
Abstract
Advances in gene-based medicine since 1990s have ushered in new therapeutic strategy of gene therapy for inborn error genetic diseases and cancer. Malignant brain tumors such as glioblastoma multiforme and medulloblastoma remain virtually untreatable and lethal. Currently available treatment for brain tumors including radical surgical resection followed by radiation and chemotherapy, have substantially improved the survival rate in patients suffering from these brain tumors; however, it remains incurable in large proportion of patients. Therefore, there is substantial need for effective, low-toxicity therapies for patients with malignant brain tumors, and gene therapy targeting brain tumors should fulfill this requirement. Gene therapy for brain tumors includes many therapeutic strategies and these strategies can be grouped in two major categories: molecular and immunologic. The widely used molecular gene therapy approach is suicide gene therapy based on the conversion of non-toxic prodrugs into active anticancer agents via introduction of enzymes and genetic immunotherapy involves the gene transfer of immune-stimulating cytokines including IL-4, IL-12 and TRAIL. For both molecular and immune gene therapy, neural stem cells (NSCs) can be used as delivery vehicle of therapeutic genes. NSCs possess an inherent tumor tropism that supports their use as a reliable delivery vehicle to target therapeutic gene products to primary brain tumors and metastatic cancers throughout the brain. Significance of the NSC-based gene therapy for brain tumor is that it is possible to exploit the tumor-tropic property of NSCs to mediate effective, tumor-selective therapy for primary and metastatic cancers in the brain and outside, for which no tolerated curative treatments are currently available.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Malignant brain tumors such as glioblastoma multiforme remain virtually untreatable and lethal [1]. Similarly in childhood brain cancers, medulloblastoma is the most common and incurable [2]. Multimodal treatment including radical surgical resection followed by radiation and chemotherapy, have substantially improved the survival rate in patients suffering from these brain cancers; however, it remains incurable in large proportion of patients [3]. Therefore, there is substantial need for effective, low-toxicity therapies for patients with malignant brain cancers, and gene therapy approach targeting brain cancer should fulfill this requirement.
During the last 20 years gene therapy research has advanced greatly. The first gene therapy clinical trial was conducted in US in 1989 in melanoma patients by Rosenberg et al [4], followed by successful treatment in 1990 of two children with severe combined immunodeficiency (ADA-SCID) using retrovirus encoding adeonsine deaminase (ADA) gene by Anderson and his colleagues [5]. According to a recent study of 2007, over 1340 gene therapy clinical trials have been completed, or are ongoing worldwide in 28 countries, and more than 70% of these trials are in cancer gene therapy [6]. Because of the complex nature of cancer, gene therapy in cancer includes many therapeutic strategies and these strategies can be grouped in two major categories: immunologic and molecular [7].
Molecular Approaches in Cancer Gene Therapy
Two gene groups that are mainly involved in cancer genesis are oncogenes and tumor suppressor genes. Oligonucletides, short nucleic acid segments, which bind and antagonize oncogenes are used in cancer treatment, while tumor suppressor genes have been widely used in cancer gene therapy to inhibit tumor growth [8]. These molecular approaches are designed to inhibit proliferation of cancer cells or to induce apoptosis (programmed cell death) of cancer cells. In addition, as a new molecular approach, RNA interference (iRNA) targeting cancer cells has received a wide attention recently [9]. Suicide gene therapy which targets cancer cells at molecular levels is also widely applied in clinical medicine [10]. Various molecular approaches in gene therapy are described as following.
-
1.
Tumor suppressor genes
The main family member of the tumor suppressor genes is the p53 gene which interferes in the signal pathways of genes responsible for regulation of cell growth and cell differentiation such as Bcl-2, caspases and IAP families [11, 12]. Mutational changes in the p53 gene were found in 40% of all cancers [13]. Transfection of wild type p53 in animals bearing tumors resulted in tumor growth inhibition and tumor regression [14, 15]. In clinical settings, intratumoral injection of adenovirus-p53 in lung cancer patients resulted in disease stabilization and tumor size reduction in more than 50% of patients [16]. Much better clinical outcomes were reported when adenovirus-p53 treatment was combined with chemotherapy [17] or radiotherapy [18].
-
2.
Anti-oncogenes
Two kinds of anti-oncogenes that bind to and inhibit biological activity of oncogenes are available: antisense oligonucleotides that bind to a specific sequence of RNA and antigene olgonnucleotide that bind to a specific sequence of DNA. Antisense oligonucleotides bind to oncogene mRNA and inhibit translation step of protein synthesis [17]. Important targets for antisense therapy are Bcl-2, c-myc and ras family oncogenes, and antisense therapy against these oncogenes in solid tumors and leukemia has resulted in tumor growth inhibition [19–21]. Antigene oligonucleotides bind to oncogene DNA forming a non-functional triple helical structure and the gene expression is blocked at the transctiption step [22]. No clinical trial has been reported but dual treatment with c-myc and c-erbB2 antigenes induced 80% cell growth inhibition in ovarian cancer cells in vitro [23].
-
3.
Short interfering RNAs (siRNAs)
A new molecular gene therapy approach that recently received worldwide attention is siRNA treatment. RNA interference was initially observed in plants and also found in animal cells showing a form of transcriptional inhibition [24]. There is a great potential of iRNA in cancer therapy and new techniques are rapidly moved to application toward this direction.
-
4.
Suicide gene therapy
This molecular therapeutic approach is based on the conversion of non-toxic prodrugs into active anticancer agents via introduction of non-mammalian or mammalian enzymes. One of the earliest suicide gene/prodrug system is the herpes simplex virus thymidine kinase (HSVtk)/ Ganciclovir (GCV) system [25, 26]. HSVtk is a viral enzyme that catalyzes the phosphrylation of nucleotide analogs such as antiviral drug GCV [27]. The phosphorylated GCV kills cancer cells via bystander effect with apoptotic or non-apoptotic mechanisms [28, 29]. Bystander effect is a mechanism by which toxic metabolites are transferred from transduced cells to neighboring cancer cells via gap juctions and apoptotic vesicles [30]. In addition to HSVtk/GCV suicide gene/prodrug system, there have been several notable suicide enzyme-prodrug systems including cytosine deaminase (CD)/5-fluorocytosine (5-FC) [31, 32], cytochrome P450/cyclophosphamide [33], carboxypeptidase/chloroethyl-mesyloxyethyl-aminobenzoyl-glutamic acid (CMDA) [34] and carboxylesterase (CE)/CPT-11 [35]. In the CD/5-FC suicide gene system, CD deaminases the non-toxic pyrimidine 5-FC to the cytotoxic 5-fluorouracil (5-FU), and 5-FU is then processed to intermetabolites that inhibit RNA processing and DNA synthesis in the cancer cells [31].
Immunologic Approach in Cancer Gene Therapy
Cancer cells are immunogenic in nature with cancer-specific antigens being intracellular molecules, thus T-cell mediated immunity is more obvious than B-cell mediated immunity [36]. Thus genetic immunotherapy aims to boost T-cell mediated immune response against cancer cells. One major immunotherapeutic approach involves the gene transfer of immune-stimulating cytokines including IL-4, IL-12 and TRAIL. Gene transfection of IL-12 cytokines into cancer cells resulted in complete regression of liver cancer and adenocarcinoma [37, 38]. Production of IL-12 by tumor cells induces the activation of immune cells such as cytotoxic T cells and natural killer cells [39].
Another important immunotherapy approach is the in vitro manipulation of antigen presenting cells (APCs) such as dendritic cells by which APCs could present tumor antigens and activate T cells. Dendritic cells are powerful APCs, and genetically engineered dendritic cells expressing hepatocellular antigen α-fetoprotein resulted in a strong immune response against liver cancer cells [40].
One of recent cancer immunotherapy approaches is direct vaccination of cancer cell antigen- encoding genes to induce immune reaction against cancer cells. Injected DNAs enter local cells such as muscle or skin cells which then produce and secrete cancer cell-specific antigens. APCs capture cancer cell antigen, migrate to the lymphatic tissues and activate the desired immune reaction against cancer cells [41, 42]. Following vaccination with α-fetoprotein gene, animals injected with α-fetoprotein-expressing tumor rejected the tumor growth and the life span of the animals was extended [43, 44].
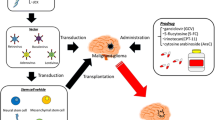
Neural Stem Cells as Gene Delivery System
Recent studies have shown that intracranially or intravenously injected neural stem cells (NSCs) or neural precursor cells migrate towards CNS locus with injury or pathology. This chemotropic property of NSCs has been utilized for cell-based therapies to treat diverse neurological diseases [45–52]. Murine and human NSCs possess an inherent tumor tropism that supports their use as a reliable delivery vehicle to target therapeutic gene products to primary and secondary invasive glioma, medulloblastoma, melanoma brain metastases and neuroblastoma throughout the brain and extracerebral loci [53–61]. We have previously utilized the immortalized human NSC line, HB1.F3, that stably expresses therapeutic genes designed to treat animal models of neurological disorders including Parkinson disease [62, 63], Huntington disease [64, 65], ALS [66], stroke [67–73] and lysosomal storage disease [74]. The novel and significant aspect of NSC-based gene therapy for brain cancer is that it may be possible to exploit the tumor-tropic property of NSCs to mediate effective, tumor-selective therapy for invasive and metastatic tumors in the brain and outside, for which no curative treatments are currently available.
Bran tumors, particularly malignant gliomas are known to release numerous cytokines, chemokines and growth factors that are capable of stimulating the directed migration of NSCs into the tumor environment. Cytokines such as stem cell factor-1 (SCF) [75, 76], monocyte chemoattractant protein-1 (MCP-1) [77], and stromal cell-derived factor-1 (SDF-1) [78, 79] are potent chemotactic molecules that stimulate NSC migration. In addition, growth factors, such as vascular endothelial growth factor (VEGF) [80], epidermal growth factor (EGF) [81] and hepatocyte growth factor (HGF) [61, 79, 82], are recognized as potent tumor-tropic agents for NSCs.
The signals required for homing and recruitment of neural stem cells to tumor sites are not well understood. Urokinase plasminogen activator (uPA) and urokinase plasminogen activator receptor (uPAR) are involved in chemotaxis and cell guidance during normal development and are upregulated in invasive tumors. In a recent study, we provided evidence that activation of uPA and uPAR in malignant solid tumors (brain, lung, prostate, and breast) augments neural and mesenchymal stem cell tropism [83]. Expression levels of uPAR on human solid tumor cell lines correlated with levels of uPA and soluble uPAR in tumor cell-conditioned media. Cytokine expression profiles of these tumor-conditioned media were determined by protein arrays. Among 79 cytokines investigated, interleukin(IL)-6, IL-8, and monocyte chemoattractant protein-1 were the most highly expressed cytokines in uPAR-positive tumors. We provided evidence that human recombinant uPA induced stem cell migration, whereas depletion of uPA from PC-3 prostate cancer cell-conditioned medium blocked stem cell migration. Furthermore, retrovirus-mediated overexpression of uPA and uPAR in neuroblastoma (NB1691) cells induced robust migration of stem cells toward NB1691 cell-conditioned media, compared with media derived from wild-type NB1691 cells [83].
Hypoxia is a critical aspect of the microenvironment in cancer including glioma and generally signifies unfavorable clinical outcome. Effective targeting of hypoxic areas in gliomas remains a significant therapeutic challenge. New therapeutic platforms using NSCs for tumor-targeted drug delivery show promise in treatment of cancers. Recently the role of hypoxia in directed migration of NSCs to glioma and identified the specific signaling molecules involved was investigated [84]. Hypoxia caused increased migration of F3 human NSCs to U251 human glioma-conditioned medium in vitro. Hypoxia led to up-regulation of CXCR4, urokinase-type plasminogen activator receptor (uPAR), VEGFR2, and c-Met receptors in F3 human NSCs. Small interfering RNA (siRNA) knockdown of hypoxia-inducible factor-1A in glioma cells blocked the hypoxia-induced migration of NSCs, which was due to decreased expression of stromal cell-derived factor-1 (SDF-1), uPA, and VEGF in glioma cells. In vivo data provided direct evidence that NSCs preferentially distributed to hypoxic areas inside intracranial glioma xenografts as well as to the tumor edge. These observations indicate that hypoxia is a key factor in determining NSC tropism to glioma and that SDF-1/CXCR4, uPA/uPAR, VEGF/VEGFR2, and HGF/c-Met signaling pathways mediate increased NSC-to-glioma tropism under hypoxia.
A better understanding of the molecular events that regulate NSC migration to brain tumors is necessary to optimize the use of NSCs as therapeutic delivery vehicles. We have recently investigated tumor-tropic property of NSCs using the immortalized HB1.F3 human NSC line, which targets human brain tumors in both in vitro and in vivo models [55, 56, 58–62]. We have demonstrated that the human glioma cell lines U251 and U87 produce HGF and VEGF, which act as potent chemoattractants for F3 human NSCs [82]. These growth factors, HGF and VEGF, stimulate receptor tyrosine kinase signaling that leads to the activation of phosphoinositide 3-kinase (PI3K), which has previously been shown to be an important regulator of directed cell migration [85–87]. Inhibition of the PI3K pathway significantly inhibited the chemotactic cell migration towards all growth factors tested (HGF, VEGF and EGF), suggesting that the growth factors produced by brain tumors converge on the PI3K signaling pathway. Collectively, these results reveal that PI3K serves as a critical convergence point for growth factor-mediated directed migration of human NSCs.
Treatment strategies for the highly invasive brain tumor, glioblastoma multiforme, require that cells which have invaded into the surrounding brain be specifically targeted. The inherent tumor-tropism of NSCs to primary and invasive tumor foci can be exploited to deliver therapeutics to invasive brain tumor cells in humans. In the context of NSC-based therapies, MRI can be used both to non-invasively follow dynamic spatio-temporal patterns of the NSC tumor targeting allowing for the optimization of treatment strategies and to assess efficacy of the therapy. Iron labeling of cells allows their presence to be visualized and tracked by MRI. In a recent study, the effect of iron loading of the therapeutic NSCs, with ferumoxide-protamine sulfate complex (FE-Pro) on viability, proliferation, migratory properties and transgene expression was investigated [88]. FE-Pro labeled NSCs were imaged by MRI at tumor sites, after intracranial administration into the hemisphere contralateral to the tumor, in a human glioma xenograft mouse model. FE-Pro labeled NSCs retain their proliferative status, tumor tropism, and maintain stem cell character, while allowing in vivo cellular MRI tracking at 7 Tesla, to monitor their real-time migration and distribution at brain tumor sites.
Suicide Gene Therapy for Brain Tumors
In the previous section, the tumor-tropic properties of NSCs that target therapeutic genes to brain cancers, such as malignant glioma, medulloblastoma and tumor metastasis are detailed. Summary of preclinical studies of both molecular (suicide gene therapy) and immunological approaches in brain cancer gene therapy is shown in Table 1.
When human NSC line HB1.F3 carrying CD enzyme gene (F3.CD) was transplanted intracranially at distant sites from the tumor, the NSCs migrate through normal tissue and selectively “home in” to the glioblastoma tumor mass and upon administration of prodrug 5-FC, 85–95% reduction in tumor volume was demonstrated (Kim, unpulished).
A previous study has shown that when murine NSC cell line C17.2 cells carrying oncolysis-promoting pro-drug activating enzyme cytosine deaminase (CD) were grafted into glioma-bearing animals followed by systemic injection of prodrug 5fluorocytosine (FC), there was an 80% reduction in resultant tumor burden [53]. Another well characterized cancer gene therapy approach has been the herpes simplex virus thymidine kinase (HSVtk)/ Ganciclovir (GCV) combination [25, 26]. In murine glioma studies, significant tumor regression was found in animalsbearing murine glioma when murine NSCs carrying HSVtk and prodrug GCV was administered [89–91].
The growth of gliomas depends on the balance of factors stimulating or inhibiting angiogenesis, tumor cell invasion and proliferation. The administration of endogenous inhibitors to experimental gliomas in animal models resulted in a significant inhibition of tumor growth.
One of such tumor growth inhibitors is PEX, a fragment of matrix metalloproteinase 2 (MMP-2), which interacts with endothelial cell integrin alpha(v)beta3 where it serves as a natural inhibitor of MMP-2 activity, thereby regulating the invasive behavior of new blood vessels [92, 93].
We have demonstrated that HB1.F3 human NSCs carrying PEX gene, “surround” the invading gliobalstoma tumor cells, “chasing down” infiltrating tumor cells, and “attack and kill” tumor cells, causing a 90% reduction in tumor volume [94]. The HB1.F3 cells were transfected by a pTracer vector with PEX. For animal studies, DiI-labeled F3.PEX cells were stereotactically injected into established glioma tumor in nude mice. Histologic analysis showed that DiI-labeled F3.PEX cells migrate at the tumor boundary and cause reduction of tumor volume. This reduction in tumor volume in treated animals was associated with a significant decrease in angiogenesis [94].
For the in vivo therapeutic proof of principal in medulloblastoma, animals bearing intracranial medulloblastoma (human Daoy medulloblastoma cell line) were inoculated ipsilaterally with F3.CD human NSC cells followed by systemic 5-FC treatment. Histological analyses showed that NSCs migrate to the tumor site leading to an 80% reduction of tumor volume. We demonstrated the potential of human NSCs as an effective delivery system to target and disseminate therapeutic agents to medulloblastomas [60]. One of the reasons for the recurrence of medulloblastoma in children after conventional treatment is inherent tendency of tumor cells to metastatize through cerebrospinal fluid leading to leptomeningeal dissemination. F3.CD human NSC cells were found to distribute diffusely to metastatic medulloblastoma cells that had spread throughout the entire spinal cord after injection in the cisterna magna, and CD gene in NSCs functioned effectively and killed tumor cells by bystander effect following 5-FC prodrug administration [61].
Patients diagnosed with metastatic cancer have almost uniformly poor prognoses. The treatments available for patients with metastatic cancer are usually not curative and have side effects that limit the therapy that can be given. The tumor-tropic property of NSCs could be utilized to selectively deliver a therapeutic gene to metastatic solid tumors, and that expression of an appropriate transgene at tumor loci might mediate cures of metastatic disease. In a recent study, HB1.F3 human NSCs were transduced by adenovirus encoding rabbit carboxylesterase (rCE), which converts CPT-11 into SN-38, a potent topoisomerase 1 inhibitor, and injected intravenously into mice bearing disseminated neuroblastoma tumors, and NSCs migrated selectively to tumor sites, then treated systemically with CPT-11, and the efficacy of treatment monitored. Mice treated with the combination NSCs expressing the CPT-11-activating enzyme CE and prodrug CPT-11 produced tumor-free survival of 100% of the mice bearing metastatic neuroblastoma for more than 6 months [55, 59].
Brain metastasis is the most common intracranial tumor type in adults including lung cancer, breast cancer and melanomas. A solitary brain metastasis can be managed by local treatment modalities such as surgery or irradiation. However, many patients with brain metastasis harbor two or more metastasis. In a recent study, MDA-MB435 human breast cancer cells were implanted into the brain of nude mice and then HB1.F3.CD human NSCs were injected into the contralateral hemisphere followed by systemic 5-FC injection. The F3.CD cells migrated selectively into the brain metastasis and resulted in significantly reduced tumor volumes [95].
Melanoma at early stages is effectively treated with surgery and radiotherapy, however metastatic disease is almost universally fatal, thus novel therapeutic approaches are needed. HB1.F3.CD human NSCs were transduced by adenovirus encoding rabbit carboxylesterase (rCE). Melanoma at early stages is effectively treated with surgery and radiotherapy, however metastatic disease is almost universally fatal, thus novel therapeutic approaches are needed. In vitro cell migration assays revealed robust migration of NSCs to conditioned media from melanoma cells. Cytokine profiles showed that IL-6, IL-8, MCP-1 and TIMP-2, known chemoattractants for stem cells, were highly expressed by melanoma cells. Exposure of melanoma cells to conditioned media from the F3.CD.rCE cells in the presence of CPT-11 increased the tumor cell-killing effect by approximately 100-fold when compared to CPT-11 alone. Our data demonstrate the rational for NSC-based enzyme/prodrug therapeutic approach to target metastatic melanoma [96].
Immunotherapy for Brain Tumors
Immunotherapy for brain cancers aim to boost T-cell mediated immune response against cancer cells. One major immunotherapeutic approach involves the gene transfer of immune-stimulating cytokines including IL-4, IL-12 and Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Infiltrating immune cells were found in brain tumors but their function can be modified by tumor-derived immuno-suppressors such as TGF-β [97], IL-10 [98] and Fas ligands [99], and earlier studies have used immune-stimulating cytokines IL-4 [100] or IL-12 [101, 102] to overcome immunosuppression by these suppressive molecules produced by cancer cells.
IL-4 is produced by activated T cells, mast cells and basophils and acts in the hematopoiesis and B cell activation [103]; IL-4 acts as a strong antitumor cytokine by activating eosinophils and macrophages and later T helper cells. In a previous study, Benedetti et al. have isolated primary fetal mouse brain cells containing NSCs (neurospheres), transduced them with a retroviral vector encoding IL-4, injected IL-4 secreting NSCs directly into glioma, and found extended survival of tumor-bearing mice [104].
IL-12 is produced by activated T cells and basophils and known for its tumorcidal activity [39]. IL-12 shows potent Th-1-mediated antitumor cyototoxic immnunity when delivered to glioma tumor mass via intratumor injection of adnovirus encoded with IL-12 gene [105]. Primary fetal mouse brain cells containing NSCs (neurospheres) were infected with an adenoviral vector bearing murine IL-12, injected into adult mouse brain bearing mouse glioma GL26 cells, and this IL-12 cell therapy resulted in prolonged survival of animals and enhanced T-cell infiltration in tumor microsatellites [106].
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) can selectively induce apoptosis in cancer cells and spare normal cells in contrast to Fas lignd and TNF-α which show sever toxic side effect in normal cells [107–109]. TRAIL is a member of TNF ligand family that binds to TRAIL receptors which contain a death domain and induce apoptotic cell death via activation of caspase-3 [110, 111]. Primary fetal mouse brain cells containing NSCs (neurospheres) were infected with an adenoviral vector bearing human TRAIL, delivered directly to glioma tumor mass via intratumor injection in adult mouse brain bearing U343 glioma cells, and this TRAIL cell therapy resulted in inhibition of tumor growth and prolonged survival of animals [112]. TRAIL is a type-2 membrane protein, and its release requires cleavage from its membrane bound location. Shah and his colleagues fused the extracellular domain of TRAIL to the extracellular domain of Flt3 ligand and this fusion protein secretable TRAIL (sTRAIL) showed higher cytotoxic effect for glioma cells than TRAIL [113, 114]. Murine NSC cell line C17.2 cells expressing combination of firefly luciferase and S-TRAIL were grafted into glioma-bearing animals, and there was a marked reduction in tumor volume [114, 115].
Type I (α/β) IFN has been known to have multiple antitumor effects including direct inhibition of tumor cell proliferation through both cell cycle arrest and induction of apoptosis [116, 117] as well as indirect antitumor activity through immunomodulation [118, 119] and inhibition of angiogenesis [117, 120]. Despite exciting preclinical results, clinical trials with IFN-α/β in tumor therapy achieved only a limited success because of its extremely short half-life upon administration and their systemic toxicity. Recently we have generated immortalized human NSCs (HB1.F3) overexpressing human interferon-β (IFN-β) by adenoviral transduction and then intravenously injected in SCID mice bearing metastatic neuroblastoma. Human NSCs expressing IFN-β displayed a high tropism for metastatic neuroblastoma in liver and kidney and targeted delivery of antitumor IFN-β, resulting in significant reduction in tumor growth [121].
As shown in earlier section, human NSCs that were retrovially transduced with cytosine deaminase (CD) gene showed remarkable ‘bystander killer effect’ on glioma cells following the application of prodrug 5-fluorocytosine (5-FC) [53, 55, 59–62]. In a recent study, human NSCs genetically modified to express both CD and IFN-β genes were found to intensify antitumor effect on experimental glioma [122]. In vitro studies demonstrated that NSCs expressing both CD and IFN-β exerted a remarkable bystander effect on human glioma cells following the application of 5-FC, as compared to parental NSCs and CD-expressing NSCs. This combination CD-IFN-β gene therapy regimen extended survival periods significantly in experimental animals. This study indicates that the toxic effect exerted by a combination of two factors, CD and IFN-β, is more effective than the effect exerted individually by either factor. These findings support the possible application of a one-two-punch combination therapy for the treatment of malignant gliomas [122, 123].
Recombinant monoclonal antibodies have emerged as important tools for cancer therapy. However, the large molecular size of antibodies limits their ability to efficiently penetrate solid tumors and precludes efficient crossing of the blood-brain-barrier into the brain. The tumor-tropic properties of human NSCs can overcome these obstacles and significantly improve cancer immunotherapy. In a recent study, F3 human NSCs were modified to secrete anti-HER2 immunoglobulin molecules (HerceptinTM, trastuzumab), a monoclonal antibody widely used to treat HER2-overexpressing breast cancer, and injected into mice bearing brain metastasis of breast cancer [124]. Human NSCs genetically modified to secrete anti-HER2 immunoglobulin molecules were found to inhibit effectively the proliferation of HER2-overexpressing breast cancer cells in vitro. In addition, immunoglobulin-secreting human NSCs exhibit preferential tropism to tumor cells in vivo, and can deliver antibodies to human breast cancer xenografts in mice. This NSC-mediated antibody delivery system has the potential to significantly improve clinical outcome for patients with breast cancer and other solid tumors.
Risk of Tumorigenesis in Immortal NSCs
Clonal immortal human NSC lines generated by expression of myc proto-oncogene show unique opportunity of providing endless supply of human stem cell populations with homogenous genetic and biological properties. These human NSCs maintain a stable karyotype and retaining biological activity and multipotent differentiation capacity [46, 47, 52]. Although tumor formation has never been observed with human NSCs in our numerous pre-clinical animal studies, it is important in determining the inherent risk of using immortal human NSCs for clinical applications. In a recent study in immortal human NSC line, silencing of c-myc expression in vivo was demonstrated and this finding is consistent with the absence of tumorigenesis [125]. In the samples prepared from grafts of human NSCs (containing a single copy of the c-myc transgene delivered by retroviral infection) in ischemia rodent brain, 62 to 100% of the transgene sites were methylated [125]. Direct modification of the DNA by methylation is an epigenetic mechanism to downregulate or silence gene expression. These results indicate that the possible risk of tumorigenesis in transplants of myc-transduced immortal NSCs in clinical setting is minimal and safe use of the cells is assured. In addition, NSCs encoded with suicide transgenes are destined to die following administration of prodrugs.
Clinical Trials
As of April 2010, according to the clinical trials.gov website, over 2050 gene therapy clinical trials have been completed, or are ongoing worldwide in 28 countries, more than 60% of these trials are in cancer gene therapy, and 81 gene therapy clinical trials currently in brain tumor gene therapy. Treatment for brain cancers involves surgical resection, chemotherapy and radiotherapy. Previously numerous gene therapy trials for brain tumors particularly for malignant glioblastoma have been conducted but the most of these trials achieved only limited success. Current status of gene therapy clinical trials for glioblastoma was reviewed by King et al. [126].
Recently (December 5, 2007), NIH Recombianant cDNA Advisory Committee (NIH RAC) has approved an application of the City of Hope Medical Center (Duarte, CA) to conduct a clinical trial in patients with recurrent high grade glioma using immortalized human NSCs that have been retrovirally transduced to express CD therapeutic transgene [127]; the human NSC cell line has been generated at my laboratory at the University of British Columbia, Vancouver, Canada [70, 128]. In animal models, the safety, feasibility, and efficacy of human NSCs to track invasive tumor cells and distant micro-tumor foci and to deliver therapeutic gene products to tumor cells, thereby providing an effective anti-tumor response overcoming obstacles facing current gene therapy strategies have been demonstrated. In this pilot study with ten patients with recurrent glioma to determine the safety and feasibility of an immortalized human NSC line (HB1.F3) that expresses the enzyme CD. NSCs will distribute throughout the primary tumor site and will co-localize with infiltrating tumor cells within 5 days. An oral prodrug (5-FC), administered on the fifth day for 7 days, will be converted to the chemotherapeutic agent 5-FU by NSCs expressing CD, which will then be secreted at the tumor site to produce an anti-tumor effect. The City of Hope Medical Center has recently established a fully characterized cGMP Master Cell Bank so that the human NSC line is readily expandable, cost-effective, and distributed to other medical centers which will allow this NSC-based therapy to be adopted widely. It took more than 2 years to clarify and implement requests by the FDA and now in the final phase of go ahead for the clinical trials from the agency.
References
Black, P. M., & Loeffler, J. (2005). Cancer of the nervous system. Oxford: Blackwell.
Packer, R. J., Cogen, P., Vezina, G., & Rorke, L. B. (1999). Medulloblastoma: clinical and biologic aspects. Neuro-Oncology, 1, 232–250.
Surawicz, T. S., McCarthy, B. J., Kupelian, V., Jukich, P., Bruner, J. M., & Davis, F. G. (1999). Descriptive epidemiology of primary brain and CNS tumors: results from the central brain tumor registry of the United States, 1990–1994. Neuro-Oncology, 1, 14–25.
Rosenberg, R. A., Aebersold, P., Cornetta, K., et al. (1990). Gene transfer into humans – Immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. The New England Journal of Medicine, 323, 570–578.
Blasé, R. M., Culver, K. W., Miller, A. D., et al. (1995). Lymphocyte directed gene therapy for ADA-SCID: initial trial results after 4 years. Science, 270, 475–480.
Edelstein, M. L., Abedi, M. R., & Wixon, J. (2007). Gene therapy clinical trials worldwide to 2007-an update. The Journal of Gene Medicine, 9, 833–842.
Brand, K. (2000). Gene therapy for cancer. In N. S. Templeton & D. D. Lasic (Eds.), Gene therapy: Thrapeutic mechanisms and stratagies (pp. 439–472). New York: Dekker.
Helene, C. (1994). Control of oncogene exression by antisense nucleic acids. European Journal of Cancer, 30A, 1721–1726.
Reynolds, A., Anderson, E., Vermeulen, A., et al. (2006). Induction of the interferon response by siRNA is cell type- and duplex length-dependent. RNA, 12, 988–993.
Mullen, C. A. (1994). Metabolic suicide genes in gene therapy. Pharmacology & Therapeutics, 63, 199–207.
Shen, Y., & White, E. (2001). p53-dependent apoptosis pathways. Advances in Cancer Research, 82, 55–84.
Opalka, P., Dickcopp, K., & Kirch, H. C. (2002). Apoptotic genes in cancer therapy. Cells, Tissues, Organs, 172, 126–132.
Greenblatt, M. S., Bennett, W. P., Hollstein, M., & Harris, C. C. (1994). Mutations in p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Research, 54, 4855–4878.
Anderson, S. C., Johnson, D. E., Engler, H., et al. (1998). P53 gene therapy in a rat model of hepatocellular carcinoma: intra-arterial delivery of recombinant adenovirus. Clinical Cancer Research, 4, 1649–1659.
Dolivet, G., Merlin, J. L., Barberi, M., et al. (2002). In vivo growth inhibitory effect of iterative wide-type p53 gene transfer in human head and neck carcinoma xenografts using glucosylated polyethyleneimmine nonviral vector. Cancer Gene Therapy, 9, 708–714.
Swisher, S. G., Roth, J. A., Nemunaitis, J., et al. (1999). Adenovirus-mediated p53 gene transfer in advanced non-small cell lung carcinoma. Journal of the National Cancer Institute, 91, 763–771.
Nemunaitis, J., Swisher, S. G., Timmons, T., et al. (2003). Adenovirus mediated p53 gene transfer in sequence with cisplatin to tumors of patients with non-small-cell lung cancer. Journal of Clinical Oncology, 18, 495–507.
Swisher, S. G., Roth, J. A., Komaki, R., et al. (2003). Induction of p53-regulated gene and tumor regression in lung cancer patients after intratumoral delivery of adenoviral p53 (INGN 2012) and radiation therapy. Clinical Cancer Research, 9, 93–101.
Marcucci, G., Byrd, J. C., Dai, G., et al. (2003). Phase 1 and pharmacodynamic studies of G3139, a Bcl-2 antisense oligonucleotide, in combination with chemotherapy in refractory or relapsed acute leukemia. Blood, 101, 425–432.
Potter, M., & Marcu, K. B. (1997). The c-myc story: where we’ve been, where we seem to be going. Current Topics in Microbiology and Immunology, 224, 1–17.
Schrovsky, O. G., Rozados, V. R., Gervasoni, S. I., et al. (2000). Inhibition of ras oncogene: a novel approach to antineoplastic therapy. Journal of Biomedical Science, 7, 292–298.
Helene, C., Thuong, N. T., & Harel-Bellan, A. (1992). Control of gene expression by triple helix-forming oligonucleotides: the antigen strategy. Annals of the New York Academy of Sciences, 660, 27–36.
Fei, R., & Shaoyang, L. (2002). Combination antigene therapy targeting c-myc and c-erbB(2) in the ovarian cancer COC cell line. Gynecologic Oncology, 85, 40–44.
Fire, A., Xu, S., Montgomery, M., et al. (1998). Potent and specific genetic interference by doubl-stranded RNA in C. elegans. Nature, 391, 874–881.
Moolten, F. L., & Wells, J. M. (1990). Curability of tumors bearing hepes simplex thymidine kinase genes transferred by retroviral vectors. Journal of the National Cancer Institute, 82, 297–300.
Kw, C., Ram, Z., Wallbridge, S., et al. (1992). In vivo gene transfer with retroviral vector producing cells for treatment of experimental brain tumors. Science, 256, 1550–1552.
Hamel, W., Magnelli, L., Korsmeyer, S. J., et al. (1996). Hepes simplex thymidine kinase/ ganciclovir-mediated apoptotic cell death of bystander cells. Cancer Research, 56, 2697–2702.
Link, C. J., Levy, J. P., McCann, L. Z., & Moorman, D. W. (1987). Gene therapy for colon cancer with the herpes simplex thymidine kinase gene. Journal of Surgical Oncology, 64, 289–294.
Filat, A. C., Carrio, M., Cascante, A., et al. (2003). Suicide gene therapy mediated by the herpes simplex virus thymidine kinase gene/ ganciclovir system: fifteen years of application. Current Gene Therapy, 3, 13–26.
Freeman, S. M., Abboud, C. N., Whartenby, K. A., et al. (1993). The ‘bystander effect’: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Research, 53, 5274–5283.
Huber, B. E., Austin, E. A., Richards, C. A., Davis, S., & Good, S. S. (1994). Metabolism of 5-FC to 5-FU in human colorectal tumor cells transduced with the cytosine deaminase gene: significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proceedings of the National Academy of Sciences of the United States of America, 91, 8302–8306.
Li, Z., Shanmugam, N., Katayose, D., et al. (1997). Enzyme/prodrug gene therapy approach for breast cancer using a recombinant adenovirus expressing E coli cytosine deaminase. Cancer Gene Therapy, 4, 113–117.
Wei, M. X., Tamiya, T., Rhee, R. J., et al. (1995). Diffusible cytotosxic metabolites contributes to the in vitro bystander effect associated with the cyclophosphamide / cytochrome P450 2B1 cancer gene therapy. Clinical Cancer Research, 1, 1171–1177.
Marais, R., Spooner, R. A., Light, Y., et al. (1996). Gene-directed enzyme prodrug therapy with mustard prodrug/carboxypeptidase G2 combination. Cancer Research, 56, 4735–4742.
Danks, M. K., Morton, C. I., Pawlik, C. A., & Potter, C. M. (1998). Overexpression of a rabbit liver carboxylesterase sensitizes human tumor cells to CPT-11. Cancer Research, 58, 20–22.
Ostrand-Rosenberg, S., Gunther, V. S., Armstrong, T. A., et al. (1999). Immunologic targets for the gene therapy of cancer. In F. C. Lattime & S. L. Gerson (Eds.), Gene Therapy of Cancer (pp. 33–48). San Diego: Academic Press.
Barajas, M., Mazzolini, G., Gnove, G., et al. (2001). Gene therapy of orthotropic hepatocellular carcinoma in rats using adenovirus coding for IL-12. Hepatology, 33, 52–61.
Shi, F., Rakhmilevich, A. L., Heise, C. P., et al. (2002). Intratumoral injection of IL-12 plasmid DNA, either naked or in complex with cationic lipid, results in similar tumor regression in a murine model. Molecular Cancer Therapeutics, 1, 949–957.
Saudemont, A., Buffenoir, G., Denys, A., et al. (2002). Gene transfer of CD154 and IL-12 cDNA induces an anti-leukemic immunity in a murine model of acute leukemia. Leukemia, 16, 1637–1644.
Vollmer, C. M., Eilber, F. C., Butterfield, L. H., et al. (1999). Alpha-fetoprotein-specific genetic immunotherapy for hepatocellular carcinoma. Cancer Research, 59, 3064–3067.
Ribas, A., Butterfield, L. H., & Economou, J. S. (2000). Genetic immunotherapy for cancer. The Oncologist, 5, 87–98.
Pardoll, D. M. (1998). Cancer vaccines. Natural Medicines, 4(suppl), 525–531.
Grimm, C. F., Ortmann, D., Mohr, L., et al. (2000). Mouse alpha-fetoprotein-specific DNA-based immunotherapy of hepatocellular carcinoma leads to tumor regression in mice. Gastroenteroly, 119, 1104–1112.
Hanke, P., Serwe, M., Dombrowski, F., et al. (2002). DNA vaccination with AFP-encoding tumors prevents growth of subcutaneous AFP-expressing tumors and does not interfere with liver regeneration in mice. Cancer Gene Therapy, 9, 346–355.
Brustle, O., & McKay, R. G. (1996). Neuronal progenotors as tools for cell replacement in the nervous system. Current Opinion in Neurobiology, 6, 688–695.
Flax, J. D., Aurora, S., Yang, C., et al. (1998). Engraftable human neural stem cells respond to developmental cues, replace neurons, and express foreign genes. Nature Biotechnology, 16, 1033–1039.
Kim, S. U. (2004). Human neural stem cells genetically modified for brain repair in neurological disorders. Neuropathology, 24, 159–171.
Lindvall, O., Kokaia, Z., & Martinez-Serrano, A. (2004). Stem cell therapy for human neurodegenerative disorders-how to make it work. Nature Medicine, 10(suppl), S42–S50.
Goldman, S. (2005). Stem and progenitor cell-based therapy of the human central nervous system. Nature Biotechnology, 7, 862–871.
Muller, F., Snyder, E. Y., & Loring, J. F. (2006). Gene therapy: can neural stem cells deliver? Nature Reviews Neuroscience, 7, 75–84.
Lee, J. P., Jeyakumar, M., Gonzalez, R., et al. (2007). Stem cells act through multiple mechanisms to benefit mice with neurodegenerative metabolic disease. Nature Medicine, 13, 439–447.
Kim, S. U., & de Vellis, J. (2009). Stem cell-based cell therapy for neurological diseases. A review. Journal of Neuroscience Research, 87, 2183–2200.
Aboody, K. S., Brown, A., Rainov, N. G., et al. (2000). Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proceedings of the National Academy of Sciences of the United States of America, 97, 12846–12851.
Aboody, K. S., Najbauer, J., Schmidt, N. O., et al. (2006). Targeting of melanoma brain metastases using engineered neural stem/progenitor cells. Neuro-Oncology, 8, 119–126.
Aboody, K. S., Bush, R. A., Garcia, E., et al. (2006). Development of a tumor-selective approach to treat metastatic cancer. PLoS ONE, 1, e123, 1–8.
Brown, A. B., Yang, W., Schmidt, N. O., et al. (2003). Intravascular delivery of neural stem cell lines to target intracranial and extracranial tumors of neural and non-neural origin. Human Gene Therapy, 14, 1777–1785.
Najbauer, J., Danks, M. K., Schmidt, N. O., Kim, S. U., & Aboody, K. S. (2007). Neural stem cell-mediated therapy of primary and metastatic solid tumors. In: Bertolotti, R., & Ozawa, K. (Eds.). Progress in gene therapy, autologous and cancer stem cell gene therapy, vol 3, (pp 1–42), London, World Scientific.
Lin, D., Najbauer, J., Salvaterra, P. M., et al. (2007). Novel method for visualizing and modeling the spatial distribution of neural stem cells within intracranial glioma. Neuroimage, 37(suppl 1), S18–S26.
Danks, M. K., Yoon, K. J., Bush, R. A., et al. (2007). Tumor-targeted enzyme/prodrug therapy mediates long-term disease-free survival of mice bearing disseminated neuroblastoma. Cancer Research, 67, 22–25.
Kim, S. K., Kim, S. U., Park, I. H., et al. (2006). Human neural stem cells target experimental intracranial medulloblastoma and deliver a therapeutic gene leading to tumor regression. Clinical Cancer Research, 12, 5550–5556.
Shimato, S., Natsume, A., Takeuchi, H., et al. (2007). Human neural stem cells target and deliver therapeutic gene to experimental leptomeningeal medulloblastoma. Gene Therapy, 14, 1132–1142.
Kim, S. U., Park, I. H., Kim, T. H., et al. (2006). Brain transplantation of human neural stem cells transduced with tyrosine hydroxylase and GTP cyclohydrolase 1 provides functional improvement in animal models of Parkinson disease. Neuropathology, 26, 129–140.
Yasuhara, T., Matsukawa, N., Yu, G., et al. (2006). Transplantation of human neural stem cells exerts neuroprotection in a rat model of Parkinson’s disease. The Journal of Neuroscience, 26, 12497–12511.
Ryu, J. K., Kim, J., Hong, S. H., et al. (2004). Proactive transplantation of human neural stem cells blocks neuronal cell death in rat model of Huntington disease. Neurobiology of Disease, 16, 68–77.
Lee, S. T., Chu, K., Park, J. E., et al. (2005). Intravenous administration of human neural stem cells induces functional recovery in Huntington’s disease rat model. Neuroscience Research, 52, 243–249.
Hwang, D. H., Lee, H. J., Seok, J. I., Kim, B. G., Joo, I. S., & Kim, S. U. (2009). Intrathecal transplantation of human neural stem cells over-expressing VEGF provides behavioral improvement, disease onset delay and survival extension in transgenic ALS mice. Gene Therapy, 16, 1234–1244.
Jeong, S. W., Chu, K., Jung, K. H., Kim, S. U., Kim, M., & Roh, J. K. (2003). Human neural stem cell transplantation promotes functional recovery in rats with experimental intracerebral hemorrhage. Stroke, 34, 2258–2263.
Chu, K., Kim, M., Jeong, S. W., Kim, S. U., & Yoon, B. W. (2003). Human neural stem cells can migrate, differentiate and integrate after intravenous transplantation in adult rats with transient forbrain ischemia. Neuroscience Letters, 343, 637–643.
Chu, K., Park, K. I., Lee, S. T., et al. (2005). Combined treatment of vascular endothelial growth factor and human neural stem cells in experimental focal cerebral ischemia. Neuroscience Research, 53, 384–390.
Lee, H. J., Kim, K. S., Kim, E. J., et al. (2007). Brain transplantation of human neural stem cells promotes functional recovery in mouse intracerebral hemorrhage stroke model. Stem Cells, 25, 211–224.
Lee, H. J., Kim, K. S., Kim, E. J., Park, I. H., & Kim, S. U. (2007). Human neural stem cells over-expressing VEGF provide neuroprotection, angiogenesis and functional recovery in mouse stroke model. PLoS ONE, 1, e156.
Lee, H. J., Park, I. H., Kim, H. J., & Kim, S. U. (2009). Human neural stem cells overexpressing glial cell line derived neurotrophic factor (GDNF) promote functional recovery and neuroprotection in experimental cerebral hemorrhage. Gene Therapy, 16, 1066–1076.
Lee, S. T., Chu, K., Jung, K. H., et al. (2008). Anti-inflammatory mechanism of intravascular neural stem cell transplantation in hemorrhagic stroke. Brain, 131, 616–629.
Meng, A., Ohashi, T., Kim, S. U., et al. (2003). Brain transplantation of genetically engineered human neural stem cells transduced with beta-glucuronidase globally corrects lysosomal storage and brain lesions in mucopolysaccharidosis VII mice. Journal of Neuroscience Research, 74, 266–277.
Erlandsson, A., Larsson, J., & Forsberg-Nilsson, K. (2004). Stem cell factor is a chemoattractant and a survival factor for CNS stem cells. Experimental Cell Research, 301, 201–210.
Sun, L., Lee, J., & Fine, H. A. (2004). Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. Journal of Clinical Investigation, 113, 1364–1374.
Widera, D., Holtkamp, W., Entschladen, F., et al. (2004). MCP-1 induces migration of adult neural stem cells. European Journal of Cell Biology, 83, 381–387.
Imitola, J., Raddassi, K., Park, K. I., et al. (2004). Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proceedings of the National Academy of Sciences of the United States of America, 101, 18117–18122.
Takeuchi, H., Natsume, A., & Wakabayashi, T. (2007). Intravenously transplanted human neural stem cells migrate to the injured spinal cord in adult mice in an SDF-1- and HGF-dependent manner. Neuroscience Letters, 426, 69–74.
Schmidt, N. O., Przylecki, W., Yang, W., et al. (2005). Brain tumor tropism of transplanted human neural stem cells is induced by vascular endothelial growth factor. Neoplasia, 7, 623–629.
Boockvar, J. A., Kapitonov, D., Kapoor, G., et al. (2003). Constitutive EGFR signaling confers a motile phenotype to neural stem cells. Molecular and Cellular Neurosciences, 24, 1116–1130.
Kendall, S. E., Najbauer, J., Johnston, H. F., et al. (2008). Neural stem cell targeting of glioma is dependent on phosphoinositide 3-kinase signaling. Stem Cells, 26, 1575–1586.
Gutova, M., Najbauer, J., Frank, R. T., et al. (2008). Urokinase plasminogen activator and urokinase plasminogen activator receptor mediate human stem cell tropism to malignant solid tumors. Stem Cells, 26, 1406–1413.
Zhao, D., Najbauer, J., Garcia, E., et al. (2008). Neural stem cell tropism to glioma: critical role of tumor hypoxia. Molecular Cancer Research, 6, 1819–1829.
Forte, G., Minieri, M., Cossa, P., et al. (2006). Hepatocyte growth factor effects on mesenchymal stem cells: proliferation, migration, and differentiation. Stem Cells, 24, 23–33.
Glaser, T., Brose, C., Franceschini, I., et al. (2007). Neural cell adhesion molecule polysialylation enhances the sensitivity of embryonic stem cell-derived neural precursors to migration guidance cues. Stem Cells, 25, 3016–3025.
Zheng, H., Fu, G., Dai, T., & Huang, H. (2007). Migration of endothelial progenitor cells mediated by stromal cell-derived factor-1alpha/CXCR4 via PI3K/Akt/eNOS signal transduction pathway. Journal of Cardiovascular Pharmacology, 50, 274–280.
Thu, M. S., Najbauer, J., Kendall, S. E., et al. (2009). Iron labeling and pre-clinical MRI visualization of therapeutic human neural stem cells in a murine glioma model. PLoS ONE, 4, e7218.
Barresi, V., Belluardo, N., Sipione, S., et al. (2003). Transplantation of prodrug-converting neural progenitor cells for brain tumor therapy. Cancer Gene Therapy, 10, 396–402.
Herrlinger, U., Woiciechowski, C., Sena-Esteves, M., et al. (2000). Neural precursor cells for delivery of replication-conditional HSV-1 vectors to intracerebral gliomas. Molecular Therapy, 4, 347–357.
Li, S., Tokuyama, T., Yamamoto, J., et al. (2005). Bystander effect-mediated gene therapy of gliomas using genetically engineered neural stem cells. Cancer Gene Therapy, 12, 600–607.
Brooks, P. C., Silletti, S., von Schalscha, T. L., Friedlander, M., & Cheresh, D. A. (1998). Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell, 92, 391–400.
Bello, L., Lucini, V., Carrabba, G., et al. (2001). Simultaneous inhibition of glioma angiogenesis, cell proliferation, and invasion by a naturally occurring fragment of human metaloproteinase-2. Cancer Research, 61, 8730–8736.
Kim, S. K., Cargioli, T. G., Machluf, M., et al. (2005). PEX-producing human neural stem cells inhibit tumor growth in a mouse glioma model. Clinical Cancer Research, 11, 5965–5970.
Joo, K., Park, I. H., Shin, J. Y., et al. (2009). Human neural stem cells target and deliver therapeutic genes to breast cancer brain metastasis. Molecular Therapy, 17, 570–575.
Gutova, M., Najbauer, J., Chen, M. Y., Kim, S. U., & Aboody, K. S. (2009). Therapeutic targeting of melanoma cells using neural stem cells expressing carboxylesterase, a CPT-11 activating enzyme. Current Stem Cell Research Therapy (in press).
Weller, M., & Fontana, A. (1995). the failure of current immunotherapy for malignant glioma. Tumor-derived TGF-β, T-cell apoptosis, and the immune privilege of the brain. Brain Research Reviews, 21, 128–151.
Hishii, M., Niita, T., Ishida, H., et al. (1995). Human glioma-derived interleukin-10 inhibits antiyumor immune responses in vitro. Neurosurgery, 37, 1160–1167.
Saas, P., Walker, P. R., Hahne, M., et al. (1997). Fas ligand expression by astrocytoma in vivo: maintaining immune privilege in the brain? Journal of Clinical Investigation, 99, 1173–1178.
Benedetti, S., Bruzzone, M. G., Pollo, B., et al. (1999). Eradication of rat malignant glioma by retrovirus-mediated, in vitro delivery of the interleukin 4 gene. Cancer Research, 59, 645–652.
Jean, W. C., Spelllman, S. R., Wallenfrieman, M., Hall, W., & Low, W. C. (1998). Interleukin-12-based immunotherapy against rat 9 L glioma. Neurosurgery, 42, 850–856.
Liu, Y., Ehtersham, M., Samoto, K., et al. (2002). In situ adenoviral interleukin-12 gene transfer confers potent and long-lasting cytotoxic immunity in glioma. Cancer Gene Therapy, 9, 9–15.
Puri, R. K., Leland, P., Kreitmen, R. J., & Pastan, I. (1994). Human neurological cancer cells express IL4 receptors which are target for the toxic effect of IL4-Pseudomonas exotoxin chimeric toxin. International Journal of Cancer, 58, 574–581.
Benedetti, S., Pirola, B., Pollo, B., et al. (2000). Gene therapy of experimental brain tumors using neural progenitor cells. Nature Medicine, 6, 447–450.
Nastala, C. L., Edington, H. D., McKinnery, T. G., et al. (1994). Recombinant IL-12 administration induces tumor regression in association with Interferon-γ production. Journal of Immunology, 153, 1697–1706.
Ehtesham, M., Kabos, P., Kabosova, A., et al. (2002). The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma. Cancer Research, 62, 5657–5663.
Rieger, J., Naumann, U., Ashkenazi, A., et al. (1998). APO2 ligand: anovel lethal weapon against malignant gliomas? FEBS Letters, 427, 124–128.
Pollack, I. F., Erff, M., & Ashkenazi, A. (2001). Direct stimulation of apoptotic signaling by soluble Apo2/tumor necrosis-related apoptosis-inducing ligand leads to selective killing of glioma cells. Clinical Cancer Research, 7, 1362–1369.
Lee, J., Elkahlun, A. G., Messina, S. A., et al. (2002). Cellular and genetic characterization of human adult bone marrow-derived neural stem cells: a potential Cellular vector. Cancer Research, 63, 8877–8889.
Hengartner, M. O. (2000). The biochemistry of apoptosis. Nature, 407, 770–776.
Corsten, M. F., & Shah, K. (2008). Therapeutic stem cells for cancer treatment: hopes and hurdles in tactical warfare. The Lancet Oncology, 9, 376–384.
Ehtesham, M., Kabos, P., Gutierrez, M. A., et al. (2002). Induction of glioblastoma apoptosis using neural stem cell-mediated delivery of tumor necrosis factor-related apoptosis-inducing ligand. Cancer Research, 62, 7170–7174.
Shah, K., Tung, C. H., Yang, K., et al. (2004). Inducible release of TRAIL fusion proteins from a proapoptotic form for tumor therapy. Cancer Research, 64, 3236–3242.
Shah, K., Bureau, E., Kim, D. E., et al. (2005). Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression. Annals of Neurology, 57, 34–41.
Tang, Y., Shah, K., Messelri, S. M., et al. (2003). In vivo tracking of neural progenitor cell migration to glioblastoma. Human Gene Therapy, 14, 1247–1254.
Stark, G. R., Kerr, I. M., Williams, B. R., et al. (1998). How cells respond to interferons. Annual Reviews of Biochemistry, 67, 227–264.
Streck, C. J., Dickson, P. V., Ng, C. Y., et al. (2006). Antitumor efficacy of AAV-mediated delivery of Interferon-beta. Cancer Gene Therapy, 13, 99–106.
Belardelli, F., Ferrantini, M., Proietti, E., et al. (2002). Interferon-alpha in tumor immunity and immunotherapy. Cytokine & Growth Factor Reviews, 13, 119–134.
Dvorak, H. F., & Gressor, I. (1989). microvascular injury in pathgenesis of interferon-induced necrosis of subcutaneous tumors in mice. Journal of the National Cancer Institute, 81, 497–502.
Yoshida, J., Mizuno, M., & Wakabayashi, T. (2004). Interferon-β gene therapy for cancer: basic research to clinical application. Cancer Science, 95, 858–865.
Dickson, P. V., Hamner, J. B., Berger, R. A., et al. (2007). Intravascular administration of tumor tropic neural progenitor cells permits targeted delivery of IFN-β and restricts tumor growth in a murine model of disseminated neuroblastoma. Journal of Pediatric Surgery, 42, 48–53.
Ito, S., Natsume, A., Shimato, S., et al. (in press). Human neural stem cells transduced with IFN-β and cytosine deaminase genes intensify bystander effect in experimental glioma. Cancer Gene Therapy.
Lee, D. H., Ahn, Y., Kim, S. U., et al. (2009). Targeting rat brainstem glioma using human neural stem cells and human mesenchymal stem cells. Clinical Cancer Research, 15, 4925–4934.
Frank, R. T., Edmiston, M., Kendal, S. E. et al. Neural stem cells as a novel platform for tumor-specific delivery of therapeutic antibodies. PLoS One, 4, e8314.
Stevanato, L., Corteling, R. L., Stroemer, P., et al. (2009). c-MycERTAM transgene silencing in a genetically modified human neural stem cell line implanted into MCAo rodent brain. BMC Neuroscience, 10, 86.
King, G. D., Curtin, J. F., Candolfi, M., et al. (2005). Gene therapy and targeted toxins for glioma. Current Gene Therapy, 5, 535–557.
Najbauer, J., Danks, M. K., Kim, S. U., et al. (2008). Neural stem cell mediated tumor-selective gene delivery: towards high grade glioma clinical trials. Molecular Therapy, 16(Suppl 1), S136.
Kim, S. U., Nagai, A., Nakagawa, E., et al. (2009). Production and characterization of immortal human neural stem cell line with multipotent differentiation property. Methods in Molecular Biology, 438, 103–121.
Acknowledgements
The author thanks Drs. K. Aboody, M. Danks, S.K. Kim, D.H. Nam, A. Natsume and H.J. Lee for their contributions to the works cited in the article. This work was supported by grants from the Canadian Myelin Research Initiative and the National R&D Program for Cancer Control, Korean Ministry for Health, Welfare and Family affairs (20090403).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kim, S.U. Neural Stem Cell-based Gene Therapy for Brain Tumors. Stem Cell Rev and Rep 7, 130–140 (2011). https://doi.org/10.1007/s12015-010-9154-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-010-9154-1