
Abstract
The generation of human induced pluripotent stem cells (hiPSCs) with a high differentiation potential provided a new source for hepatocyte generation not only for drug discovery and in vitro disease models, but also for cell replacement therapy. However, the reported hiPSC-derived hepatocyte-like cells (HLCs) were not well characterized and their transplantation, as the most promising clue of cell function was not reported. Here, we performed a growth factor-mediated differentiation of functional HLCs from hiPSCs and evaluated their potential for recovery of a carbon tetrachloride (CCl4)-injured mouse liver following transplantation. The hiPSC-derived hepatic lineage cells expressed hepatocyte-specific markers, showed glycogen and lipid storage activity, secretion of albumin (ALB), alpha-fetoprotein (AFP), urea, and CYP450 metabolic activity in addition to low-density lipoprotein (LDL) and indocyanin green (ICG) uptake. Similar results were observed with human embryonic stem cell (hESC)-derived HLCs. The transplantation of hiPSC-HLCs into a CCl4-injured liver showed incorporation of the hiPSC-HLCs into the mouse liver which resulted in a significant enhancement in total serum ALB after 1 week. A reduction of total serum LDH and bilirubin was seen when compared with the control and sham groups 1 and 5 weeks post-transplantation. Additionally, we detected human serum ALB and ALB-positive transplanted cells in both the host serum and livers, respectively, which showed functional integration of transplanted cells within the mouse livers. Therefore, our results have opened up a proof of concept that functional HLCs can be generated from hiPSCs, thus improving the general condition of a CCl4-injured mouse liver after their transplantation. These results may bring new insights in the clinical applications of hiPSCs once safety issues are overcome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
From the first human liver transplantation in 1992 [1] until now, orthotopic liver transplantation (OLT) has been the only effective cure for end stage liver patients as well as many genetic liver disorders [2]. However in addition to the risk of surgery itself, there are two major problems with OLT: lack of a suitable donor and the risk of organ rejection [3]. Therefore, alternative therapeutic approaches, such as cell therapy, are needed to overcome these problems. Hepatocyte transplantation has been shown to be effective and minimally invasive, but there is a worldwide shortage for human livers [1, 4]. Many other cell sources have been proposed to generate or replace human hepatocytes [5, 6]. The discovery of human induced pluripotent stem cells (hiPSCs) [7, 8] have raised hopes for curing diseases with poor prognoses. Rather than their use in regenerative medicine, hiPSCs or their derivatives can serve as good tools in drug toxicity and drug discovery. In fact, they can provide us with a “disease in culture” [5].
The wide differentiation potential of these cells provides an invaluable potential for contributing to the field of regenerative medicine. hiPSCs avoid the ethical concerns surrounding human embryonic stem cell (hESC) research and development, therefore these cells can serve as an unlimited cell source for advancement of patient-specific pluripotent stem cell lines and the generation of lines suitable for clinical applications. It has been demonstrated that hepatocyte-like cells (HLCs) can be produced from hiPSCs [9–13], using protocols similar to human embryonic stem cells (hESCs). The efficacy of these approaches has been demonstrated using induced pluripotent stem cells of diverse origins. However, the reported hiPSC-HLCs have not been well characterized when compared with hESC-HLCs and cell transplantation, as the most promising clue of cell function was not performed. Here we used a three-step, developmental-based protocol for efficient derivation of HLCs from hiPSCs and hESCs. The differentiated cells were subsequently evaluated for mRNA and protein hallmarks at each step and functional assays were performed at the last stage. Finally, we transplanted our terminally differentiated cells into a carbon tetrachloride (CCl4)-injured mouse liver. Histopathology results showed successful incorporation of transplanted cells into mice livers and according to serological data there was a significant improvement in the conditions of the mice. These data may shed light on future clinical applications for hiPSC-HLCs.
Materials and Methods
Culture and Hepatic Lineage Differentiation of Human Induced Pluripotent Stem Cells
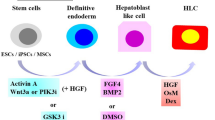
Royan hiPSC1 [14] at passages 40–50 and the hESC line, Royan H5 [15] at passages 110–120, were used in these experiments. Cells were expanded and passaged under feeder-free culture conditions in hESC medium that contained 100 ng/ml basic fibroblast growth factor (bFGF) as previously described [16]. We changed the medium every other day, until day seven. The hepatic lineage differentiation procedure is outlined in Fig. 1a using a stepwise, three stage protocol based on liver development.

Stepwise protocol for derivation of hiPSC-HLCs and hiPSC-HLCs. Illustration protocol for differentiation of hESCs and hiPSCs into HLCs (a). Temporal morphological change of hESCs and hiPSCs after differentiation induction (b). In both hiPSC (up) and hESC (down) cultures, cells are shown to change from round, small pluripotent cells to large, granulated and polygonal hepatocytes with a high cytoplasm to nucleus ratio. There are several bi-nucleated cells in the culture. Scale bar: 100 μm. (c) Scanning electron microscopy of differentiated hiPSC-HLCs. Well-developed apical microvilli can be easily visualized. Cells are connected tightly in their interface. Scale bar, left: 50 μm, right 10 μm
Briefly, five day-old colonies were fed with RPMI1640 (Invitrogen, 51800–035) supplemented with 1 mM nonessential amino acids (Invitrogen, 11140–050), 2 mM L-glutamine (Invitrogen, 25030–164) and 0.1 mM β-mercaptoethanol (Sigma-Aldrich, M7522). The knockout serum replacement (KOSR, Invitrogen, 10828–028) concentration was reduced from 20% to 10% and then to 0% within the following 2 days, of which this step was meant to prime cells for endoderm formation. Media was changed to RPMI1640 supplemented with 100 ng/ml Activin A (R & D Systems, Inc., 338-AC) for 3 days to induce a definitive endoderm lineage (Stage 1). The insulin-transferrin-selenite (ITS, Invitrogen, 41400–045) concentration was 0% for the first 24 h, then increased to 0.5% and 1.5% for the next 2 days after priming. Cells were then cultured for 8 days in the same basal media that contained 10% KOSR supplemented with 10 ng/ml FGF-4 (R & D Systems, Inc., 235-F4) and 10 ng/ml HGF (R & D Systems, Inc., 294-HG; Stage 2). During the last 2 days of Stage 2, 50% RPMI and 50% HCM were used. Cell differentiation was followed by culture for ten additional days in HCM (Lonza, Inc., CC-3198) that contained 10% KOSR, 1 mM nonessential amino acids, L-glutamine, 0.1 μM dexamethasone (Dex, Sigma-Aldrich, D-2915), and 10 ng/ml oncostatin M (OSM, R & D Systems, Inc., 295OM; Stage 3).
Evaluation of Differentiated Hepatic-lineage Cells
Immunofluorescence Staining
Cells were fixed in 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 for 10 min and blocked in 10% goat serum (SAFC Bioscience, 12306 C) in phosphate buffered saline (PBS, pH = 7.4) for 1 h at 37°C. Cells were incubated with primary antibodies (Supplementary Table 1) overnight at 4°C, after which they were washed and incubated with PE/FITC-conjugated secondary antibodies for 1 h at 37°C. Nuclei were counterstained with DAPI. The cells were analyzed with a fluorescent microscope (IX71, Olympus, Japan).
Flow Cytometry Analysis
Hepatic lineage directed differentiated cells underwent dissociation in 0.05% trypsin-EDTA (Invitrogen, 25200–072). Then, cells were washed twice in PBS (supplemented with 1% heat-inactivated FBS, 0.1% sodium azide and 2 mM EDTA) and then fixed in 4% paraformaldehyde for 15 min. For permeabilization, Triton X-100 [0.1% (v/v), Merck, 11869] was used for 10 min at room temperature. Nonspecific antibody binding was blocked for 15 min at 4°C with a combination of 10% heat-inactivated goat serum in staining buffer. For each analysis, 1–5 × 105 cells were used per sample. Cells were incubated with primary antibodies [or the appropriate isotype matched controls (Supplementary Table 1)] overnight at 4°C. Primary and secondary antibodies were the same as immunofluorescence staining. Flow cytometric analysis was performed with a BD-FACS Calibur Flow Cytometer (FACS Calibur, BD, USA). The experiments were replicated at least three times. Acquired data was analyzed with WinMDI12.9 software.
Quantitative RT-PCR Analysis
Total RNA was isolated using the RNeasy Kit (Qiagen, 74104). Two micrograms of total RNA was used for the reverse transcription reaction with the RevertAid First Strand cDNA Synthesis Kit (Fermentas, K1621) and random hexamer primer (Fermentas), according to the manufacturer’s instructions. Quantitative PCR reactions were run in duplicate with the Power SYBR Green Master Mix (Applied Biosystems, 4364344) and analyzed with the 7500 real-time PCR system (Applied Biosystems, 7500, USA). The samples were collected from at least three independent experiments. Expression values were normalized to the average expression of the housekeeping gene [ActB by the Comparative CT Method (2-ΔΔct)]. The primer sequences are presented in Supplementary Table 2.
Scanning Electron Microscopy
LHLC samples were washed with PBS, pre-fixed by 2.5% glutaraldehyde in 0.1 M PBS for 2 h and postfixed by 1% osmium tetroxide for 1.5 h. After dehydration in an ethanol series, the samples were air-dried, mounted on stab and gold-coated using a sputter coater (Balzers SCD 004, Germany). The samples were examined with a Zeiss DSM 940A scanning electron microscope (Germany).
Uptake of Low-density Lipoprotein
The cell’s ability to uptake low-density lipoprotein (LDL) was examined using 10-dioctadecyl-10-3,3,30,30-tetramethyl-indo-carbocyanine perchlorate that had been conjugated into acetylated LDL, (LDL Uptake Cell-Based Assay Kit, Cayman Chemical, Item Number 10011125). The assay was performed according to the manufacturer’s instructions. Cells were visualized using a fluorescent microscope (IX71, Olympus, Japan).
Cytochrome P450 Activity and Inducibility
Cytochrome P450-dependent pentoxyresorufin o-dealkylase activity (PROD) was evaluated using pentoxyresorufin substrates. Pentoxyresorufin is o-dealkylated by cytochrome P450 and changes a nonfluorescent compound into resorufin, a fluorescent compound [17]. To evaluate the inducibility of cytochrome P450, day 23 differentiated cells were exposed to sodium phenobarbital for 3 days and subsequently washed. Next, an incubation mixture that contained 7-pentoxyresorufin substrate (Sigma-Aldrich, P9049) and dicumarol (Sigma-Aldrich, M1390) in HBSS was added and plates incubated at 37°C in a 5% CO2 incubator for 30 min. Nuclei were counterstained with DAPI and in situ assessment and detection of resorufin was performed by fluorescent microscopy (IX71, Olympus, Japan). The percent of positive cells of three independent experiments before and after induction for each line were counted.
Periodic Acid-Schiff Staining
Glycogen storage of hiPSC-derived HLCs was evaluated by Periodic acid-Schiff (PAS) staining at day 23. Culture dishes that contained the cells were fixed with 4% paraformaldehyde, oxidized in 1% periodic acid for 5 min and washed. Cells were then treated with Schiff’s reagent for 15 min with subsequent color development in dH2O for 5–10 min followed by assessment with a light microscope (IX50, Olympus, Japan).
Indocyanine Green Uptake and Release
Differentiated cells at day 23 were incubated with indocyanin green (ICG, CardioGreen, Sigma-Aldrich, 12633) in basal medium for 1 h at 37°C. Uptake of ICG was detected with light microscopy (IX50, Olympus, Japan). ICG elimination from the positive cells was verified 6 h later.
Secretion Analysis
Forty-eight hour conditioned media obtained from the fully differentiated hiPS cells was collected at day 23 and stored at −20°C until assayed. The conditioned media were assayed for alpha-fetoprotein (AFP) secretion using a Chemiluminescence Immunoassay Kit (Pishtaz-Teb, Iran); for ALB secretion, a mouse ALB ELISA Kit (Bethyl, E99-134); and for urea secretion, a Colorimetric Assay Kit (Pars Azmun, Iran) according to the manufacturers’ recommendations. Secretion was normalized to the total cell number. Samples were collected from at least three independent experiments.
Oil Red Staining
Cells were assessed for lipid vesicles storage by oil red staining. Differentiated cells were fixed with 4% paraformaldehyde and then incubated for one h with oil red. Afterwards, cells were washed and analyzed with a light microscope (IX50, Olympus, Japan).
Cell Preparation for Transplantation
Differentiated cells at stage 3 or day 23 were prepared for cell transplantation. Cells that detached from the plate by trypsin/EDTA treatment were counted and labeled using the PKH67 Fluorescent Cell Linker Kit (Sigma-Aldrich, PKH67GL) according to the manufacturer’s recommendation and suspended in PBS.
Animal Experimental Model
Four groups of nine mice each were divided into the following groups: normal, sham, control and transplanted. For the first and fifth week analyses, 4 and 5 animals per group were used. Liver fibrosis was induced in 8 week-old male NMRI mice by intraperitoneal injections of 1.0 ml/kg dose of CCl4 twice weekly for 4 weeks. The normal group received no CCl4. After 4 weeks and eight injections of CCl4 in the transplanted group, 1 × 106 PKH-labeled cells were infused slowly via the tail vein. The sham group, only received a PBS (200 μl) injection via the tail vein. Again, the toxin was administered following transplantation to induce a high selective pressure for transplanted cells to engraft and differentiate [18]. All animals in the control, sham and transplanted groups received intraperitoneal injections of cyclosporin (Sandimmune, Novartis Pharmaceuticals, Switzerland) at a dose of 20 mg/kg/24 h per injection, beginning 5 days prior to transplantation and continuing after transplantation. At the end of the procedure, animals were sacrificed under deep anesthesia and their blood serum collected. Additionally, liver samples were taken for histopathologic analysis as well as cell engraftment and immunohistofluorescence evaluations. All in vivo experiments were performed at the end of 1 and 5 weeks post cell transplantation.
Liver Histopathology
Liver tissues were processed per standard protocol for paraffin embedding and sectioned into 6 μm sections. The sections were stained with masson trichrome (MT) according to standard protocol.
To analyze the extent of liver fibrosis, randomly picked fields of MT sections were captured from each animal using a 10× objective lens. Image analysis and percentages of fibrotic areas were measured with Image J software (National Institutes of Health). A total of 30 random fields per group were analyzed independently and the percentages of these fibrotic areas in the livers were calculated by dividing the total blue/green-colored area by the total areas of the livers [19, 20].
Engrafted Cells into the Liver
Recipient liver tissues were fixed in 4% paraformaldehyde and then soaked in 30% sucrose for 24 h at 4°C. Liver tissues were embedded in Tissue-Tek OCT compound and 8 μm thick sections were taken by cryostat at −20°C. The sections were mounted on glass slides and nuclei staining were performed using 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI, Sigma D-8417). The specimens were examined with a fluorescent microscope (BX51, Olympus) and randomly picked fields of sections were captured from each animal, using a 20× objective lens.
Immunofluorescence for Transplanted Cells
To evaluate hepatocyte differentiation of engrafted cells in the recipient livers, human ALB was used as a marker of transplanted cells. Cryosections of liver tissues from mice in the cell therapy groups were taken as described above. The sections were rinsed with dH2O, blocked with rabbit serum and then incubated with primary antibody goat anti-ALB (Santa Cruz, 1:200, sc-46293) overnight at 4°C. After rinsing, sections were incubated with secondary antibody rabbit anti-goat IgG-Texas Red (Santa Cruz, cs-3919, 1:200) for 1 h at 37°C. Double positive cells (PKH+/ALB+) in the liver were quantified using a fluorescent microscope (BX51, Olympus). A total of ten different areas per liver section were independently analyzed. The percentages of double positive cells in comparison to the total cells and in comparison to all PKH + cells of the fields were examined [21] 5 weeks after cell transplantation.
Serum Albumin, LDH and Bilirubin Measurements
Serologic analysis was performed using commercial kits (all from Pars Azmun, Iran) to measure serum albumin (ALB) levels by the bromocresol green method; serum lactate dehydrogenase enzyme activities by the DGKC method; and serum bilirubin levels by the photometric DCA method, according to the manufacturer’s instructions.
Statistical Analysis
Results were expressed as mean ± standard deviation (SD). Functional analyses of HLCs in vitro were done with the independent sample t-test. One-way repeated measures analysis of variance (ANOVA) followed by the Tukey post hoc test multiple group comparison analyzed group differences of the in vivo resultant data. The mean difference was significant at the p < 0.05 level. In the case of quantification by Image J software, a total of 30 fields for each group was assayed (fibrosis and cell count).
Results
Generation of HLCs from hESCs and hiPSCs
hESC and hiPSC colonies were cultured in feeder-free conditions at a high concentration of bFGF on Matrigel (Fig. 1a). Cells in these colonies were compact and expressed high nucleus to cytoplasm ratios as well as prominent nucleoli (Fig. 1b). Our stepwise protocol was composed of the following: cell priming and definitive endoderm specification, hepatic induction and expansion, and hepatic maturation (Fig. 1a). During these stages, we compared hiPSC1 with the previously established Royan H5 hESC line, which has been proven to have the ability of differentiation toward a hepatic lineage. The results indicated that hiPSC1 also has the ability to differentiate into HLCs. At day five of the hESC culture, we replaced hESC medium with RPMI for 2 days supplemented with 10% KOSR, which was then decreased to 0%. Although this treatment caused no obvious morphological changes it seemed to protect cells against activin-induced cell death [22], medium replacement or KOSR removal and increased cell viability up to 50% (data not shown) over the next 3 days. Cells were subsequently treated with activin A for 3 days in the absence of serum or serum replacement. For the next 7 days, cells were placed in RPMI that contained 10% KOSR supplemented with FGF-4 and HGF.
Morphologically, the cells gradually transformed into less dense, flatter cells with prominent nuclei (day 5, Stage 1) and then to a spiky shape (day 13, Stage 2, Fig. 1b). Finally, the medium was changed to HCM medium supplemented with OSM and Dex for an additional 10 days. Cells exhibited the characteristic hepatocyte morphology of a polygonal shape, distinct round nuclei and large cytoplasm that contained various granules (Stage 3, Fig. 1b). Bi-nucleated cells were readily visible in Stage 3.
The hiPSC-LHCs at Stage 3 were also examined by scanning electron microscopy (Fig. 1c). Ultrastructural analysis showed that the apical surface of the differentiated cells contained numerous microvilli. HLCs were polygonal and in contact with one another.
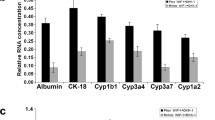
To confirm that the differentiated hESCs and hiPSCs developed into endoderm and then hepatocytes, we performed qRT-PCR analysis on the cells at various differentiation stages with primers specific to genes of the hepatic lineages (Fig. 2). The expression levels of endodermal marker genes (SOX17 and FOXA2) for both cell lines were at their highest level in Stage 1, which suggested the induction of an endodermal fate at this stage. While the early endodermal marker genes were gradually down-regulated during additional differentiation, expression of the hepatocyte gene, HNF4α was up-regulated through days 5 to 23 (Fig. 2). The expressions of mature hepatic marker genes, ALB, G6P, and CYP3A4 peaked at day 23 (Fig. 2). In total, these expressions were indicative of the transition from an endodermal to a hepatic fate.

Gene expression analysis. Relative mRNA expression pattern of hiPSCs and hESCs in the endoderm (S1), hepatic induction (S2) and hepatic maturation (S3) have been examined during differentiation. PLC: positive control. S: stage
To further determine the lineage information along the course of differentiation, we performed immunofluorescence staining on hiPSCs colonies at different differentiation stages (Fig. 3a). We observed that expression of the pluripotency marker, OCT4, was down-regulated by the initiation of differentiation and undetectable at Stage 3, whereas cells which expressed SOX17, CXCR4 and FOXA2 were mainly detected in Stage 1, all of which confirmed qRT-PCR results (Fig. 3a). The early hepatic cells that expressed AFP and CK18 began at Stage 2 and continued to Stage 3, respectively (Fig. 3a). ALB expression followed the same pattern, whereas AAT and CYP1A1 were only detected in Stage 3 (Fig. 3a). Few SOX7-positive cells were detected at Stage 1, which indicated these populations failed to differentiate to extra-embryonic endodermal cells.

Protein expression analysis by immunofluorescence staining. (a) Expression pattern of pluripotency (OCT4), endoderm (SOX7, SOX17, CXCR4, FOXA2), hepatic induction (CK18, AFP, ALB) and hepatic maturation (AAT, CYP1A1) proteins during the differentiation time span. The results indicate down-regulation of pluripotency and endoderm markers and up-regulation of hepatic maturation markers during this time. Scale bar: 100 μm. Quantitative protein expression of (double SOX17+/CXCR4+) in endoderm (b, S1), and ALB in hepatic induction (c, S2), and ALB in hepatic maturation (d, S3). The number of double positive cells for S1 and single positive cells for S2 and S3 are shown on the panels. The data shows efficient induction of definitive endoderm followed by induction of hepatic fate (ALB expression) and further maturation (increase in ALB expression level)
To further confirm the above results, we performed a quantitative protein expression assay. Cells at each stage were examined for expression of protein hallmarks. Flow cytometry analysis showed that differentiated cells on day five from hESC and hiPSC expressed both SOX17 and CXCR4 as markers of definitive endoderm (55% and 87% in hESC and hiPSC derivatives, respectively; Fig. 3b). Cells were then examined for ALB expression at days 13 and 23 (Fig. 3d-e). ALB was expressed in day 13 differentiated cells as a marker for hepatic induction from definitive endoderm [hESCs (20%) and hiPSCs (30%)] and further increased in day 23 differentiated cells as the protein hallmark of mature hepatocytes [hECSs (45%) and hiPSCs (74%)].
Despite the expression data, hiPSC-HLCs should have the capability to perform hepatic functions for further use in the clinic and industry settings. Therefore, we next examined the functional properties of hESC/hiPSC-derived differentiated cells. Conditioned media previously incubated with differentiating cells at Stages 1, 2 and 3 were collected for hepatocyte functional tests, which included ALB and AFP synthesis and ureagenesis (Fig. 4b-d). Both generated HLCs exhibited ALB (p < 0.05), AFP (p < 0.01), and urea synthesis (p < 0.01) that significantly increased in parallel with the duration of hepatic differentiation, which indicated increased hepatic maturation over the course of additional culture.

Functional analysis of hiPSC/hESC-HLCs. (a) Qualitative functions of the cells in the hepatic maturation phase (S3) including LDL-uptake, oil red staining, ICG-uptake, PAS staining and PROD activity. The results indicate in vitro functionality of the cells. Scale bar: 100 μm. Quantitative functions including (b) ALB, (c) AFP and (d) urea secretion during the time span of differentiation and (e) PROD activity of hiPSC/hESC-HLCs before and after phenobarbital induction. Quantitative functional analysis shows increase in secretion ability of the cells during the time span of differentiation. PROD activity has increased about 1.5 times in response to phenobarbital induction
These HLCs at Stage 3 also exhibited other characteristics of hepatocytes that included LDL conjugated to DyLight™ 549and ICG uptake (Fig. 4a). Accumulation of lipid droplets and glycogen storage activity were observed by oil red and PAS staining, respectively (Fig. 4a). Additionally, hESC/hiPSC-HLCs exhibited CYP450 metabolic activity as demonstrated by PROD activity before and after phenobarbital induction (Fig. 4e). About 50% of the cells were able to perform the function in each cell line and the number of positive cells increased up to 1.5 fold after phenobarbital induction. These results showed that hiPSC-HLCs were functional in vitro.
Analysis of qRT-PCR, flow cytometry and functional results showed that the expression levels of hepatic genes and their functions varied among the HLCs of hiPSCs and hESCs (Figs. 2, 3 and 4), which indicated that pluripotent stem cells responded differently to exogenous stimulation.
Transplantation of HLCs into a Mouse Model of a CCl4-injured Liver
To evaluate in vivo functionality and the therapeutic potential of our generated hiPSC-HLCs, we compared the animal survival rate, total LDH, total bilirubin, and mouse and human ALB levels in blood serum from CCl4-injured mice in the control, sham and transplant groups. Additionally, we examined liver sections from all groups for the percent of fibrotic area plus the amount of homing and cell fate of the transplanted cells, 5 weeks after transplantation (Figs. 5, 6 and 7).

Evaluation of transplantation of hiPSC-HLCs, 5 weeks post-transplantation for recovery of the CCl4 injured model. The survival curve of injured mice who underwent intravenous cell transplantation (a). All animals received Cyclosporine 5 days before cell transplantation and CCl4 administration started 4 weeks prior to transplantation and continued until the end of the survey (b). Representative photomicrographs of masson trichrome-stained mouse livers in different experimental groups, fibrous area stained green-blue. (c) Analysis of fibrotic percent by Image J software. a: p < 0.05 vs. other groups

Serological analyses of blood serum in different animal groups during 1 and 5 weeks post-treatment. Total levels of LDH and bilirubin decreased 1 and 5 weeks after cell transplantation. Total albumin increased 1 week post cell transplantation; however, it reduced after 5 weeks. a: p < 0.05 vs. other groups

The engraftment of hiPSCs-HLCs in recipient livers with functional integration in the CCl4 injured mouse model 5 weeks post-transplantation. (a) Cells were labeled with PKH (green color) prior to transplantation. After 5 weeks, transplanted cells were detected in the mouse host liver by fluorescence microscopy. The nuclei were counterstained with DAPI (blue color) to calculate homing cells. Liver sections were stained with an anti-ALB antibody. Pictures shown are representative for different experiments. (b) Quantification of engrafted cells
Figure 5a shows all hiPSC-HLCs and normal mice survived till week five, whereas only three animals survived in the control and sham groups by this time. Investigation of histology results demonstrated that liver fibrosis increased significantly in CCl4-treated mice when compared with normal mice, with thick bundles of collagen that surrounded the lobules which lead to fibrous septa (Fig. 5b). Evaluation of liver sections stained with Mason-trichorom by Image J software 1 and 5 weeks post-cell transplantation showed signs of decreased fibrosis when compared with the control and sham groups, but was not significant (Fig. 5c).
Serum analyses in mice who received HLCs, showed a significant reduction of serum total LDH and total bilirubin when compared with the control and sham groups (p < 0.05). There was an increase in total ALB (p < 0.05) followed by a decrease 1 and 5 weeks post-cell transplantation (Fig. 6).
Before transplantation, we labeled cells with PKH to assess homing of the transplanted cells. Subsequently liver sections were examined by fluorescent microscope, which demonstrated incorporation of the cells into the host livers. Transplanted cells were detected in the background of the mouse liver by PKH fluorescence (Fig. 7a). The absolute numbers of PKH-positive cells in recipients’ livers were counted 5 weeks post-cell transplantation. Within the recipient livers, there were 2.0 ± 0.7% of transplanted cells visualized (67 PKH + cells out of 3358 counted cells, Fig. 7b). To further explore their fate, transplanted cells were evaluated in corresponding serial sections. Double PKH+/ALB+ labeled cells were used as markers of functional activity of the transplanted cells (Fig. 7a). Albeit rare, 0.3% of total cells were ALB + and 18% (12 out of 67) were double PKH+/ALB+, and were located in the parenchyma and perivenous regions of the livers of all recipients (Fig. 7b). Additionally, human ALB was detected at one (30.5 ± 2.0 mg/dl) and five (36.33 ± 4.10 mg/dl) weeks post-transplantation in the serum of transplanted animals. These data suggested a functional integration of the transplanted cells within the mouse livers.
Additionally, we did not find tumor in the hiPSC-HLCs transplanted recipients during our study.
Discussion
Access to a pure terminally differentiated hepatocyte population with therapeutic potential for transplantation to liver failure patients has been the objective of several studies. Here we demonstrated the hepatic lineage differentiation potential of hiPSCs following stepwise application of growth factors and removal of serum in comparison with hESCs, as well as further transplantation of hiPSC-HLCs to a CCL4-injured mouse model. Based on our knowledge, this is the first report of hiPSC-HLCs administration in an animal model of liver injury.
According to our results, the examined hiPSC line followed the same differentiation pattern as the hESC line, and this result agreed with other hESCs [for review see [5]] and hiPSC studies [9–13]. HLCs from both pluripotent stem cell types exhibited a polygonal epithelial morphology and large cytoplasm that contained various granules and expressed hepatocyte-specific genes at both the mRNA and protein levels. Additionally, as some of the known functions of mature hepatocytes, the differentiated cells were able to secrete ALB, AFP, and urea into the medium. The differentiated HLCs also showed cytochrome p450 activity, uptake of organic anion ICG and LDL in a fluorescence form (LDL conjugated to DyLight™ 549). Glycogen and lipid storage, two other characteristics of hepatocytes, were visible on both types as detected by PAS and oil red staining, respectively. However, it seemed that hiPSC1 exhibited a differentiation propensity into hepatic lineage. A marked propensity to differentiate into specific lineages was exhibited in various hESC lines [23] and recently in hiPSCs [24]. This needs further investigation and repeating the experiment with increased hiPSC and hESC cell lines.
Although clinical applications of hiPSC-HLCs still seem to be far from reach, the availability of these cells and relative ease of derivation of hepatocytes from these cells allows for the opportunity to test drugs on a range of genetic backgrounds. Moreover, generation of disease-specific hiPSC-HLCs can provide scientists with an in vitro model of liver disorder for testing new drugs or studying the mechanism of such disorders. Rashid et al. [25] have reported the generation of hiPSCs with α1-antitrypsin deficiency, familial hypercholesterolemia and glycogen storage disease type 1a. Those cells demonstrated key pathological characteristics of the diseases, such as aggregation of misfolded α1-antitrypsin in the endoplasmic reticulum, deficient LDL receptor–mediated cholesterol uptake and increased lipid and glycogen accumulation. Recently, we have produced liver-specific patient hiPSCs with tyrosinemia, glycogen storage disease, progressive familial hereditary cholestasis, and from two siblings with Crigler-Najjar syndrome that had the potential to differentiate into functional HLCs [26].
This study also assessed the therapeutic effects of hiPSC-HLCs in the CCL4-injured mouse liver model over 5 weeks post-cell transplantation. When compared with the control and sham groups, the transplanted group had a better survival rate, and reduced levels of LDH and bilirubin. The significant difference in serologic markers between the cell transplanted group and control/sham groups was possibly due to the recovery potential of HLCs. Transplanted cells could re-establish the altered liver architecture and possibly stop additional cell death which can be detected by restored LDH and bilirubin levels. On the other hand, mouse ALB increased during the first week post-cell transplantation. This was a possible effect of HLCs on mice hepatocytes, which enabled them to increase capabilities such as ALB secretion. However, this was not a steady effect that continued until the end of 5 weeks, which may have been due to long-term CCL4 exposure in the animals and its negative effects on transplanted cells. Human ALB was also detected in serum 1 and 5 weeks after HLC transplantation, which has shown that HLCs could secret ALB in vivo and be active in their new niche. CCl4-treated mice had more fibrotic areas when compared to the normal group. The histological improvement was not significant in the HLC transplanted group 1 or 5 weeks post-cell transplantation. However, HLC transplantation ameliorated collagen excess and improved liver histologic architecture when CCL4 was removed, which was the main cause of the matrix imbalance. Several PKH+/ALB + (18%) cells showed survival and integration of engrafted HLCs into the host tissue at 5 weeks following transplantation and assistance with recovery of liver function. It has been shown that transplanted hESC-HLCs improved recovery in liver failure animal models [27–29]. Recently, it has been also demonstrated that mouse iPSC-derived hepatocytes were able to regenerate the liver after transplantation and in a two-third partial hepatectomy in mice with fumarylacetoacetate hydrolase deficiency [29].
In conclusion, our results open up a proof of concept that functional HLCs can be generated from hiPSCs and improve the fibrotic mouse model after their transplantation. Further research of this nature and investigation of the in vivo functionality of hiPSC-HLCs will bring new insights into the clinical applications of these cells in regenerative and/or individualized medicine. However, further investigations for producing safe iPSCs, more efficient differentiation protocols, long term analysis and larger animal models are necessary.
References
Mazaris, E. M., Roussos, C. T., & Papalois, V. E. (2005). Hepatocyte transplantation: a review of worldwide clinical developments and experiences. Experimental and Clinical Transplantation, 3, 306–315.
Miro, J. M., Laguno, M., Moreno, A., & Rimola, A. (2006). Management of end stage liver disease (ESLD): what is the current role of orthotopic liver transplantation (OLT)? Journal of Hepatology, 44, S140–145.
Lorenzini, S., Isidori, A., Catani, L., et al. (2008). Stem cell mobilization and collection in patients with liver cirrhosis. Alimentary Pharmacology and Therapeutics, 27, 932–939.
Lee, S. W., Wang, X., Chowdhury, N. R., & Roy-Chowdhury, J. (2004). Hepatocyte transplantation: state of the art and strategies for overcoming existing hurdles. Annals of Hepatology, 3, 48–53.
Asgari, S., Pournasr, B., Salekdeh, G. H., Ghodsizadeh, A., Ott, M., & Baharvand, H. (2010). Induced pluripotent stem cells: a new era for hepatology. Journal of Hepatology, 53, 738–751.
Pournasr, B., Farzaneh, Z., Shahsanvani, M., & Baharvand, H. (2010). Liver development and In vitro differentiation of embryonic stem cells to hepatocytes. Yakhteh Medical Journal (Cell Journal), 11, 348–373.
Takahashi, K., Tanabe, K., Ohnuki, M., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131, 861–872.
Yu, J., Vodyanik, M. A., Smuga-Otto, K., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science, 318, 1917–1920.
Liu, H., Ye, Z., Kim, Y., Sharkis, S., & Jang, Y. Y. (2010). Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology, 51, 1810–1819.
Touboul, T., Hannan, N. R., Corbineau, S., et al. (2010). Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology, 51, 1754–1765.
Si-Tayeb, K., Noto, F. K., Nagaoka, M., et al. (2010). Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology, 51, 297–305.
Song, Z., Cai, J., Liu, Y., et al. (2009). Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Research, 19, 1233–1242.
Sullivan, G. J., Hay, D. C., Park, I. H., et al. (2010). Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology, 51, 329–335.
Totonchi, M., Taei, A., Seifinejad, A., et al. (2010). Feeder- and serum-free establishment and expansion of human induced pluripotent stem cells. International Journal of Developmental Biology, 54, 877–886.
Baharvand, H., Ashtiani, S. K., Taee, A., et al. (2006). Generation of new human embryonic stem cell lines with diploid and triploid karyotypes. Development, Growth & Differentiation, 48, 117–128.
Pakzad, M., Totonchi, M., Taei, A., Seifinejad, A., Hassani, S. N., & Baharvand, H. (2010). Presence of a ROCK inhibitor in extracellular matrix supports more undifferentiated growth of feeder-free human embryonic and induced pluripotent stem cells upon passaging. Stem Cell Reviews, 6, 96–107.
Tzanakakis, E. S., Hsiao, C. C., Matsushita, T., Remmel, R. P., & Hu, W. S. (2001). Probing enhanced cytochrome P450 2B1/2 activity in rat hepatocyte spheroids through confocal laser scanning microscopy. Cell Transplantation, 10, 329–342.
Piryaei, A., Valojerdi, M. R., Shahsavani, M., & Baharvand, H. (2011). Differentiation of bone marrow-derived mesenchymal stem cells into hepatocyte-like cells on nanofibers and their transplantation into a carbon tetrachloride-induced liver fibrosis model. Stem Cell Reviews, 7, 103–118.
Marumoto, Y., Terai, S., Urata, Y., et al. (2008). Continuous high expression of XBP1 and GRP78 is important for the survival of bone marrow cells in CCl4-treated cirrhotic liver. Biochemical and Biophysical Research Communications, 367, 546–552.
Sakaida, I., Terai, S., Yamamoto, N., et al. (2004). Transplantation of bone marrow cells reduces CCl4-induced liver fibrosis in mice. Hepatology, 40, 1304–1311.
Terai, S., Sakaida, I., Yamamoto, N., et al. (2003). An in vivo model for monitoring trans-differentiation of bone marrow cells into functional hepatocytes. Journal of Biochemistry, 134, 551–558.
Mfopou, J. K., & Bouwens, L. (2008). Hedgehog signals in pancreatic differentiation from embryonic stem cells: revisiting the neglected. Differentiation, 76, 107–117.
Osafune, K., Caron, L., Borowiak, M., et al. (2008). Marked differences in differentiation propensity among human embryonic stem cell lines. Nature Biotechnology, 26, 313–315.
Kim, D. S., Lee, J. S., Leem, J. W., et al. (2010). Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell Reviews, 6, 270–281.
Rashid, S. T., Corbineau, S., Hannan, N., et al. (2010). Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. The Journal of Clinical Investigation, 120, 3127–3136.
Ghodsizadeh, A., Taei, A., Totonchi, M., et al. (2010). Generation of liver disease-specific induced pluripotent stem cells along with efficient differentiation to functional hepatocyte-like cells. Stem Cell Reviews, 6, 622–632.
Basma, H., Soto-Gutierrez, A., Yannam, G. R., et al. (2009). Differentiation and transplantation of human embryonic stem cell-derived hepatocytes. Gastroenterology, 136, 990–999.
Agarwal, S., Holton, K. L., & Lanza, R. (2008). Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells, 26, 1117–1127.
Espejel, S., Roll, G. R., McLaughlin, K. J., et al. (2010). Induced pluripotent stem cell-derived hepatocytes have the functional and proliferative capabilities needed for liver regeneration in mice. The Journal of Clinical Investigation, 120, 3120–3126.
Acknowledgements
This study was funded by a grant provided from Royan Institute and the Iranian Council of Stem Cell Technology. We also thank Mehran Rezaei, Arefeh Ghodsizadeh, Zahra Farzaneh and Abbass Piriayei for their critical comments. Additionally, we would like to thank the Electron Microscopy Lab at the University of Tehran.
Conflict of interest
None of the authors have any conflicts of interest to disclose and all authors support submission to this journal.
Author information
Authors and Affiliations
Corresponding author
Additional information
Samira Asgari and Mohsen Moslem are contributed equally in this work.
Electronic Supplementary Materials
Below is the link to the electronic supplementary material.
Supplementary Table 1
Primary and secondary antibodies used for immunofluorescence staining and flow cytometry. (DOCX 39 kb)
Supplementary Table 2
List of primers used in this study. (DOCX 38 kb)
Rights and permissions
About this article
Cite this article
Asgari, S., Moslem, M., Bagheri-Lankarani, K. et al. Differentiation and Transplantation of Human Induced Pluripotent Stem Cell-derived Hepatocyte-like Cells. Stem Cell Rev and Rep 9, 493–504 (2013). https://doi.org/10.1007/s12015-011-9330-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-011-9330-y