
Abstract
Dramatic advances have been made in the understanding of cancer over the past decade. Prime among those are better appreciation of the biology of cancer and the development of targeted therapies. Despite these improvements, however, most tumors remain refractory to anti-cancer medications and frequently recur. Cancer Stem Cells (CSCs), which in some cases express markers of pluripotency (e.g., Oct-4), share many of the molecular features of normal stem cells. These cells have been hypothesised to play a role in tumor resistance and relapse. They exhibit dependence on many primitive regulatory pathways and may be best viewed in the context of embryonic signaling pathways. In this article, we review important embryonic signaling cascades and their differential expression in CSCs. We also discuss these pathways as actionable targets for novel therapies in hopes that eliminating cancer stem cells will lead to an improvement in overall survival for patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A recurring clinical observation in the management of hematologic malignancies has been the discordance between early measures of clinical response and long-term outcome. In many types of cancers (i.e. indolent lymphoma, multiple myeloma), there is no association between the achievement of objective early responses and duration of overall survival [1, 2]. Patients may experience dramatic initial clinical responses to cytotoxic medications only to relapse and succumb to their disease several months or years later. Among the factors that contribute to disease recurrence are epigenetic modulation, microRNAs, epithelial-mesenchymal transition, and micrometastases. Yet another mediator of relapse, accounting for the paradox between response and survival, has to do with understanding of cancer’s hierarchical differentiation structure. It was Rudolf Virchow who has first foreseen that cancer may develop in some embryonic remnants in the adult as an “embryonic development of cancer” [3]. And indeed, human cancers are composed of phenotypically-heterogenous cells that resemble different stages of tissue development. Of the variety of cells in a given tumor, only a small fraction maintains tumorigenic capacity and may give rise to the entire spectrum of cells. Cancer cells of pluripotent nature have been identified in many tumors and their unique biological properties have given rise to the CSC (Cancer Stem Cell) concept. According to this model, primordial cancer-initiating cells that suffer a “critical insult” expand and form the biological origin for the rest of the (differentiated) tumor. These transit-amplifying cells, as they are often termed, exhibit self-renewal capacity and multilineage potential, similar to normal stem cells. CSCs also appear to be relatively resistant to anticancer therapies, and are therefore believed to be partly responsible for disease relapse after conventional-dose chemotherapy. Cytotoxic therapy primarily targets differentiated cancer cells (and thereby may lead to diminishment in tumor bulk), yet its failure to eliminate the rare and biologically-distinct pool of CSCs may render it less likely to affect overall survival. Persistence of CSCs has therefore been hypothesised to explain why complete treatment responses translate only infrequently into long-term clinical remission or cure for the majority of patients.
The Clinical Relevance of Cancer Stem Cells
The central biological question concerning CSCs pertains to their precise relationship and contribution to treatment resistance and relapse. Recent data have shed light on the clinical relevance of CSCs. Following the administration of chemotherapy to patients with breast cancer and acute myeloid leukemia (AML), minimal residual disease (MRD) was found to be enriched for cancer cells with a stem cell phenotype [4–6]. For patients with AML, the presence of CSCs after treatment correlates with shorter progression-free survival and the long-term outcome has been shown to be dictated by the level of residual disease [5]. These insights point at the biological significance of CSCs, not only for substantiating the CSC concept, but also, more practically, as a potential predictive and therapeutic tool.
Interestingly, cancer stem cells express high levels of tumor-associated antigens. Melanoma stem cells, for instance, have a homogenous expression of cancer-testis antigens (CTA) that makes them susceptible to various immunotherapeutic strategies [7]. Similarly, glioma stem cells have a high density of tumor-associated surface antigens, unlike that of differentiated glial cells. Effective targeting of glial and melanoma stem cells by focusing on CTA genes is therefore a strategy that is being currently investigated [8]. In our essay, however, we focus on critical embryonic signaling pathways that are central to cellular growth and survival and the biological ramifications of their biochemical suppression.
CSCs Are Becoming Priority Experimental Targets in Anti-Tumor Therapeutics
Selective inhibition of CSCs (sparing normal stem cells), applied in conjunction with treatments that target differentiated tumor cells, may improve long-term outcomes of cancer patients. For that to be achieved, however, the precise molecular distinction between normal stem cells and CSCs has to be delineated. Embryonic signaling pathways, the likes of Notch, Wnt, and Hedgehog, offer an ideal opportunity for cascade-specific molecular inhibition as they are fundamental to (cancer and normal) stem cell maintenance and growth.
The Notch Pathway: a Communication System Controlling Cell Fate
Signaling through the Notch pathway has critical implications for embryogenesis, cellular differentiation, proliferation, and apoptosis [9]. Local cell interactions between Notch molecules are fundamental for development of a wide range of organ and tissue functions. These include genesis of breast and nervous system, acquisition of normal hematopoiesis, and generation of adequate immune regulation [10]. The binding of a Notch ligand to its receptor is translated into a multitude of transcriptional regulatory events that alter the expression of hundreds of genes, with significant phenotypic consequences. These regulatory signals profoundly affect cells’ proliferation and survival. By determining their fate, the Notch pathway directs individual cells to form multicellular three-dimensional structure with characteristic dimensions.
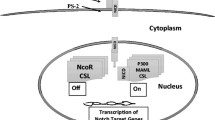
The Notch signaling cascade depends on an interaction between a specific group of surface-bound ligands and transmembrane receptors (Fig. 1). Mammalian Notch ligands are classified into two structurally distinct families: Delta-like ligands (DLLs) 1, 3, and 4, and Jagged ligands 1 and 2 [11]. Pairing of ligand to membrane-spanning receptor (notch 1–4) induces a conformational change of the receptor culminating in a series of cleavage events by gamma-secretase (presenilin) and members of the ADAM protease family [10]. Intracellular and extracellular fragments of the receptor are generated, with the former yielding an active molecule, the notch intracellular domain (niCD) that is released into the cytoplasm. niCD then translocates into the nucleus where it modulates gene expression [12].

The Notch signaling pathway (a) assumes a key role in the regulation of proliferation, differentiation and survival of cells and is vital for normal tissue development and homeostasis. Efforts to alter its pathway have focused on the inhibition of ligand-induced receptor activation, suppression of a membrane-bound enzyme complex (γ-secretase) responsible for generation of NiCD, and interference with nuclear co-activators (MAML) The Wnt signaling pathway (b) regulates cellular morphology, proliferation and motility, and influences cell fate. The activated pathway’s hallmark molecule, β-catenin, leads to gene transcription and a resultant cell growth and survival. Investigational compounds include antibodies with (Wnt) ligand-neutralizing or (Fz) receptor-blocking properties, agents that inhibit (the PDZ domain of) DVL, and those that act at the response-element binding level to diminish gene expression. The Hedgehog signaling pathway (c) controls the proliferation and differentiation of cells, tissue polarity and maintenance of stem cells. Hh-specific modulators include ligand (sonic Hh) inhibitors, receptor (smo) antagonists, and suppressors of the zinc finger protein, Gli. HIF [1, 2] (d) proteins are crucial mediators of hypoxic response. They serve as transcriptional factors and promote angiogenesis and anaerobic metabolism, especially under hypoxic circumstances. Pharmacological suppression of HIF-1α, using echinomycin, limits the growth of lymphoma cancer stem cells (to a greater extent than it affects the growth of normal hematopoietic stem cells) in mice. It also reverses the negative-self-feedback of Hes1, a target gene of Notch
Notch-regulated biological processes (cell differentiation, proliferation, and apoptosis) contribute to the development of cancer. The specific role of the Notch pathway in malignant transformation has therefore been extensively studied in various hematopoietic and solid tumors [13–15]. Understanding of Notch regulation and its context-specific interactions with close pathways is required if Notch-targeted therapeutics are to be designed. Moreover, defining the role of Notch signaling in CSCs is particularly important given the unique function of Notch in cellular development.
Evidence implicating Notch in tumor biology is most rigorous in gliomas, embryonal brain tumors, and breast cancers [16–18]. In Ductal Carcinoma in Situ (DCIS), signaling through Notch has been shown to affect cell survival and self-renewal. Multilineage spheroids (“mammospheres”), grown from epithelial cells of DCIS, serve as a novel laboratory marker of stem cell activity. Formation of these mammospheres is inhibited after exposure to Gamma-Secretase Inhibitors (GSIs) or a Notch-4 monoclonal antibody, both agents that interfere with Notch signaling [19]. The role of Notch signaling in human breast cancer is further substantiated by studies of interleukin-6 (IL-6), an important player in the pathophysiology of cancer. When mammospheres derived from invasive breast cancer tissue are exposed to IL-6, there is profound upregulation of Notch ligand Jagged-1 [20]. By increasing the expression of Jagged-1 mRNA, IL-6 may contribute to mammosphere formation and self-renewal. Moreover, the dual effect of IL-6 and Notch-3 results in upregulation of the carbonic-anhydrase IX (CA-IX) gene, a hypoxia survival element [20]. Both the mammospheres and the cell lines show an increase in their degree of invasiveness and hypoxia resistance following exposure to IL-6 [20]. These findings couple IL-6 with the Notch pathway, associating the two with acquisition of malignant features in breast cancer cells.
Insights about Notch signaling in CSCs, in the context of breast cancer, have also been derived from research of Her2/Neu-overexpressing tumors. Her2 has been shown to support maintenance of CSCs in breast cancer [21]. Targeting of Her-2-positive CSCs is achieved by tratsuzumab, a monoclonal antibody against Her-2. Since the Her2 promoter contains Notch-binding sequences, a biological relationship between the two systems has been suggested. Indeed, cells that overexpress Her2 display enhanced Notch signaling [21]. After administration of a small interfering RNA or a GSI, breast carcinoma cell lines show decreased expression of Her2 and diminished sphere formation [21]. Notch and Her2, dominant pathways at the core of the cancerous breast process, seem to intertwine. The exact nature of their inter-communication, however, has not been fully elucidated. Further meticulous investigation of their biology is mandated for possible selective targeting of breast CSCs in the future.
The Wnt/β-Catenin Pathway: a Multifunctional Signaling Cascade
Wnt signaling is essential for embryonic development and tissue homeostasis in adult tissues. Similar to the Notch pathway, it controls proliferation of differentiated cells, determines their eventual fate, and regulates self-renewal of stem cells [22]. The functional versatility of the Wnt pathway is impressive, considering its role in directing the development of various organ systems, including the cardiovascular, lung, renal, and central nervous systems [23].
Wnt proteins consist of a family of secreted glycoproteins that act as ligands for the Frizzled (Fz) transmembrane receptor [24] (Fig. 1). Wnt homologs interact with 10 known mammalian Fz receptors resulting in a broad spectrum of cellular responses. Cytoplasmic β-catenin, the hallmark of Wnt pathway activation, is normally maintained in low level owing to proteasome-mediated degradation. Such steady-state is made possible through the action of a multiprotein “destruction” complex that consists of adenomatous polyposis coli (APC), axin, and glycogen synthase kinase-3β (GSK-3β) [25]. Wnt signaling is also modulated by Secreted Frizzled-Related Proteins (sFRP), soluble factors that act either as stimulators or (most commonly) as antagonists of the pathway. Upon binding of Wnt proteins to the receptor complex, disheveled (Dvl), a protein downstream of the receptor, is phosphorylated (to its active state). It is primarily the activity of Dvl as inhibitor of GSK-3β that leads to accumulation of β-catenin in the cytoplasm [25]. β-catenin then translocates to the nucleus, where it forms a complex with members of the T cell factor (TCF) lymphoid enhancer factor family of transcription factors. Co-activators of transcription (cyclic AMP response element-binding protein, p300) are recruited to activate the basal transcription machinery [26, 27]. A host of target genes are subsequently expressed.
Impaired regulation of the Wnt pathway plays a role in neoplastic proliferation. Somatic mutations in this pathway have been associated with colorectal cancer, while germline mutations have been linked to hereditary diseases [22]. In breast cancer, negative regulators of Wnt signaling (e.g. Frizzled-related protein 1) are often inactivated, and positive regulators (e.g. Dvl) are overexpressed [28, 29]. Wnt signaling has been hypothesized to influence stem cell differentiation, although the extent to which it contributes to proliferation and multipotency of these cells remains to be defined. In chronic myelogenous leukemia (CML), an intriguing association has been found between progression from chronic phase to blast crisis and increased β-catenin levels [28]. Resistance to imatinib in CML has similarly been correlated with increased levels of β-catenin [30]. The mechanism for drug resistance relates to the effect of Wnt/β-catenin signaling in enhancing transcription of multidrug resistance genes (MDR-1, ABCG2, and ABCA3) in stem cells [31].
Given the involvement of the Wnt pathway in oncogenesis, attempts are being made to attenuate the transcriptional activity of β-catenin. Natural compounds offer unique insights into the signaling cascade and may be utilized therapeutically. For instance, nonsteroidal anti-inflammatory drugs (NSAIDs) lead to degradation of TCFs (independent of COX-2 suppression) and thus inhibit the Wnt pathway [32–34]. This may partly explain the chemopreventive effects of NSAIDs against colorectal cancer. Other compounds thought to inhibit carcinogenesis are derivatives of vitamin A and the active form of vitamin D. In the clinical setting, a form of retinoic acid (all-trans retinoic acid; ATRA) is used in the treatment of acute promyelocytic leukemia. The precise nature by which vitamins affect the Wnt signaling pathway is not entirely clear. However, there is evidence that nuclear receptors that are activated by vitamins compete with TCFs, and that they may increase synthesis of proteins that suppress Wnt signaling [35].
Active pursuit of inhibitors to the Wnt pathway is under way. A small molecule inhibitor, FJ9, has recently been shown to suppress transduction of extracellular signals to downstream factors [36]. FJ9 works by blocking the interaction between the Fz receptor and PDZ, a crucial domain of Dvl that is responsible for protein-protein interactions. By this mechanism, it limits accumulation of cytoplasmic β-catenin and subsequent nuclear events [36]. Human melanoma cell lines grown with FJ9 show heightened levels of apoptosis, and mouse xenograft models inhibit tumor growth following exposure to the same compound [36].
The Hedgehog Pathway: from Gradient to Morphology
A highly conserved pathway, Hedgehog (Hh) plays a crucial role in embryogenesis and morphogenesis of specific organs. Cellular functions contributed to by the Hh pathway are varied and include maintenance of patterning, tissue polarity, and stem cell support [37]. What allows a single morphogen to elicit distinct molecular responses and affect a diversity of aspects of organ development is still a mystery. However, the unique nature of Hh signaling – being both short- and long-range, direct and indirect, and concentration-dependent – may be part of the explanation. The dose-dependent effects of morphogens in the Hh pathway are particularly crucial for development of the nervous system, where proteins act from a distance to establish different cell identities in the ventral spinal cord. Processing, secretion and transport of proteins contribute to generation of the Hh gradient, which is an important requisite for cell growth and survival.
Hh signaling is initiated by the binding of a Hh ligand (Sonic, Indian, or Desert) to the transmembrane receptor Ptch1 [38] (Fig. 1). In the absence of Hh ligand, Ptch1 inhibits the adjacent receptor smoothened (smo), presumably by suppressing its localization to the primary cilium [39]. When Ptch1 is occupied by one of the mentioned ligands, the inhibition of Ptch1 on smo is relieved. Smo then migrates from the plasma membrane to the primary cilium, and mediates activation of the glioma-associated (GLI) family of zinc fingers transcriptional regulators [38]. The balance of activation versus repression of the latter dictates target gene transcription.
Aberrant regulation of Hh signaling has been recognised as a contributor to tumorigenesis in various tissues, including the colon, lung, skin and prostate [40]. Mutations in Ptch1 were first discovered in Gorlin syndrome, a rare hereditary disorder characterized by skeletal abnormalities and increased propensity to develop basal cell carcinoma (BCC) and medulloblastoma [41]. These and other mutations in various components of the Hh cascade (i.e smo, suppressor of fused) have since been found in patients with BCC and medulloblastoma, substantiating the role of this pathway in oncogenesis. Interestingly, most human cancers do not manifest activating mutations of Hh pathway components. Rather, it is overexpression of ligands that results, via autocrine or juxtacrine signaling, in heightened pathway activity. In addition, paracrine Hh signaling has been receiving increasing attention, as the role of the local microenvironment is now better appreciated in multiple myeloma and lymphoma.
Research of hematologic malignancies has also corroborated the role of the Hh pathway in regulation of CSCs. Activation of Hh signaling in multiple myeloma has been shown to trigger CSC proliferation whereas suppression of the pathway has been found to induce terminal differentiation and restrict self-renewal [41, 42]. In CML, smo has been found to augment the number of CML stem cells and its loss has been associated with stem cell depletion [43, 44]. Induction of CML and renewal of pathologic stem cells are therefore thought to be mediated by components of the Hh pathway. Metastatic spread of solid tumors also depends on the Hh pathway. In both epithelial cells of human colon carcinomas and their stem cells, high Hh signature has been associated with dissemination of CSCs, and shown to induce epithelial-to-mesenchymal transition (EMT) [45].
The most extensively researched inhibitor of the Hh pathway is Cyclopamine, a steroidal alkaloid derived from a plant. Cyclopamine binds to smo and leads to its deactivation [46]. It has been shown to restrict the growth of tumors in numerous in vivo and in vitro models [47]. Vismodegib, also an inhibitor or smo, has been administered to patients with different solid malignancies as part of clinical trials [48]. A clinical benefit has been found for patients with locally advanced or metastatic BCC (55 % clinical response rate) and for those with meduloblastoma. Vismodegib has been approved by the FDA for treatment of advanced or metastatic BCC. Two other inhibitors of smo, PF04449913 and lDe225, are being investigated in a phase 1 trials in patients with CML and BCC, respectively [10]. iPi926, an inhibitor of the Hh pathway that is a derivative of cyclopamine, is being evaluated in a randomised, double-blind, phase-II clinical trials for advanced-stage solid tumors [10].
Hypoxia-Inducible Factor 1 and 2: the Orchestrator of Hypoxia Response
One of the key drivers of cancer cell biology is hypoxia. Cancer cells adapt to hypoxic microenvironments by acquiring gene mutations that prove advantageous by facilitating angiogenesis, tissue invasion, nutrient metabolism and metastasis. Even more critical is the effect of low oxygen on CSCs. Hypoxia alters the expression of stem cell markers in various tumor cells and shifts the balance between stem cell maintenance and differentiation. If CSCs have a unique hypoxia response, its pinpointing could possibly lead to development of strategies that specifically target those cells. A recent series of experiments investigated that biological conundrum.
In the cancer niche, oxygen level is translated into an angiogenic signature through complex protein interaction and downstream transcriptional modulation of hundreds of genes. The master regulator of these responses to hypoxia is a transcriptional family of proteins named Hypoxia-Inducible Factor (HIF). The HIFs function as heterodimers and are composed of α subunit, whose expression depends on oxygen level, and β subunit, which is constitutively synthesized (Fig. 1). Upon normoxia, prolyl residues on the α subunit of HIF undergo hydroxylation (in an O2-dependent step). This hydroxylation increases the affinity of HIF to the Von-Hippel Lindau (VHL) protein, the interaction with which promotes ubiquitilation and degradation of HIF. In contrast, hypoxic states do not result in an interaction between HIFα and VHL (as the α subunit is not hydroxylated), leading instead to dimerization of both subunits of HIF and subsequent activation of genes responsible for cell survival, motility, and metabolism.
Li et al. recently mapped the hypoxia gene response profile in glioma stem cells (GSC) and non-stem cells in culture [49]. They showed that GSC, compared with non-stem cells, differentially express HIF2α mRNA (and produce higher levels of HIF2α protein) under both hypoxic and normoxic conditions. HIF2α mRNA generation was blocked by treatment with actinomycin D, an inhibitor of transcription, supporting the notion that increased mRNA level in GSC is the result of enhanced transcription rather than increased transcript stability. Also specific to GSC (in contrast to non-stem cells), in the same study, was the impairment of cell growth following knockdown of HIF2α, which emphasises the importance of this protein for GSC growth and survival. Knockdown of HIF2α inhibited self-renewal and proliferation in vitro and diminished tumor initiating potential in vivo.
A study by Wang et al. connected the intriguing dots of CSCs, hypoxia, and embryonic signaling pathways in lymphoma [50]. The researchers found that a subset of c-Kit+Sca-1+ lymphoma CSCs in mice have high levels of HIF1α protein and that they downregulate VHL under normoxic conditions. Maintenance of the same lymphoma stem cell subset required signaling through HIF1α. When cells in culture were exposed to echinomycin, a peptide antibiotic that intercalates into DNA to inhibit binding of HIF1α, it suppressed the colony forming unit of lymphoma CSCs, but not that of normal hematopoietic progenitor cells. The investigators then compared the sensitivity of their (c-Kit+Sca-1+) lymphoma CSCs and normal hematopoietic (c-Kit+Sca-1+) cells to echinomycin. They found that the former were much more sensitive to the drug than the latter, suggesting that it might be possible to selectively eliminate CSCs while minimising untoward effects on normal hematopoietic progenitor cells. Finally, they uncovered an interesting cross-talk between HIF1 and the Notch signaling pathway: the expression of Hes1, a target gene of Notch was increased in the c-Kit+Sca-1+ lymphoma subset. HIF1α appeared to block a negative feedback autoregulation of Hes1, thus allowing self-renewal of CSCs (Fig. 1). Knocking down HIF1α resulted in decreased expression of Hes1. Moreover, a dominant-negative regulator of Notch signaling decreased the percentage of c-Kit+Sca-1+ cells. Subsequent transplantation of these cells into syngeneic mice resulted in delayed development of lymphoma.
The findings of Li et al. and Wang et al. raise important questions [49, 50]. First, would the extensive overlap between embryonic signaling pathways alter the efficacy of selective pharmacological inhibition at the stem cell level, and increase the likelihood of drug resistance and relapse? The HIF cascade is embedded within myriad collaterals, so it is reasonable to expect some degree of in-vivo compensation by cross-talk circuits (Notch, Wnt, and Hedgehog), and even between HIF1α (predominant in lymphoma and AML) and HIF2α (predominant in glioma). Secondly, how specific could HIF1α/ HIF2α targeting be given our current understanding of stem cell pathobiology? Novel interventions should focus on a crucial cellular function that, biologically-speaking, is more characteristic of CSC than of normal stem/non-stem cell. However, the mentioned pathways execute essential roles in homeostatic tissue repair and regeneration, making strict elimination of CSCs a challenging goal. Finally, the long-term adverse effects of even a modest suppression of the normal hematopoietic pool remain unclear. Would higher rates of malignancy, marrow aplasia, or autoimmune disorders be seen? Further studies will be required to answer these unknowns and gradually translate CSC insights into useful clinical wisdom.
Conclusions
A growing body of evidence shows that CSCs play an important role in tumor resistance and relapse. In a variety of cancer settings, residual disease enriched by CSCs has been associated with worse prognosis, strengthening the notion that these cells should be selectively targeted if long-term clinical remissions are to be achieved. Intensive scientific efforts are currently underway, with the attempt to better understand cancer-related embryonic pathways and their distinctive expression patterns in CSCs versus in non-cancer stem cells. These insights will help us achieve a more granular comprehension of biologically-conserved cascades, their functionality in various tumor environments, and their pharmacologic predisposition. As our mastery of the biology of CSCs improves, we may be better able to effectively manipulate some of these these pathways - extracting the root of the cancer rather than its branches - while hoping for improvements in patient outcomes.
References
Horning, S. J., & Rosenberg, S. A. (1984). The natural history of initially untreated low-grade non-Hodgkin’s lymphomas. The New England Journal of Medicine, 311, 1471–1475.
Durie, B. G., Jacobson, J., Barlogie, B., & Crowley, J. (2004). Magnitude of response with myeloma frontline therapy does not predict outcome: importance of time to progression in southwest oncology group chemotherapy trials. Journal of Clinical Oncology, 22, 1857–1863.
Virchow, R. (1855). Editorial archive fuer pathologische. Anatomie und Physiologie fuer klinische Medizin., 8, 23–54.
Jordan, C. T., Guzman, M. L., & Noble, M. (2006). Cancer stem cells. The New England Journal of Medicine, 355, 1253–1261.
Gerber, J. M., Smith, B. D., Ngwang, B., et al. (2011). The clinical relevance of acute myeloid leukemia stem cells. Blood, 118, 240a.
Creighton, C. J., Li, X., Landis, M., et al. (2009). Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences, 106, 13820–13825.
Sigalotti, L., Covre, A., et al. (2008). Cancer testis antigens in human melanoma stem cells: expression, distribution, and methylation status. Journal of Cellular Physiology, 215(2), 287–291. doi:10.1002/jcp.21380.
Yawata, T., Nakai, E., et al. (2010). Enhanced expression of cancer testis antigen genes in glioma stem cells. Molecular Carcinogenesis, 49(6), 532–544. doi:10.1002/mc.20614.
Artavanis-Tsakonas, S., Rand, M. D., & Lake, R. J. (1999). Notch signaling: cell fate control and signal integration in development. Science, 284, 770–776.
Takebe, N., Harris, P. J., Warren, R. Q., & Ivy, S. P. (2011). Targeting cancer stem cells by inhibiting Wnt, notch, and hedgehog pathways. Nature reviews Clinical oncology, 8, 97–106.
Dontu, G., et al. (2004). Role of notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Research, 6, R605–R615.
Kadesch, T. (2000). Notch signaling: a dance of proteins changing partners. Experimental Cell Research, 260, 1–8.
Allenspach, E. J., Maillard, I., Aster, J. C., & Pear, W. S. (2002). Notch signaling in cancer. Cancer Biology & Therapy, 1, 466–476.
Koch, U., & Radtke, F. (2007). Notch and cancer: a double-edged sword. Cellular and Molecular Life Science, 64, 2746–2762.
Roy, M., Pear, W. S., & Aster, J. C. (2007). The multifaceted role of notch in cancer. Current Opinion in Genetics & Development, 17, 52–59.
Fan, X., Khaki, L., Zhu, T. S., et al. (2010). Notch pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells, 28, 5–16.
Fan, X., Matsui, W., Khaki, L., et al. (2006). Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Research, 66, 7445–7452.
Farnie, G., & Clarke, R. B. (2007). Mammary stem cells and breast cancer-role of notch signalling. Stem Cell Reviews, 3, 169–175.
Farnie, G., Clarke, R. B., Spence, K., et al. (2007). Novel cell culture technique for primary ductal carcinoma in situ: role of notch and epidermal growth factor receptor signaling pathways. Journal of the National Cancer Institute, 99, 616–627.
Sansone, P., Storci, G., Tavolari, S., et al. (2007). IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. The Journal of Clinical Investigation, 117, 3988–4002.
Magnifico, A., Albano, L., Campaner, S., et al. (2009). Tumorinitiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are trastuzumab sensitive. Clinical Cancer Research, 15, 2010–2021.
Clevers, H. (2006). Wnt/β-catenin signaling in development and disease. Cell, 127(3), 469–480.
Grigoryan, T., Wend, P., Klaus, A., & Birchmeier, W. (2008). Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of betacatenin in mice. Genes & Development, 22, 2308–2341.
Angers, S., & Moon, R. T. (2009). Proximal events in Wnt signal transduction. Nature Reviews Molecular Cell Biology, 10, 468–477.
Takahashi-Yanaga, F., & Kahn, M. (2010). Targeting Wnt signaling: can We safely eradicate cancer stem cells? Clinical Cancer Research, 16, 3153–3162.
Hecht, A., Vleminckx, K., Stemmler, M. P., van Roy, F., & Kemler, R. (2000). The p300/CBP acetyltransferases function as transcriptional coactivators of β-catenin in vertebrates. The EMBO Journal, 19, 1839–1850.
Takemaru, K. I., & Moon, R. T. (2000). The transcriptional coactivator CBP interacts with β-catenin to activate gene expression. The Journal of Cell Biology, 149, 249–254.
Nagahata, T., Shimada, T., Harada, A., et al. (2003). Amplification, up-regulation and over-expression of DVL-1, the human counterpart of the drosophila disheveled gene, in primary breast cancers. Cancer Science, 94, 515–518.
Ugolini, F., Adélaïde, J., Charafe-Jauffret, E., et al. (1999). Differential expression assay of chromosome arm 8p genes identifies frizzled-related (FRP1/FRZB) and fibroblast growth factor receptor 1 (FGFR1) as candidate breast cancer genes. Oncogene, 18, 1903–1910.
Jamieson, C. H., Weissman, I. L., & Passegue, E. (2004). Chronic versus acute myelogenous leukemia: a question of self-renewal. Cancer Cell, 6, 531–533.
Hirschmann-Jax, C., Foster, A. E., Wulf, G. G., Goodell, M. A., & Brenner, M. K. (2004). A distinct “side population” of cells with high drug efflux capacity inhuman tumor cells. Proceedings of the National Academy of Sciences, 101, 14228–14233.
Chan, T. A. (2002). Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. The Lancet Oncology, 3, 166–174.
Thun, M. J., Henley, S. J., & Patrono, C. (2002). Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. Journal of the National Cancer Institute, 94, 252–266.
Baron, J. A., Cole, B. F., Sandler, R. S., et al. (2003). A randomized trial of aspirin to prevent colorectal adenomas. The New England Journal of Medicine, 348, 891–899.
Shah, S., Hecht, A., Pestell, R., & Byers, S. W. (2003). Trans-repression of β- catenin activity by nuclear receptors. The Journal of Biological Chemistry, 278, 48137–48145.
Fujii, N., You, L., Xu, Z., et al. (2007). An antagonist of dishevelled proteinprotein interaction suppresses β-catenin-dependent tumor cell growth. Cancer Research, 67, 573–579.
Ingham, P. W., & McMahon, A. P. (2001). Hedgehog signaling in animal development: paradigms and principles. Genes & Development, 15, 3059–3087.
Fuccillo, M., Joyner, A. L., & Fishell, G. (2006). Morphogen to mitogen: the multiple roles of hedgehog signalling in vertebrate neural development. Nature Reviews Neuroscience, 7, 772–783.
Stone, D. M., et al. (1996). The tumor-suppressor gene patched encodes a candidate receptor for sonic hedgehog. Natur., 384, 129–134.
Ingham, P. W., & McMahon, A. P. (2011). Hedgehog signaling in animal development: paradigms and principles. Genes & Development, 15, 3059–3087.
Hahn, H., et al. (1996). Mutations of the human homolog of drosophila patched in the nevoid basal cell carcinoma syndrome. Cell, 85, 841–851.
Peacock, C. D., Wang, Q., Gesell, G. S., Corcoran-Schwartz, I. M., Jones, E., Kim, J., et al. (2007). Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proceedings of the National Academy of Sciences, 104, 4048–4053.
Zhao, C., Chen, A., Jamieson, C. H., Fereshteh, M., Abrahamsson, A., Blum, J., et al. (2009). Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature, 458, 776–779.
Liu, S., Dontu, G., Mantle, I. D., Patel, S., Ahn, N. S., Jackson, K. W., et al. (2006). Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Research, 66, 6063–6071.
Varnat, F., et al. (2009). Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Molecular Medicine, 1, 338–351.
Taipale, J., et al. (2000). Effects of oncogenic mutations in smoothened and patched can be reversed by cyclopamine. Nature, 406, 1005–1009.
McMillan, R., & Matsui, W. (2012). Molecular pathways: the hedgehog signaling pathway in cancer. Clinical Cancer Research, 18, 4883–4888.
LoRusso, P. M., Rudin, C. M., Reddy, J. C., Tibes, R., Weiss, G. J., Borad, M. J., et al. (2011). Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clinical Cancer Research, 17, 2502–2511.
Li, Z., Bao, S., Wu, Q., Wang, H., Eyler, C., Sathornsumetee, S., et al. (2009). Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell, 15, 501–513.
Wang, Y., Lui, Y., Malek, S. N., Zheng, P., & Liu, Y. (2011). Targeting HIF1a eliminates cancer stem cells in hematological malignancies. Cell Stem Cell, 8, 399–411.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Authors declare that there are no relevant competing interests. Also, no funding was used in preparation of this manuscript.
Author Contribution
OO and BDS are responsible for the conception and design of the manuscript. OO wrote the first draft. BDS critically revised the draft. OO and BDS finalised a final version and approved it.
Rights and permissions
About this article

Cite this article
Oren, O., Smith, B.D. Eliminating Cancer Stem Cells by Targeting Embryonic Signaling Pathways. Stem Cell Rev and Rep 13, 17–23 (2017). https://doi.org/10.1007/s12015-016-9691-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-016-9691-3