic markers, such as type II procollagen, Acan, Sox9 at the mRNA (Fig. 2a, b, c) and corresponding protein levels, but showed differences at the protein level (Fig. 3a, c). Although there was inter-strain variability, BM-MSCs seemed to synthesize a higher amount of type II procollagen, but a lower amount of Sox9 than UCB-MSCs (Fig. 3a, c, e). The expression of type II procollagen in undifferentiated MSCs is characteristic as it is not synthesized in bone (Fig. 3e). Moreover, there was a lack of expression of the mature type II collagen as only immature forms of this collagen isotype are significantly expressed by both types of MSC, procollagen and pC/pN alpha1(II) (Fig. 3a, e). These data suggest a reduced or delayed type II procollagen processing. The mRNA level and protein amounts of type I collagen (undesirable for cartilage regeneration) was also significantly higher in BM-MSCs than in UCB-MSCs (Fig. 2d, e; Fig. 3a), an indicator of active synthesis, with also a decreased or delayed procollagen processing. Thus, the mRNA Col2a1:Col1 ratios tended to be higher in UCB-MSCs (Fig. 2g, h) than in BM-MSCs. BM- and UCB-MSCs also differed in osteogenic and hypertrophic marker expression. The expression of Col10a1 and AlpI were higher in UCB-MSCs and the expression of Bglap and Runx2 were lower than in BM-MSCs (Fig. 2i, j, k, l). The same patterns were observed at the protein level regarding osteocalcin (Bglap) but not Runx2, which was expressed variably across strains (Fig. 3d). The serine protease overexpressed in OA pathogenesis, Htra1, was expressed similarly in both MSC types at the mRNA level, but expression was higher in UCB-MSCs at the protein level (Fig. 2f; Fig. 3a). The main conclusions are supported by statistical analyses performed on the quantification of the blots (Supplemental Table 2). Finally, we also studied the proliferative markers Ki67 and Pcna (Fig. 2m, n; Fig. 3b). We did not observe any differences at the mRNA level, however, Pcna seemed to show lower protein expression in UCB-MSCs, which correlates with the population doubling levels (Fig. 1b).

Comparison of mRNA expression in BM- and UCB-MSCs at their basal level. MSCs were cultured in monolayer until P4 in amplification medium and their mRNA was extracted as described in the Materials and Methods. The mRNAs were estimated using RT-qPCR after normalization relative to the β-actin reference gene. The Col2a1:Col1a1 ratio is shown. The results are represented as box plots (median, quartiles, extreme values) and the significance of the values between BM- and UCB-MSCs was tested using a Mann-Whitney test (* p < 0.05); n = 4. AU: arbitrary units

Comparison of protein expression in BM and UCB-MSCs at their basal level. MSCs were cultured in monolayer until P4 in amplification medium and proteins were extracted as described in Materials and Methods. The total protein extracts, from 4 different strains of both types of MSCs, were separated by electrophoresis on a 7.5% acrylamide gel under denaturing conditions. The proteins were then transferred to a PVDF membrane, which was incubated with the antibodies indicated on the right. The molecular weight marker (MW kDa) is indicated on the left. Ostc: osteocalcin. For the cropped blots displayed, the full-length blots with different exposure times are included in the Supplementary Information section (Supplemental Figs. 3–7). Statistical analyses of the main differences are shown in supplemental Table 2
Comparison of the Chondrogenic Potential of MSCs
To determine the best source of MSCs for cartilage tissue engineering, we analysed their chondrogenic differentiation in normoxia and hypoxia. We first studied the normoxic condition (Figs. 4 and 5) and assessed the levels of mRNAs and proteins typical and atypical of cartilage every week from the beginning to the end of the differentiation protocol. Typical hyaline cartilage-associated molecules, except for Col9a1, were quickly upregulated upon chondrogenic treatment: we observed an increase in their mRNA levels (Col2a1, Acan, Col11a1, Prg4, Snorc) and protein amounts (type II and IIB collagens) from D7 for both MSC types (Fig. 4a, b, c). Nevertheless, the expression of these molecules was even higher at D28. Although we did not observe any significant differences in the induction of the mRNA levels between BM- and UCB-MSCs and their respective D0, UCB-MSCs expressed a higher amount of type II collagen protein from D7 to D28 (Fig. 4a, b, c; supplemental Table 4). The proliferation marker Pcna was still expressed at D7 in both MSC types, but then decreased until D28 in UCB-MSCs, whereas its expression was no longer observed after D14 in BM-MSCs (Fig. 4c).

Kinetic study of specific mRNA and protein markers of articular hyaline cartilage during chondrogenic differentiation of BM- and UCB-MSCs in normoxia. Equine MSCs derived from BM or UCB were amplified and seeded in collagen sponges at P4 (n = 5). They were then grown in normoxia (21% O2) for 7, 14, 21 and 28 days (D7, D14, D21 and D28, respectively) in the presence of incomplete chondrogenic medium enriched with BMP-2 (50 ng/ml) and TGF-β1 (10 ng/ml) (B + T). The D0 condition corresponds to stem cells cultured in monolayer at P4 with the amplification medium and the equine articular cartilage (eAC) condition corresponds to the protein extracted from healthy cartilage. mRNA levels were estimated by RT-qPCR after normalization relative to the β-actin reference gene (A). The results are represented as box plots (median, quartiles, extreme values) and the significance of the values, compared to the D0 condition, was tested using a Mann-Whitney test (* p < 0.05). The total protein extracts were separated by electrophoresis on a 7.5% acrylamide gel under denaturing conditions (B, C). The proteins were then transferred to a PVDF membrane which was incubated with the antibodies indicated on the right. The molecular weight marker (MW kDa) is indicated on the left. The eAC sample and the other samples were loaded and run in the same gel, but the exposure time of the membrane was longer for the eAC lane (B). For the cropped blots displayed, the full-length blots with different exposure times are included in the Supplementary Information section (Supplemental Fig. 8). Statistical analyses of the main differences are shown in supplemental Table 4. AU: arbitrary units

Kinetic study of atypical mRNA and protein of hyaline articular cartilage during chondrogenic differentiation of BM- and UCB-MSCs in normoxia. Equine MSCs derived from BM or UCB were amplified and seeded in collagen sponges at P4 (n = 5). They were then grown in normoxia (21% O2) for 7, 14, 21 and 28 days (D7, D14, D21 and D28, respectively) in the presence of incomplete chondrogenic medium enriched with BMP-2 (50 ng/ml) and TGF-β1 (10 ng/ml) (B + T). The D0 condition corresponds to stem cells cultured in monolayer at P4 in amplification medium and the equine articular cartilage (eAC) condition corresponds to the protein extracted from healthy cartilage. mRNA levels were estimated using RT-qPCR after normalization relative to the β-actin reference gene (A). The results are represented as box plots (median, quartiles, extreme values) and the significance of the values, compared to the D0 condition, was tested using a Mann-Whitney test (* p < 0.05). The total protein extracts were separated by electrophoresis on a 7.5% acrylamide gel under denaturing conditions (B). The proteins were then transferred to a PVDF membrane which was incubated with the antibodies indicated on the right. The molecular weight marker (MW kDa) is indicated on the left. The eAC sample and the other samples were loaded and run in the same gel, and the exposure time of the membrane was the same for all the lanes. For the cropped blots displayed, the full-length blots with different exposure times are included in the Supplementary Information section (Supplemental Fig. 9). Statistical analyses of the main differences are shown in supplemental Table 6. AU: arbitrary units
In both MSC types, the cartilage-atypical type I collagen increased from D7 to D21 – to a greater extent in UCB-MSCs at the protein level – but then decreased until D28 only for UCB-MSCs (Fig. 5b; supplemental Table 6). These observations correlate with Col2a1:Col1 mRNA ratios which tended to be most strongly increased in BM-MSCs, although it significantly increased in both MSC types, compared with their respective D0 controls (Fig. 5a). For BM- and UCB-MSCs, HtrA1 had the highest protein expression at D14 and then decreased (Fig. 5b; supplemental Table 6). However, the protein expression of HtrA1 remained higher in UCB-MSCs from D0 to D28. Although the expression of Mmp13 remained unchanged during chondrogenic differentiation from D0 in both MSC types, the mRNA expression of Runx2 and AlpI increased for both MSC types as of D7 and, regarding AlpI, the increase tended to be stronger in BM-MSCs (Fig. 5a). Additionally, type X collagen increased at the protein level from D14 to D28 in UCB-MSCs only (Fig. 5b). The mRNA amounts of the osteogenic marker Bglap remained unchanged compared with their D0 respective controls for both BM- and UCB-MSCs (Fig. 5a).
In conclusion, all the results obtained in Figs. 4 and 5 are summarized in supplementary Tables 3 and 5 respectively.
In hypoxia, when chondrogenic differentiation was performed both MSC sources showed – as in normoxia – a rapid increase in Col2a1, Acan, Col11a1, Prg4, Snorc and Col9a1 (Supplemental Fig. 10A). Differences between BM- and UCB-MSCs were also observed: UCB-MSCs produced higher amounts of type II collagen and Pcna (Fig. 6a; supplemental Table 7) and, in contrast to normoxia, Pcna increased until D28.

Kinetic study of specific (a) and atypical (b) protein markers of hyaline articular cartilage during chondrogenic differentiation of BM- and UCB-MSCs in hypoxia. Equine MSCs derived from BM or UCB were amplified and seeded in collagen sponges at P4 (n = 5). They were then grown in hypoxia (3% O2) for 7, 14, 21 and 28 days (D7, D14, D21 and D28, respectively) in the presence of incomplete chondrogenic medium enriched with BMP-2 (50 ng/ml) and TGF-β1 (10 ng/ml) (B + T). The D0 condition corresponds to stem cells cultured in monolayer at P4 with the amplification medium and the equine articular cartilage (eAC) condition corresponds to the protein extracted from healthy cartilage. The total protein extracts were separated by electrophoresis on a 7.5% acrylamide gel under denaturing conditions. The proteins were then transferred to a PVDF membrane which was incubated with the antibodies indicated on the right. The molecular weight marker (MW kDa) is indicated on the left. For the cropped blots displayed, the full-length blots with different exposure times are included in the Supplementary Information section (Supplemental Figs. 11 and 12). Statistical analyses of the main differences are shown in supplemental Table 7
Over all, regarding type I collagen in hypoxia, we observed patterns similar to those in normoxia. Type I collagen increased from D7 to D21, although to greater degree in UCB-MSCs, and thus the Col2a1:Col1a and Col2a1:Col1a2 ratios tended to be most strongly increased in BM-MSCs, but only until D21 (Supplemental Fig. 10B, Fig. 6b; supplemental Table 7). However, after D21, type I collagen protein amounts decreased only in UCB-MSCs (Fig. 6b; supplemental Table 7). Htra1 also increased in its mRNA steady-state amounts in both MSC types and its protein expression was higher in UCB-MSCs, although it decreased from D14 to D28 (Supplemental Fig. 10b, Fig. 6b; supplemental Table 7). Induction of AlpI mRNA tended to be higher in BM-MSCs, whereas the expression of Runx2 was similar in BM- and UCB-MSCs (Supplemental Fig. 10B). Type X collagen was expressed at higher protein amounts at D14 in UCB-MSCs, decreasing thereafter (Fig. 6b), whereas no effect was observed at the mRNA level (Supplemental Fig. 10B). In contrast, type X collagen was not observed at the protein level in BM-MSCs. The induction of Bglap mRNA remained unchanged compared with their respective D0 controls for both BM- and UCB-MSCs (Supplemental Fig.10B).
Previous data has demonstrated that the optimal chondrogenic differentiation lasts 28 days, for both MSC types (Fig. 4 to Fig. 6). Nevertheless, the two different oxygen conditions led to slight differences in the MSC phenotype. Therefore, we aimed to determine the best oxic conditions for each MSC type to compare their chondrogenic differentiation when they were cultured in their respective optimal oxic conditions.
Analysis of mRNA levels, typical or atypical of the chondrocyte phenotype, did not allow to determine which oxic condition was the best, because the analysed markers did not vary for either MSC type (Supplemental Fig. 13A and B). On the other hand, the analysis of protein expression showed differences (Figs. 7a and b; supplemental Table 8). Although the total amount of type II collagen seemed to be the same regardless of the oxic condition, the proportion of the different maturation forms varied (in a similar fashion) for both MSC types at D28. We observed more pC and procollagen forms of type II collagen in hypoxia, whereas the pN form was more abundant in normoxia. Type IIB collagen followed a similar pattern, with pN forms disappearing in hypoxia in favour of pro forms (Fig. 7a). These findings indicate that both types of MSC are actively producing type II collagen since the precursor isoforms are expressed at a high level. Concerning type I collagen, the mature form strongly decreased in hypoxia for both MSC sources (Fig. 7b).

Effect of the oxygen condition on the chondrogenic differentiation of BM- and UCB-MSCs on specific (a) and atypical (b) protein markers of hyaline articular cartilage. Equine MSCs derived from BM or UCB were amplified and seeded in collagen sponges at P4 (n = 5). They were then grown in hypoxia (H, 3% O2) or normoxia (N, 21% O2) for 14 and 28 days (D14 and D28, respectively) in the presence of incomplete chondrogenic medium enriched with BMP-2 (50 ng/ml) and TGF-β1 (10 ng/ml) (B + T). The total protein extracts were separated by electrophoresis on a 7.5% acrylamide gel under denaturing conditions. The proteins were then transferred to a PVDF membrane which was incubated with the antibodies indicated on the right. The molecular weight marker (MW kDa) is indicated on the left. For the cropped blots displayed, the full-length blots with different exposure times are included in the Supplementary Information section (Supplemental Figs. 14 and 15). Statistical analyses of the main differences are shown in supplemental Table 8
Finally, we performed immunohistochemistry (IHC) analyses to visualize the ECM. The collagen sponges were homogeneously filled with neo-synthesized ECM for both MSC types (Figs. 8, 9, 10, and 11). However, especially for BM-MSCs, cells did not have a uniform distribution throughout the sponge, with a larger number of cells localised at the periphery.

IHC analysis of type I collagen expression in the neosynthesized cartilaginous substitute. Equine MSCs derived from BM or UCB were amplified and seeded in collagen sponges at P4 (n = 5). They were then grown in hypoxia (3% O2) or normoxia (21% O2) for 14 and 28 days (D14 and D28, respectively) in the presence of incomplete chondrogenic medium enriched with BMP-2 (50 ng/ml) and TGF-β1 (10 ng/ml) (B + T). Serial paraffin sections (4 μm) of the sponges at D14 and D28 were analyzed by IHC to evaluate type I collagen distribution. 1 cm = 150 μm. Magnifications (× 2.5) are displayed in the top left corner box. Blue arrow: type I collagen fibres of the biomaterial

IHC analysis of type IIB collagen expression in the neosynthesized cartilaginous substitute. Equine MSCs derived from BM or UCB were amplified and seeded in collagen sponges at P4 (n = 5). They were then grown in hypoxia (3% O2) or normoxia (21% O2) for 14 and 28 days (D14 and D28, respectively) in the presence of incomplete chondrogenic medium enriched with BMP-2 (50 ng/ml) and TGF-β1 (10 ng/ml) (B + T). Serial paraffin sections (4 μm) of the sponges at D14 and D28 were analyzed by IHC to evaluate type IIB collagen distribution. 1 cm = 150 μm. Magnifications (× 2.5) are displayed in the top left corner box

Evaluation of aggrecan expression by IHC in the neosynthesized cartilaginous substitute. Equine MSCs derived from BM or UCB were amplified and seeded in collagen sponges at P4 (n = 5). They were then grown in hypoxia (3% O2) or normoxia (21% O2) for 14 and 28 days (D14 and D28, respectively) in the presence of incomplete chondrogenic medium enriched with BMP-2 (50 ng/ml) and TGF-β1 (10 ng/ml) (B + T). Serial paraffin sections (4 μm) of the sponges at D14 and D28 were analyzed by IHC to evaluate aggrecan distribution. 1 cm = 150 μm. Magnifications (× 2.5) are displayed in the top left corner box

Global distribution of type I and IIB collagens and aggrecan in the neosynthesized cartilaginous substitute. Equine MSCs derived from BM or UCB were amplified and seeded in collagen sponges at P4 (n = 5). They were then grown in hypoxia (3% O2) or normoxia (21% O2) for 14 and 28 days (D14 and D28, respectively) in the presence of incomplete chondrogenic medium enriched with BMP-2 (50 ng/ml) and TGF-β1 (10 ng/ml) (B + T). Serial paraffin sections (4 μm) of the sponges at D14 and D28 were analyzed by IHC to evaluate type I and type IIB collagens, and aggrecan distribution. 1 cm = 150 μm
Regardless of the MSC type or the oxygen condition, at D14 and D28, type I collagen was distributed along a gradient decreasing from the periphery to the inner areas of the neo-synthesized cartilaginous substitute (Figs. 8 and 11). In the centre of the biological substitute, labelling was not observed in the vicinity of the nucleus of the cells, which may indicate a cessation of type I collagen synthesis in these cells. Type I collagen fibres of the biomaterial were also stained. For the BM-MSCs, staining intensity increased from D14 to D28, unlike the neo-synthesized cartilaginous substitute obtained with UCB-MSCs, which appeared to have the same staining intensity over time.
Just like type I collagen, type IIB collagen was distributed according to a gradient which steepened at D28 for both types and to a greater extent for UCB-MSCs (Figs. 9 and 11). The staining appeared to increase in hypoxia only for BM-MSCs, particularly at the periphery of the neocartilage substitute. Regardless of the oxygen condition and in contrast to type I collagen, a strong staining signal was observed near the nucleus for both MSC types, but this staining was stronger for UCB-MSCs. For BM-MSCs, this observation was not as intense at D28.
Aggrecan is also distributed according to a gradient (Figs. 10 and 11). At D14, a stronger staining is observed in close vicinity of the cells, whatever the MSC source or the oxic condition. Furthermore, the staining seems to be accentuated from D14 to D28 for BM-MSCs, whereas the staining remains unchanged for UCB-MSCs, whatever the oxic condition.
Discussion
MSCs are multipotent stem cells that can differentiate into mesodermal cells. These cells thus offer new perspectives for cartilage cell and/or tissue engineering. Nevertheless, the differentiation capacities of MSCs are correlated with their age and tissue source. Previous studies have demonstrated the usefulness of BM-MSCs and UCB-MSCs for producing hyaline-like cartilage [14, 15]. In this study, we compared BM- and UCB-MSCs to determine the best source of MSCs for generating neo-cartilage in vitro. Because equine BM-MSCs can be isolated from young horses, we used the equine model to avoid potential differentiation differences between BM- and UCB-MSCs due to their age. In addition, using equine cells helps meet the challenge to develop effective therapies to treat horse chondral lesions; furthermore, the studies performed in the horse are transferable to humans.
Following isolation, we characterized putative MSCs according to international CD criteria [18]. The isolated cells, regardless of their source, showed typical MSC features. Nevertheless, UCB-MSCs seemed to grow more slowly, in terms of the population doubling level and the Pcna protein amounts. The higher proliferative capacity of BM-MSCs is in favour of their use for cartilage tissue engineering, primarily because a large number of cells is required. Similarly, BM- and UCB-MSCs displayed different differentiation potentials: adipogenic treatments were not able to induce the formation of lipid droplets in UCB-MSCs, and more interestingly, in the aim of cartilage tissue engineering, the Alcian bleu staining was stronger in UCB-MSCs. Several studies have demonstrated that MSCs should be considered as the sum of sub-populations sharing common features and there may be some variation, particularly in the expression of their CDs [19, 20]. Differences in CD expression can be associated with different differentiation capacsities [21]. The study of CDs revealed a higher proportion of CD73+ cells in UCB-MSCs, which may suggest a different distribution of the sub-populations between BM- and UCB-MSCs. The higher proportion of CD73+ cells seems to suggest that UCB-MSCs have a strong potential to mimic native cartilage ECM [22]. In contrast to expected results and to international criteria used to characterize MSCs, our cells were CD105-negative. Previous immuno-phenotyping analyses performed on equine MSCs corroborate our results [23]. The absence of CD105 can be attributed to the use of trypsin to detach the cells during the procedure. Trypsin can damage some cell surface proteins, thus hampering their detection by flow cytometry, whereas other proteins are unaffected [24]. This assumption is supported by the study of Braun et al. that showed a positive CD105 signal in equine MSCs derived from adipose tissue, using accutase instead of trypsin/EDTA [25].
At their basal undifferentiated state, UCB-MSCs synthesized a lower amount of all the immature forms of collagens studied, but a higher amount of Sox9, which may suggest a higher chondrogenic potential, especially because the mRNA Col2a1:Col1 ratios tended to be higher, too.
Based on our previous studies [15, 26], we used normoxic and hypoxic 3D cultures in the presence of chondrogenic factors (BMP-2, TGF-β1) to induce the chondrogenesis of MSCs. Whatever the oxic condition and in correlation with the Alcian blue staining during the MSCs characterization, UCB-MSCs synthesized higher levels of type II and IIB collagens than BM-MSCs, but also type X and I collagens. Thus, UCB-MSCs showed a stronger response to the differentiation protocol, because their collagen protein amounts were lower at D0. However, in UCB-MSCs, regardless of the oxygen level, the amounts of type I collagen protein decreased after 21 days. Furthermore, the hypoxic condition led to a decrease in type X collagen and HtrA1 after 14 days, but still remained higher than in BM-MSCs. Nonetheless, in both types of MSCs, type II and IIB collagen increased until D28. If we consider cell differentiation as a dynamic and ever-evolving process, the decrease in molecules atypical of hyaline cartilage indicates that a switch in cell behaviour occurred. Thus, the UCB-MSCs phenotype had not stabilized by the end of our chondrogenic differentiation protocol, and seemed to still be in chondrogenic differentiation, owing to the continuing expression of undesirable, atypical cartilage molecules and Pcna. Furthermore, chondrocytes from hyaline articular cartilage are not proliferative when skeletal maturity is reached [27]. Another indication that the chondrocyte phenotype had not yet stabilized in UCB-MSCs is the stronger presence of type II collagen near their nuclei, revealing strong anabolic activity even after 28 days; stabilized hyaline articular chondrocytes should have low anabolic activity. Alternatively, the decrease in the expression of atypical hyaline cartilage molecules may be due to the selection of specific sub-populations. This difference between BM- and UCB-MSCs can be linked to the potential different sub-populations found in each MSC source, as attested by CD73 expression.
Beyond the phenotype of differentiated cells, the quality of the neo-cartilage ECM produced is crucial. The ECM must be abundant to fill a cartilage defect in situ. For both MSC types, the newly synthesized ECM was homogenously distributed throughout the construct. UCB-MSCs produced significantly higher amounts of ECM. The fact that type I collagen staining was not stronger at D28 for UCB-MSCs correlates with the results of western blots (Figs. 5b and 6b). Nevertheless, when considering type IIB collagen, the neosyntesized cartilaginous substitute with UCB-MSCs showed a sharper gradient than BM-MSCs at D28. This difference in the organization of the ECM may be due to the different state of cell differentiation from the periphery to the center that could lead to weaker mechanical properties. Futhermore, the ECM produced in UCB-MSCs also contained atypical molecules that BM-MSCs had not synthesized, such as type X collagen and HtrA1, and a larger amount of type I collagen in western-blot. These latter results are the major reason for the choice of BM-MSCs as a better cell type for cartilage tissue engineering. UCB-MSCs may likely be a good candidate, but they need to undergo a complete chondrogenic differentiation program with an optimized protocol to decrease the synthesis of molecules atypical to hyaline cartilage. Furthermore, although the neo-synthesized cartilaginous substitute is composed of a uniform ECM, the IHC study revealed that various important proteins had a gradient distribution from the periphery to the center of the neo-cartilage. Applying the chondrogenic differentiation protocol in standardized and dynamic cultures grown in perfusion bioreactors may help avoid the heterogenous distribution of nutrients and oxygen, as demonstrated by Mayer et al. [28].
Various studies have compared the chondrogenic potential of MSCs in various species. Nevertheless, the differentiation efficiency of a cell type can be modulated by the differentiation protocol, which can be optimal for one MSC source but not for another, and the assessment itself may also influence how differentiation is gauged. Thus, a study comparing the mechanical properties of equine BM- and UCB-MSCs determined that UCB-MSCs were the best cell type for cartilage tissue engineering [29]. The study of mechanical properties of neo-synthesized cartilage is a useful strategy for assessing the quality of neo-cartilage, because the major function of hyaline cartilage is to absorb shocks. Nevertheless, mechanical studies should not be restricted to compressive forces because cartilage undergoes other types of forces, such as frictional forces, and additionally, this tissue must be constituted of a suitable matrix for nutrient diffusion. Moreover, cartilage features depend on matrix composition, distribution and arrangement. Therefore, biomechanical and histological approaches should be combined to assess neo-cartilage quality.
Finally, the cartilage matrix is the result of the mRNA and protein synthesis in the resident cell type of the tissue, the chondrocytes. In cartilage tissue engineering, particularly when using MSCs, ECM composition is the cumulation of mRNA and protein synthesis processes that occur in the cells, which go through various differentiation stages and thus adapt their mRNA and protein levels throughout differentiation.
Ultimately, at this point of our study and according to our protocol, BM-MSCs appear to be the most attractive cell type for producing a hyaline-like cartilage in vitro, as summarized in Table 2. Nevertheless, we have to be careful about a possible use in vivo. Indeed, the atypical type I collagen synthesis throughout the differentiation protocol remained but could be prevented by culturing MSCs in perfusion bioreactors in dynamic conditions that may help to produce a neo-cartilage substitute in vitro with higher hyaline quality and superior function.
References
Brandt, K. D., Dieppe, P., & Radin, E. L. (2008). Etiopathogenesis of osteoarthritis. Rheumatic Diseases Clinics of North America, 34, 531–559.
Mobasheri, A., Kalamegam, G., Musumeci, G., & Batt, M. E. (2014). Chondrocyte and mesenchymal stem cell-based therapies for cartilage repair in osteoarthritis and related orthopaedic conditions. Maturitas, 78, 188–198.
Brooks, P. M. (2006). The burden of musculoskeletal disease-a global perspective. Clinical Rheumatology, 25, 778–781.
Brittberg, M., Lindahl, A., Nilsson, A., Ohlsson, C., Isaksson, O., & Peterson, L. (1994). Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. The New England Journal of Medicine, 331, 889–895.
Benya, P. D., Padilla, S. R., & Nimni, M. E. (1978). Independent regulation of collagen types by chondrocytes during the loss of differentiated function in culture. Cell, 15, 1313–1321.
Demoor, M., Ollitrault, D., Gomez-Leduc, T., Bouyoucef, M., Hervieu, M., Fabre, H., Lafont, J., Denoix, J.-M., Audigié, F., Mallein-Gerin, F., Legendre, F., & Galera, P. (2014). Cartilage tissue engineering : Molecular control of chondrocyte differentiation for proper cartilage matrix reconstruction. Biochimica et Biophysica Acta, 1840, 2414–2440.
Dewan, A. K., Gibson, M. A., Elisseeff, J. H., & Trice, M. E. (2014). Evolution of autologous chondrocyte repair and comparison to other cartilage repair techniques. BioMed Research International, 2014, 1–11.
Pittenger, M. F., Mackay, A. M., Beck, S. C., Jaiswal, R. K., Douglas, R., Mosca, J. D., Moorman, M. A., Simonetti, D. W., Craig, S., & Marshak, D. R. (1999). Multilineage potential of adult human mesenchymal stem cells. Science, 284, 143–147.
Hass, R., Kasper, C., Böhm, S., & Jacobs, R. (2011). Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Communication and Signaling: CCS, 9, 12.
McCarthy, H. E., Bara, J. J., Brakspear, K., Singhrao, S. K., & Archer, C. W. (2012). The comparison of equine articular cartilage progenitor cells and bone marrow-derived stromal cells as potential cell sources for cartilage repair in the horse. Veterinary Journal, 192, 345–351.
Kanawa, M., Igarashi, A., Ronald, V. S., Higashi, Y., Kurihara, H., Sugiyama, M., Saskianti, T., Pan, H., & Kato, Y. (2013). Age-dependent decrease in the chondrogenic potential of human bone marrow mesenchymal stromal cells expanded with fibroblast growth factor-2. Cytotherapy, 15, 1062–1072.
Kern, S., Eichler, H., Stoeve, J., Klüter, H., & Bieback, K. (2006). Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells, 24, 1294–1301.
Lo, W. C., Chen, W. H., Lin, T. C., Hwang, S. M., Zeng, R., Hsu, W. C., Chiang, Y. M., Liu, M. C., Williams, D. F., & Deng, W. P. (2013). Preferential therapy for osteoarthritis by cord blood MSCs through regulation of chondrogenic cytokines. Biomaterials, 34, 4739–4748.
Branly, T., Bertoni, L., Contentin, R., Rakic, R., Gomez-Leduc, T., Desancé, M., Hervieu, M., Legendre, F., Jacquet, S., Audigié, F., Denoix, J.-M., Demoor, M., & Galéra, P. (2017). Characterization and use of equine bone marrow mesenchymal stem cells in equine cartilage engineering. Study of their hyaline cartilage forming potential when cultured under hypoxia within a biomaterial in the presence of BMP-2 and TGF-ß1. Stem Cell Rev,5, 611–630.
Desancé, M., Contentin, R., Bertoni, L., Gomez-Leduc, T., Branly, T., Jacquet, S., Betsch, J.-M., Batho, A., Legendre, F., Audigié, F., Galéra, P., & Demoor, M. (2018). Chondrogenic differentiation of defined equine mesenchymal stem cells derived from umbilical cord blood for use in cartilage repair therapy. International Journal of Molecular Sciences, 19, 537.
Rakic, R., Bourdon, B., Demoor, M., Maddens, S., Saulnier, N., & Galéra, P. (2018). Differences in the intrinsic chondrogenic potential of equine umbilical cord matrix and cord blood mesenchymal stromal/stem cells for cartilage regeneration. Scientific Reports, 8, 13799.
Aubert-Foucher, E., Mayer, N., Pasdeloup, M., Pagnon, A., Hartmann, D., & Mallein-Gerin, F. (2014). A unique tool to selectively detect the chondrogenic IIB form of human type II procollagen protein. Matrix Biology, 34, 80–88.
Dominici, M., Le Blanc, K., Mueller, I., Slaper-Cortenbach, I., Marini, F., Krause, D., Deans, R., Keating, A., Prockop, D. J., & Horwitz, E. (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy, 8, 315–317.
Pérez-Silos, V., Camacho-Morales, A., & Fuentes-Mera, L. (2016). Mesenchymal stem cells subpopulations: Application for orthopedic regenerative medicine. Stem Cells International, 2016(3187491), 1–9.
McLeod, C. M., & Mauck, R. L. (2017). On the origin and impact of mesenchymal stem cell heterogeneity: New insights and emerging tools for single cell analysis. Eur. Cells Mater., 34, 217–231.
Li, H., Ghazanfari, R., Zacharaki, D., Lim, H. C., & Scheding, S. (2016). Isolation and characterization of primary bone marrow mesenchymal stromal cells. Ann. N.Y. Acad. Sci.,1370, 109–118.
Arufe, M. C., De la Fuente, A., Fuentes, I., de Toro, F. J., & Blanco, F. J. (2010). Chondrogenic potential of subpopulations of cells expressing mesenchymal stem cell markers derived from human synovial membranes. Journal of Cellular Biochemistry, 111, 834–845.
De Schauwer, C., Piepers, S., Van de Walle, G. R., Demeyere, K., Hoogewijs, M. K., Govaere, J. L., Braeckmans, K., Van Soom, A., & Meyer, E. (2012). In search for cross-reactivity to immunophenotype equine mesenchymal stromal cells by multicolor flow cytometry. Cytometry. Part A, 81, 312–323.
Hackett, C. H., Flaminio, M. J., & Fortier, L. A. (2011). Analysis of CD14 expression levels in putative mesenchymal progenitor cells isolated from equine bone marrow. Stem Cells and Development, 20, 721–735.
Braun, J., Hack, A., Weis-Klemm, M., Conrad, S., Treml, S., Kohler, K., Walliser, U., Skutella, T., & Aicher, W. K. (2010). Evaluation of the osteogenic and chondrogenic differentiation capacities of equine adipose tissue-derived mesenchymal stem cells. American Journal of Veterinary Research, 71, 1228–1236.
Branly, T., Contentin, R., Desancé, M., Jacquel, T., Bertoni, L., Jacquet, S., Mallein-Gerin, F., Denoix, J.-M., Audigié, F., Demoor, M., & Galéra, P. (2018). Improvement of the chondrocyte-specific phenotype upon equine bone marrow mesenchymal stem cell differentiation: Influence of culture time, transforming growth factors and type I collagen siRNAs on the differentiation index. International Journal of Molecular Sciences, 19, 435.
Archer, C. W., & Francis-West, P. (2003). The chondrocyte. The International Journal of Biochemistry & Cell Biology, 35, 401–404.
Mayer, N., Lopa, S., Talò, G., Lovati, A. B., Pasdeloup, M., Riboldi, S. A., Moretti, M., & Mallein-Gerin, F. (2016). Interstitial perfusion culture with specific soluble factors inhibits type I collagen production from human osteoarthritic chondrocytes in clinical-grade collagen sponges. PLoS One, 11, e0161479. https://doi.org/10.1371/journal.pone.0161479.
White, J. L., Walker, N. J., Hu, J. C., Borjesson, D. L., & Athanasiou, K. A. (2018). A comparison of bone marrow and cord blood mesenchymal stem cells for cartilage self-assembly. Tissue Engineering. Part A, 24, 1262–1272.
Acknowledgements
We are grateful to Marylin Guillamin for her help for flow cytometry analyses and to Marie-Christine Enos for her administrative and technical support.
Role of the Funding Sources
This research was funded by the French National Research Agency (ANR) and by the Regional Council of Basse-Normandie through the ANR TecSan PROMOCART program to P.G. (917RB020 and 917RB072 respectively), by the French Ministry of Research and Technology to PG, by three ERDF (European Regional Development Funds) grants to P.G. and M.D. (HIPPOCART 1 no. 2897/33535, 917RB148; HIPPOCART 917CB174, EQUISTEM 917CB210), by a Regional Council of Basse-Normandie program to PG and MD (HIPPOCART N° 2013-AGRI-236/13P07492, 917CB166), by Fonds Eperon to PG and MD (EQUISTEM, N80–2014, 917CB194), by the GIS CENTAURE-equine research (EQUISTEM-G program to PG, 014CJ061), by ERDF and Regional Council of Basse-Normandie grant to P.G. and M.D. in the CPER Centaure program (2015–2020, Centaure Biotechnologies 2015 N° 917CB213&Centaure PREACH 2016–17). CENTAURE is an European project co-funded by the Normandy County Council, European Union in the framework of the ERDF-ESF operational program 2014–2020. These funding sources had no involvement in the study design, collection, analysis, or in interpretation of the data, in the writing of the manuscript, or in the decision to submit the manuscript for publication.
RC was supported by a PhD scholarship co-financed by the Fonds Eperon and the Regional Council of Basse-Normandie, and TB by a PhD fellowship from the French Ministry of Research and Technology. MDes was recipient from a PhD fellowship from the Regional Council of Basse-Normandie. MC was supported by an ERASMUS program (ERASMUS center of Parma University, Italy).
Author information
Authors and Affiliations
Contributions
RC and TB were involved in study design, in data collection/assembly and analysis, and performed experiments. TB, RC, MD, MDes and FA were involved in collecting BM- and UCB-MSCs. MC and MDes performed experiments, collected and analysed the data. FA analysed the data. PG and MD were involved in study conception and design, in supervision of the study, in data analysis and interpretation, and provide the financial support. RC wrote the original draft and participated to the responses to the reviewers with PG. PG and TB were involved in writing-reviewing, realized the critical revision of the article and the final approval. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Disclosure Statement
No conflict of interests is declared by all the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 24.6 mb)
Rights and permissions
About this article

Cite this article
Contentin, R., Demoor, M., Concari, M. et al. Comparison of the Chondrogenic Potential of Mesenchymal Stem Cells Derived from Bone Marrow and Umbilical Cord Blood Intended for Cartilage Tissue Engineering. Stem Cell Rev and Rep 16, 126–143 (2020). https://doi.org/10.1007/s12015-019-09914-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-019-09914-2