
Abstract
Embryonic stem cell (ESC) research is a promising area of investigation with enormous therapeutic potential. We have injected murine wild type (WT) ESCs into a variety of mutant murine blastocysts, which are predisposed to develop a human-like disease, such as muscular dystrophy or the embryonic lethal “thin myocardial syndrome”. In this review, we summarize data indicating that partial incorporation of ESCs is sufficient to prevent disease from occurring. We also present data indicating that blastocyst incorporation of ESCs may aid in the prevention of heart failure in stressed WT mice. In some cases, the rescue observed is predominantly non-cell autonomous and relies on the production of secreted factors from the ES-derived cells, but in others, cell replacement is required. Thus, congenital or acquired disease can be pre-emptively averted in mice by developmental injection of ESCs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Embryonic stem cell (ESC) research is considered an area of study with significant promise for the future treatment of congenital and acquired diseases. ESCs have the capacity to differentiate into a wide range of the cell types that compose the human body. This unique ability makes ESCs important candidates for study in the emerging era of regenerative medicine [1–4]. Our laboratory utilizes ESCs to study the mechanisms by which congenital or acquired diseases can be corrected.
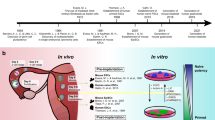
The general approach that we use is to inject wild-type (WT) mouse ESCs into early mouse blastocysts that harbor a mutation. The mutant blastocysts that do not receive ESC treatment are predisposed to develop a disease that recapitulates a human disease. As the ESCs are originally derived from blastocysts, injection of normal ESCs into mutant blastocysts results in a mutual recognition. As a result of the injection of ESCs, the developing animal will be composed of WT cells (derived from the ESCs) and mutant cells (derived from the blastocysts). These mice are termed “chimeras” (Fig. 1). While blastocyst injection is a commonly utilized technique, our laboratory’s protocol employs an important modification. As opposed to the injection of mutant ESCs into WT blastocysts to generate knock-out and knock-in mice, we generate chimeras via the injection of WT ESCs into mutant blastocysts. The question we ask is also different: can we prevent disease from occurring and elucidate the underlying molecular mechanisms that govern these ESC-mediated corrections? Via protein assays and genomic arrays, we have been able to examine differences in expression between WT, mutant and chimeric mice and identify candidate molecules for continued study. These candidates will be eventually tested by their ability to replace the therapeutic capability of the ESCs.

Approach. The approach involves injection of 15 wild type (WT) ROSA26 LacZ-marked embryonic stem cells (ESCs) into mutant blastocysts, which are predisposed to develop a disease. As a result of incorporation of ESCs, chimeric embryos/mice containing cells derived from the ESCs and from the blastocyst are generated
In this review, we summarize data indicating that partial incorporation of ESCs at blastula stage is sufficient to exert corrections in various mouse models of human disease. Interestingly, in some cases the rescue observed is predominantly non-cell autonomous and relies on the production of secreted factors, but in others, cell replacement, or cell autonomy, is the predominant corrective process.
Rescue of the “Thin Myocardial Syndrome”
Our first model to test was the “thin myocardial syndrome”, which is exemplified by the inhibitor of DNA binding (Id) knockout (KO) mice [5]. The Id proteins (Id1 to Id4) are dominant negative antagonists of basic helix-loop-helix (bHLH) transcription factors and regulate differentiation in multiple lineages [6]. Previous studies have shown that Id1 to Id4 are expressed in embryonic tissues during development in partially overlapping patterns, in particular in the heart [7, 8]. Within the developing heart, Id genes are expressed specifically in nonmyocardial layers [9]. The Id genes are expressed in the epicardium, in the endocardium, in the endothelium and in endocardial cushion. However, the primary affected target of the Id proteins is the myocardium, which does not express Id proteins but is adjacent to these nonmyocardial structures, suggesting cross-communication between the layers, likely via the control of secreted factors [9, 10]. We studied the requirement of Id1-3 genes during embryogenesis. Loss of one Id gene (Id1 or Id2 or Id3) does not result in any developmental defect, but double KO embryos (Id1/Id2 or Id1/Id3 or Id2/Id3) in any combination display multiple cardiac defects and die by mid-gestation development, which is consistent with the redundant role of Id genes. In these double KO embryos, the myocardial wall is thin, because of a defect in proliferation, the trabeculae have disorganized sheets of myocytes surrounded by an impaired endocardial cell lining, the endocardial cushion is hypoplastic and the ventricular septum is impaired. Because of the prominent cardiac phenotype, these embryos die in utero (Fig. 2) [9, 10].

Rescue of the “thin myocardial syndrome”. Double Id1/Id3 knockout embryos bear a prominent phenotype in the developing heart that causes early fetal demise. Because of the lack of proliferative Id signals originating in the non-myocardial structures (endocardium, epicardium), the embryos bear a thin myocardium and a disorganized endocardial lining. Restoration of Id signaling by ESC incorporation in chimeras (WT/Id1Id3) normalizes the thickness of the myocardium and organizes the endocardium. Two paracrine proliferative factors are implicated: IGF1 and Wnt5a
With the initial goal of extending the viability of the Id1/Id3 double KO embryos for 1-2 days, we injected ROSA26 LacZ-marked WT ES cells into mutant (Id1/Id3 KO) blastocysts. With surprise, the resultant embryos survived to birth (7–8 days after the mid-gestation lethality of the double KO embryos), and a portion of these newborn pups survived to adulthood. None of the defects observed in the Id double KO embryos were observed in the embryos that received ES cells, as all the structures were rescued (Fig. 2). When we examined the hearts of the resultant adult mice, they were normal histopathologically and functionally. Intriguingly, only a small fraction of the chimeric hearts originated from the WT ES cells, just 20–30%. In other words, 70–80% of the chimeric hearts were composed of Id1/3 double KO mutant cells. Thus, a small fraction of WT ES cells in the chimeras can rescue cardiac function and morphology. Two possibilities can explain this observation. One is that only the WT cells are functional and the heart survives with a small percentage of normal cells. A more provocative possibility is that the Id KO cells, despite harboring the lethal mutation, are induced to behave as WT cells by the surrounding ES derived cells. To distinguish between these two possibilities, we performed a microarray analysis. Of the genes dysregulated in the Id KO hearts, 85% were restored to WT levels throughout the chimeric hearts. Therefore we conclude that the fraction of the heart (originated from the WT ES cells) induced the majority of the heart (originated from the Id1/3 KO blastocyst) to behave normally, likely via the secretion of healing factors. Our next step was to identify the nature of the healing factors [9, 10].
To address this central question, we performed a microarray analysis with RNA from epicardial cultures (which are one source of Id proteins) of WT, KO and chimeric hearts. We became interested in insulin-like growth factor-1 (IGF-1), because IGF-1 was downregulated in the Id KO cultures (versus WT) and was normalized in the chimeric cultures (versus WT). In addition, IGF-1 promotes cardiac myocyte proliferation, is released into the bloodstream, IGF-1 and Id gene expression patterns overlap, and the loss of IGF-1 or Id genes has remarkable similarities [11–13]. To study if IGF-1 could replace the rescue effect of the ES cells, we injected mouse recombinant IGF-1 into the mother (intraperitoneal injection) harboring Id1/3 double KO embryos, predisposed to die at mid-gestation. With surprise again, we noticed that the embryos developed to term, but in this case they died soon thereafter. When we examined the IGF-1-rescued embryos at mid-gestation, the proliferative defect that leads to a thin myocardium was corrected (Fig. 2) [9]. However, the rescue was partial, as some of the structural defects (ventricular septal defects, compromised trabeculation) failed to be corrected by IGF-1 administration [9]. We then reasoned that in addition to IGF-1, the ES cells could be supplying paracrine (short-range) factors, which could be required to achieve a full rescue of the cardiac phenotype. We next turned our attention to Wnt5a, which was overproduced in the chimeric epicardial cultures (versus WT) (Fig. 2). The fact that the chimeric heart had more Wnt5a than the WT heart suggested a “neomorphic” effect of the ES cells. In these sense, the ES derived cells, surrounded by a mutant environment (Id KO cells) would produce disproportionate levels of Wnt5a to compensate for a reduction in Id dose (Fig. 2). As opposed to IGF-1, Wnt5a is a lipid-modified glycoprotein that cannot travel long-distances [14]. In addition, loss of Wnt5a leads to ventricular septal defects [15], and Wnt signaling is required for cardiac development [16]. But Wnt5a cannot be injected maternally, as it cannot travel long-distances to reach the embryo. If Wnt5a is a short-range signal protein involved in the corrections, it should correct gene expression profiles in the Id KO hearts. In a control experiment, WT and KO heart explants were cocultured with murine embryonic fibroblasts (MEFs), and subsequently arrayed. In this control condition (MEFs-no Wnt5a), over 600 genes were dysregulated in the Id KO explants (relative to WT explants). Next we cocultured WT and KO heart explants with MEFs that overexpress Wnt5a (MEFs-Wnt5a), to mimic a chimeric environment, and observed that 85% of the dysregulated genes were normalized (relative to WT explants). Therefore, overexpression of Wnt5a corrects gene expression profiles.
The take-home message of this section is that a few ES cells incorporated in the developing heart prevent a lethal disease in a non-cell autonomous manner, by secretion of healing factors [5, 9, 10].
Rescue of Duchenne Muscular Dystrophy
Our second model focuses on Duchenne muscular dystrophy (DMD), which is an X-linked congenital disease that results from a spontaneous mutation in the dystrophin gene [17]. Most sufferers tend to be males but heterozygote females show mild symptoms. DMD is recapitulated in mdx mice [18]. Dystrophin, the 2nd largest protein in the proteome, is pivotal for the stabilization of the dystrophin-glycoprotein complex (DGC). The DGC is a complex consisting of many proteins (sacoglycans, dystrobrevin, syntrophin, nNOS, etc) that spans the sarcolemma of muscular tissue (skeletal, cardiac and smooth muscles) and acts as a stable connection between the extracellular matrix and the cytoskeleton [19]. When dystrophin is not present, the DGC is destabilized and thereby more prone to stress-induced damage from normal everyday muscular contractions. This results in the progressive degradation of DGC members and the ensuing muscular degeneration forces patients into a wheel chair as early as ten years old. Over time, the patient outcome worsens as the muscular tissues continue to degenerate and death normally occurs in the late teens or early twenties, primarily due to the dystrophic heart and diaphragm (Fig. 3).

Rescue of muscular dystrophy. Absence of dystrophin in mdx mice destabilizes the dystrophin-glycoprotein complex in the skeletal muscle, resulting in cycles of degeneration/regeneration, necrosis, mononuclear invasion and central nucleation. In addition, mdx mice are very lean. Incorporation of 10–30% of ESCs supplies dystrophin to most of the skeletal fibers. As a result, the chimeric muscle (WT/mdx) is not dystrophic. Chimeric mice also regain fat mass, which in turn produces hypertrophic factors to help fibers not containing dystrophin in the WT/mdx chimeras
Our lab wanted to determine whether early injection of ROSA26 LacZ-marked WT ESCs into mutant mdx blastocysts can improve pathology and function. We hypothesized that partial restoration of dystrophin in all tissues could uncover novel corrective effects of dystrophin and prevent disease at morphological and functional levels. We then proceeded to produce chimeric mice with varying percentages of WT ESC (dystrophin-positive) incorporation. As a negative control, we also produced chimeras with the injection of mdx ESCs (dystrophin-negative) into mdx blastocysts. The degree of chimerism was determined via Xgal staining of tail biopsies at 3-weeks of age and from tissues at the time of sacrifice, which was 4 months. β-galactosidase activity was detected in all major tissues of the chimeric mice, including brain, liver, heart and skeletal muscles. When we injected mdx ESCs (dystrophin-negative) into mdx blastocysts, the resulting chimeras (mdx/mdx) showed no correction of gene expression profiles (microarray) and displayed the same dystrophic phenotype as mdx mice. However, when we examined the WT/mdx tissues, global corrections were observed in chimeras with 10–30% WT ESC incorporation due to the increased expression of dystrophin. As for the WT/mdx chimeras with <10% incorporation, they were almost identical to the mdx phenotype and showed little, if any corrections. For example, histological analysis of 10–30% chimeric skeletal muscles such as pectoralis, quadriceps and tibialis anterior showed a reversal of the necrosis and mononuclear invasion that is paramount in the mdx phenotype (as well as <10% skeletal muscle tissues) (Fig. 3) [20]. Functional improvements were established by investigating stress outputs for tetanic contractions and twitch contractions from the extensor digitorum longus muscle. Consistent with the aforementioned histological corrections, stress outputs of the 10–30% WT/mdx chimeras were comparable to WT stress outputs and the <10% WT/mdx chimeras resembled the mdx mice [20].
Skeletal muscle hypertrophy and disorganization is a common effect associated with the phenotype of DMD patients [21]. In our 10–30% chimeras, we observed improved pathology of WT/mdx chimeric skeletal tissues to near wild type levels. In our chimeras, this occurred in a dose-dependent manner as the level of WT ESC incorporation increased. For example, the thickness of the WT/mdx chimera diaphragms (which is comparable to WT diaphragms) was at least twice as thin as mdx diaphragms. Whereas, mdx tissues are recognized by their disorganized structure, the WT/mdx chimera diaphragms had a more organized structure, with a smaller amount of fibers across the width of the tissue as well as more uniform cross-sectional areas of those fibers [20].
Overproduction of dystrophin occurs in the skeletal muscle but not in the heart. This difference in expression can be attributed to the fact that skeletal muscle cells fuse and form a syncytium consisting of a multinucleated fiber, whereas cardiac tissue does not. For example, heterozygote DMD hearts only have 50% of dystrophin levels due to X-chromosome inactivation, but all skeletal muscle fibers contain dystrophin. Concordant with this observation, western blot analysis of skeletal muscles (quadriceps and pectoralis) in our 10–30% WT/mdx chimeras, showed 70–100% of dystrophin levels. And as expected, dystrophin levels in the hearts of these same mice only reached 10–30% of WT levels via western blot and immunohistochemical analysis. It can be concluded that dystrophin levels are elevated in skeletal muscle relative to heart muscle because WT ESC-derived myonuclei have the ability to overproduce dystrophin to make up for the inability of the other mdx myonuclei in the same fiber, to produce this key structural protein and/or reduce its degradation. Further western blotting also showed that in skeletal tissues with high levels of dystrophin, other DGC members such as dystrobrevin, syntrophin and nNOS were present at much higher levels suggesting that dystrophin prevents the destabilization and the resultant degradation of DGC member proteins. In summary, each cardiac cell is responsible for itself but that is not the case in skeletal muscle because there are compensatory mechanisms due to its own syncitium that facilitate its own rescue [20].
Skeletal muscle has the distinct property to regenerate via activation of satellite cells. The WT ESCs we injected were shown to contribute to the mosacism of the myogenic population and successfully produces Xgal positive, multinucleated myotubes. The major function of the satellite cells is to repair, revitalize, and mediate skeletal muscle tissue and growth by differentiating into skeletal myocytes [22]. Unfortunately, the regenerative capacity of the satellite cells cannot compensate for the structural damage associated with DMD, resulting in the loss of muscle fibers and an increase in fibrosis over time. When skeletal fibers are regenerating, they become centrally nucleated and this is widespread in mdx tissues. Our 10–30% WT/mdx chimeras showed little central nucleation (diaphragm and tibialis anterior muscles), indicating a significant slowdown in the demand for skeletal fiber regeneration. To further this point, we performed immunohistochemistry for embryonic myosin heavy chain (eMHC), which is a marker for regeneration. We found that eMHC was greatly reduced in the skeletal muscle of the 10–30% WT/mdx chimeras. Together, these data show that WT/mdx muscle shows significantly less regeneration than mdx tissue [20].
Along with the corrections within the skeletal fibers, the localized area outside the fibers also benefited from the WT ESC injections. For example, mdx mice are normally very lean compared to WT, but our 10–30% WTmdx chimeras showed nearly a 4-fold improvement in abdominal fat, almost equal to WT mice. This fat increase was not due to increased adipocyte size but actually resulted from an increased adipocyte population. The fat does not produce dystrophin, but the recovery in the amount of fat depends on the presence of dystrophin from dystrophin-expressing tissues (i.e. skeletal muscle). Compared to WT fat, this chimeric fat showed an upregulation of skeletal and cardiac contractile genes such as myosin, actin, tropomyosin and several troponin isoforms. And finally, follistatin-like 1 and 3, which both promote hypertrophy [23], were shown to be highly upregulated in WT/mdx fat relative to WT fat. Thus, the fat is highly dependent on the recovery of dystrophin-expressing tissues, which in turn could be secreting hypertrophic factors that could impinge on the recovery of the skeletal muscle. This suggests a positive feed-back mechanism of tissue communication (in this case fat/skeletal muscle), mediated perhaps by a novel mechanism of factor secretion (Fig. 3) [20].
Rescue of Central Nervous System Demyelination Disease
Recently, other labs also utilized this same model of injecting LacZ-labeled WT ESCs into mutant blastocysts in order to study the pathways responsible for disease rescue [24]. They employed the mutant homozygous shiverer mouse (shi) that harbors a large deletion in the myelin basic protein (MBP) gene, and as a result, suffer from severe demyelination throughout the central nervous system (CNS). As a result, their phenotype is characterized by tremors as early as postnatal day 10 and hindlimb ataxia soon thereafter. Death ensues in a few months. This group was able to produce 48 WT/shi chimeras and their phenotypes were assessed for the aforementioned tremors and ataxia up until the mice were sacrificed at nine weeks. Following sacrifice, the presence of MBP was determined via both genotyping and immunofluorescence. The genotype of the rescued shi chimeras showed both WT and shi bands. Immunofluorescence analysis of the rescued shi mice showed a widespread and strong distribution of MBP throughout the brain sections. However, the chimeras that were not rescued showed regional gaps and abnormal patterns of MBP expression throughout the brain. Furthermore, the motor functionality of these chimeras correlated with the levels of MBP distribution throughout the brain. This group noted that they did not quantify the level of chimerism via LacZ staining due to variable expression levels of the β-gal reporter following ESC differentiation. In spite of this, they report that they were able to observe co-localization of LacZ with other proteins such as Olig1, GFAP and NeuN, suggesting that the injected ESCs contributed to not only oligodendrocytes but also astrocytes and neurons.
Engineered Myocardial Infarction-Resistant Phenotype
Can pre-natal ESC transplantation yield resistance to post-natal myocardial infarction? It has recently been established that heart muscle continuously rejuvenates during lifespan through renewal of cardiomyocytes [25], yet in the context of large-scale destruction—as it occurs following ischemic insult—the regenerative potential is insufficient to ensure organ salvage. Indeed, acquired heart disease is a leading cause of morbidity and mortality despite advances in our understanding of cardioprotective processes [26]. A preemptive intervention capable to engineer tolerance would offer prevention to tissue at risk avoiding the anticipated progression towards debilitating heart failure.
To test whether pre-natal incorporation of ESCs could augment stress tolerance and ensure lifelong protection, pluripotent stem cells were microsurgically introduced at the blastocyst stage of murine embryo development to generate stochastic integration throughout differentiating lineages [27]. Using microinjection, ESCs, obtained from the ROSA26 line constitutively expressing the LacZ reporter, were placed into the blastocoele of a C57BL/6J (WT) embryo. Surgical transfer of derived blastocysts into the uterus of pseudopregnant females yielded full-term chimera. Within offspring, tissues derived from ROSA26 ESCs were traced by the LacZ transgene (Xgal staining) demonstrating that transplantation of ESCs into pre-implantation blastocysts generated a mosaic embryo with sustained engraftment of stem cell-derived tissue into adulthood. Noticeably, chimera displayed central obesity, associated with dominant depots of subcutaneous and visceral fat, but were otherwise devoid of obesity-related morbidity (Fig. 4). Throughout the one year follow-up, non-chimera and chimera exhibited similar cardiac performance. Importantly, however, upon permanent ligation of the left anterior coronary artery and despite comparable ischemic insult, middle-age non-chimeric hearts rapidly deteriorated with irreversible decline in contractile function, in contrast to chimera that displayed progressive recovery of heart performance (Fig. 4). Contrary to the vulnerable non-chimera, 12–14 months old age-matched chimera were remarkably resistant to imposed myocardial infarction and regained within 1-month contractile performance at pre-occlusion levels. In fact, preemptive transplantation of ESCs at the embryonic stage halted development of cardiomyopathy traits in the adult chimera, averting progression of disease which manifested in failing non-chimera as electrical remodeling and ventricular enlargement with fibrosis (Fig. 4). In contrast, activation of the tissue repair process in the chimeric cohort was characterized by an increased stem cell load in adipose tissue and upregulated markers of biogenesis Ki67, c-Kit and Sca-1 in the myocardium. Favorable outcome in infarcted chimera translated into an overall benefit in workload capacity and survival. In this way, stem cell transplant into pre-implantation embryo yielded a myocardial infarction-tolerant adult phenotype mitigating the clinically relevant end-points of the heart failure syndrome [27].

Myocardial infarction-resistant phenotype. Permanent ligation of the left anterior coronary artery in normal (WT) hearts results in myocardial infarction, with a prominent damaged (fibrotic) area in the left ventricular wall, cardiomegaly and decreased ejection fraction. Incorporation of WT ESCs in chimeras augments the stem cell pools in the heart and in the fat. Unlike the WT hearts, the chimeric hearts (WT/WT) resist the external insult, as they are not damaged and have normal size and function
The beneficial effect of chimeric tissue in the adult heart under imposed stress provides the initial evidence of preventive regenerative medicine in the setting of myocardial infarction implemented through prophylactic intervention. Delivery of exogenous embryonic stem cells into the blastocoel of an early embryo joined native to non-native progeny to yield composite, chimeric blastocysts with a finite blastomere number. In the context of the developing embryo, microsurgical transfers allow effective and sustained integration of independent sources of pluripotent progenitors that are in principle competent to differentiate into all lineages. In view of the physical restrictions imposed by the inner cell mass, the dual population of co-existing native and implanted progenitor cells creates the opportunity for competitive selection and titration of overall chimerism to meet developmental requirements [28]. Predetermined chimerism through embryo manipulation allowed assessment of the therapeutic impact of pre-natal enrichment with non-native progenitors upon cardiac stress challenge in the adulthood. The pattern of tissue formation optimized to accommodate the high-demand of initial cardiogenesis reinforced a failure-safe blueprint that was maintained in offspring throughout lifespan. Although the spectrum of contributing cell types and their origin remains to be defined, the favorable outcome in infarcted chimera corresponded to reparative features of cardiac biogenesis characterized by increased mitotic activity and tissue-specific progenitor cell load associated with reduced fibrosis [27]. Moreover, the surplus of adipose tissue that developed in chimera contained elevated levels of non-hematopoietic mesenchymal progenitor pools, in line with the therapeutic potential of adult stem cells in the setting of myocardial infarction [27]. Preemptive stem cell-based intervention in utero thus provides a strategy to engineer tolerance, and prevent incidence of life-threatening organ failure in the adult. In this way, prenatal transplantation of embryonic stem cells expands the scope of traditional retrospective therapy to the previously unexplored prospective protection.
Final Remarks and Future Directions
We have employed a simple approach (blastocyst injection of WT embryonic stem cells) to correct disease state phenotypes in a variety of mouse models of human disease. In some cases the corrections are achieved predominantly by secretion of healing factors (the “thin myocardial syndrome” or Id KO embryos). Some of the factors in this new chimeric secretome are overproduced (relative to WT) to compensate for the absence of the Id1 and Id3 proteins, whose loss triggers the embryonic lethal phenotype. These compensatory factors (i.e. Wnt5a) were named “neomorphic” factors. In other paradigms (shiverer mice), the corrections are achieved strictly in the structures that are replaced by the WT ESC-derived cells. Other models need both cell and non-cell autonomous mechanisms, like Duchenne muscular dystrophy. In the latter, not only the missing protein (dystrophin) has to be replenished, but also some secreted factors, like follistatin-like proteins from neighboring adipose tissue, are thought to contribute to the corrections via enhancement of the hypertrophic state of the chimeric skeletal muscle. Intriguingly, the stem cells not only prevent disease in mice that bear a genetic mutation, as they also induce changes in the cardiac and adipose stem cell pools in a WT/WT mouse. As a result of these “neomorphic” changes in a wild type environment, the chimeric heart (unlike the normal heart) resists extreme external insults like a permanent coronary artery occlusion.
The studies described above are not aimed for direct translation into humans, rather, they are aimed at identifying novel mechanisms of disease correction. We exploit the ability of the ESCs to deliver defined factors that may ultimately be used to treat congenital and acquired diseases. The identification of corrective molecules derived from the ESCs may have tremendous implications for the treatment of human disease, as they could replace the beneficial effects of the ESCs.
Recently, a novel method for the generation of pluripotent stem cells has emerged. These are somatic cells (from skin fibroblasts for example) subjected to dedifferentiation by the addition of four defined transcription factors (Sox2, Oct3/4, cMyc and Klf4). These cells, named induced pluripotent stem cells (iPSCs), resemble ESCs in many aspects. The iPSCs have the potential to differentiate into a wide range of cell types. As in the case of the ESCs, the iPSCs can generate chimeras when injected into blastocysts. Importantly, human iPSCs can be derived from patients, a feature that cannot be paralleled yet with ESCs [29–35]. It would be intriguing to test the corrective behavior of the iPSCs in our mouse models of human disease, for example, whether the iPSCs secrete healing factors or supply dystrophin to most of the skeletal muscle tissue.
References
Odorico, J. S., Kaufman, D. S., & Thomson, J. A. (2001). Multilineage differentiation from human embryonic stem cell lines. Stem Cells, 19(3), 193–204.
Laflamme, M. A., & Murry, C. E. (2005). Regenerating the heart. Nature Biotechnology, 23(7), 845–856.
Rossant, J. (2008). Stem cells and early lineage development. Cell, 132(4), 527–531.
Murry, C. E., & Keller, G. (2008). Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell, 132(4), 661–680.
Chien, K. R., Moretti, A., & Laugwitz, K. L. (2004). Development. ES cells to the rescue. Science, 306(5694), 239–240.
Ruzinova, M. B., & Benezra, R. (2003). Id proteins in development, cell cycle and cancer. Trends in Cell Biology, 13(8), 410–418.
Jen, Y., Manova, K., & Benezra, R. (1996). Expression patterns of Id1, Id2, and Id3 are highly related but distinct from that of Id4 during mouse embryogenesis. Developmental Dynamics, 207(3), 235–252.
Jen, Y., Manova, K., & Benezra, R. (1997). Each member of the Id gene family exhibits a unique expression pattern in mouse gastrulation and neurogenesis. Developmental Dynamics, 208(1), 92–106.
Fraidenraich, D., Stillwell, E., Romero, E., et al. (2004). Rescue of cardiac defects in id knockout embryos by injection of embryonic stem cells. Science, 306(5694), 247–252.
Fraidenraich, D., & Benezra, R. (2006). Embryonic stem cells prevent developmental cardiac defects in mice. Nature Clinical Practice Cardiovascular Medicine, 3(Suppl 1), S14–S17.
Powell-Braxton, L., Hollingshead, P., Warburton, C., et al. (1993). IGF-I is required for normal embryonic growth in mice. Genes and Development, 7(12B), 2609–2617.
Powell-Braxton, L., Hollingshead, P., Giltinan, D., Pitts-Meek, S., & Stewart, T. (1993). Inactivation of the IGF-I gene in mice results in perinatal lethality. Annals of the New York Academy of Sciences, 692, 300–301.
Liu, J. P., Baker, J., Perkins, A. S., Robertson, E. J., & Efstratiadis, A. (1993). Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell, 75(1), 59–72.
Cadigan, K. M., & Nusse, R. (1997). Wnt signaling: a common theme in animal development. Genes and Development, 11(24), 3286–3305.
Oishi, I., Suzuki, H., Onishi, N., et al. (2003). The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells, 8(7), 645–654.
Schneider, V. A., & Mercola, M. (2001). Wnt antagonism initiates cardiogenesis in Xenopus laevis. Genes and Development, 15(3), 304–315.
Hoffman, E. P., Brown, R. H., Jr., & Kunkel, L. M. (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 51(6), 919–928.
Stedman, H. H., Sweeney, H. L., Shrager, J. B., et al. (1991). The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature, 352(6335), 536–539.
Durbeej, M., & Campbell, K. P. (2002). Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Current Opinion in Genetics and Development, 12(3), 349–361.
Stillwell, E., Vitale, J., Zhao, Q., et al. (2009). Blastocyst injection of wild type embryonic stem cells induces global corrections in mdx mice. PLoS ONE, 4(3), e4759.
Hardiman, O. (1994). Dystrophin deficiency, altered cell signalling and fibre hypertrophy. Neuromuscular Disorders, 4(4), 305–315.
Peault, B., Rudnicki, M., Torrente, Y., et al. (2007). Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Molecular Therapy, 15(5), 867–877.
Walsh, K. (2009). Adipokines, myokines and cardiovascular disease. Circulation Journal, 73(1), 13–18.
Low, H. P., Greco, B., Tanahashi, Y., et al. (2009). Embryonic stem cell rescue of tremor and ataxia in myelin-deficient shiverer mice. Journal of the Neurological Sciences, 276(1–2), 133–137.
Bergmann, O., Bhardwaj, R. D., Bernard, S., et al. (2009). Evidence for cardiomyocyte renewal in humans. Science, 324(5923), 98–102.
Terzic, A., Moore, R. L., & Waldman, S. A. (2007). Acquired and innate cardioprotection. Journal of Applied Physiology, 103(4), 1436–1437.
Yamada, S., Nelson, T. J., Behfar, A., Crespo-Diaz, R. J., Fraidenraich, D., & Terzic, A. (2009). Stem cell transplant into preimplantation embryo yields myocardial infarction-resistant adult phenotype. Stem Cells, 27(7), 1697–1705.
Nelson, T. J., Martinez-Fernandez, A., & Terzic, A. (2009). KCNJ11 knockout morula re-engineered by stem cell diploid aggregation. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 364(1514), 269–276.
Takahashi, K., Tanabe, K., Ohnuki, M., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131(5), 861–872.
Rodolfa, K. T., & Eggan, K. (2006). A transcriptional logic for nuclear reprogramming. Cell, 126(4), 652–655.
Yu, J., Vodyanik, M. A., Smuga-Otto, K., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science, 318(5858), 1917–1920.
Meissner, A., Wernig, M., & Jaenisch, R. (2007). Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nature Biotechnology, 25(10), 1177–1181.
Soldner, F., Hockemeyer, D., Beard, C., et al. (2009). Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell, 136(5), 964–977.
Park, I. H., Arora, N., Huo, H., et al. (2008). Disease-specific induced pluripotent stem cells. Cell, 134(5), 877–886.
Freund, C., & Mummery, C. L. (2009). Prospects for pluripotent stem cell-derived cardiomyocytes in cardiac cell therapy and as disease models. Journal of Cellular Biochemistry, 107(4), 592–599.
Acknowledgments
We thank Dr. Robert Benezra (Memorial Sloan-Kettering Cancer Center) for support with initial studies. This work was supported by the National Institutes of Health (D.F., A.T.), American Heart Association (D.F.), Muscular Dystrophy Association (D.F.), New Jersey Commission on Science and Technology (D.F.), UMDNJ Foundation (D.F.), American Society for Clinical Pharmacology and Therapeutics (A.T.), Marriott Heart Disease Research Program (A.T.), Marriott Foundation (A.T.), Ted Nash Long Life Foundation (A.T.), Ralph Wilson Medical Research Foundation (A.T.), and Mayo Clinic Clinician-Investigator Program (A.T.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Joel S. Schneider and Joseph M. Vitale contributed equally to this work
Rights and permissions
About this article
Cite this article
Schneider, J.S., Vitale, J.M., Terzic, A. et al. Blastocyst Injection of Embryonic Stem Cells: A Simple Approach to Unveil Mechanisms of Corrections in Mouse Models of Human Disease. Stem Cell Rev and Rep 5, 369–377 (2009). https://doi.org/10.1007/s12015-009-9089-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-009-9089-6