
Abstract
Modulation of nuclear factor KappaB (NF-κB) activation may play a role in regulating inflammatory conditions associated with coronary artery disease (CAD). MicroRNA-146a (miR-146a) primarily targets interleukin-1 receptor-associated kinase 1 (IRAK-1) and tumour necrosis factor receptor associated factor 6 (TRAF-6), which results in inhibition of NF-κB via the TLR pathway. This study investigated the influence of the miR-146a GC rs2910164 on miR-146a expression in young South African Indians with CAD. CAD patients and controls were genotyped by PCR–RFLP and miRNA-146a levels were measured by qPCR. IRAK-1, TRAF-6 and NF-κB expression was determined by Western blot. No differences in genotypic frequency was found (GG: 45 vs. 47 %, GC: 46 vs. 41 %, CC: 9 vs. 12 %) in controls and patients respectively (odds ratio = 1.025; 95 % confidence interval 0.6782–1.550; p = 0.9164). Significantly higher levels of miR-146a was associated with CAD patients with the CC genotype (6.25-fold increase relative to controls and patients with the wildtype variant, p < 0.0001). Significantly lower levels of IRAK-1 (0.38 ± 0.02; p = 0.0072) and TRAF-6 (0.44 ± 0.02; p = 0.0146) was found in CAD patients with the CC genotype. The lowest levels of NF-κB and C-reactive protein were found in patients with the homozygous C allele compared to the heterozygous GC and wildtype variants. We propose a role for miR-146a in TLR signalling through a negative feedback mechanism involving the attenuation of NF-κB by down-regulation of IRAK-1 and TRAF-6. Our observations implicate miR-146a as a target for lowering inflammation in CAD patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Micro-RNAs (miRs) are a class of small non-coding RNA that negatively regulate the expression of cognate target genes [1, 2]. MiRs attach to the 3′-untranslated region (3′-UTR) of their target messenger RNAs (mRNA) through their highly conserved seed sequence (2–7 nucleotides) [3, 4]. A single miR can potentially target hundreds of mRNA transcripts, and can thus modulate transcriptional programs that affect entire cellular networks [5, 6]. Genetic alterations in miR sequences affect precursor processivity, and thus maturation and expression of miRs. In addition, changes in the seed sequence alter binding kinetics between the mature miR and 3′-UTR target. It is conceivable that since miRs regulate magnitude of targets, small changes in genetic sequence will have large implications for transcriptional regulation and ultimately for the development of disease [7]. Single nucleotide polymorphisms (SNPs) have been well investigated in exons [8, 9], but little is known about their effect in miR and role in disease conditions. The miR-146a rs2910164 has been widely investigated in rheumatoid arthritis, tuberculosis and several cancers [1, 7, 10].
Single nucleotide polymorphisms are the most common type of genetic variation in the human genome and have been well investigated in protein coding genes, which affect the functioning of proteins and consequently render susceptibility to disease [8, 9]. There are limited studies focusing on the relationship between miR-146a expression and cardiovascular diseases [2, 11–13]. miR-146a is involved in regulating the innate immune, and inflammatory response pathways [14]. A common G/C SNP (rs2910164) located within the crucial seed sequence of miR-146a [15] causes a change from a G:U pair to a mismatched C:U in the stem region and was shown to affect the expression of mature miR-146a [1, 9]. Whether genetic variants of miR-146a confer differences in expression of miR-146a in CAD is unclear [1, 16, 17]. If such differences do occur, whether they are enough to produce significantly different regulatory effects on major mRNA targets remains unknown.
miR-146a primarily targets IRAK-1 and TRAF-6, which are involved in TLR and IL-1 receptor (IL-1R) signalling [18]. Upon stimulation of the TLR, the adaptor proteins myeloid differentiation primary-response gene 88 (MyD88) and TLR domain-containing adaptor-inducing interferon-β (TRIF) are recruited from the cell cytoplasm to the receptor, resulting in the activation of two independent branches of TLR signalling [19]. In the MyD88-dependent branch, MyD88 recruits IRAK-1, which in turn phosphorylates and activates TRAF-6. This chain of events leads to the phosphorylation and degradation of inhibitory kappa B (IκB), allowing NF-κB to enter the cell nucleus and activate the transcription of inflammatory genes [7, 19]. The effect of dysregulated miR-146a expression on its target molecules IRAK-1 and TRAF-6 and their downstream functions in the TLR signalling pathway remain unclear. It is likely that alterations in miR-146a expression caused by genetic variation in miR-146a, such as SNPs may affect TLR signalling.
CAD has been the leading cause of fatality worldwide since 1990, and this trend is expected to continue in the future [20, 21]. At present, developing countries such as South Africa and India contribute a large share to the global burden of CAD and are expected to account for 85 % by 2020 [20, 22]. Most South African Indians are descendants of indentured labourers who were brought from India between 1860 and 1911 [23]. Indians in India and South Africa show early onset of CAD, one to two decades earlier than other ethnic groups [24], indicating a genetic link. The highest death rates for CAD in South Africa occurs in Indians [25].
The present study aimed to investigate the influence of miR-146a rs2910164 on miR-146a expression in young South African Indian CAD patients. This is the first investigation of the miR-146a SNP in this population group.
Methods
Patient Recruitment and Sample Collection
A total of 106 young Indian male CAD patients and 100 age-, race- and sex-matched controls were enrolled following institutional ethical approval (BE154/010). A full pathology report of clinical markers was assessed by routine laboratory testing at Global Clinical and Viral Laboratory (Durban, South Africa)—a fully accredited South African National Laboratory. The following parameters were tested: Haematology (Roche Sysmex 1800XT), Chemistry (Beckman Coulter DXC600), Endocrinology and hsCRP (Siemens Centaur XP), Serology (BD Biosciences FACS Calibur), as per international standards to obtain levels of total cholesterol, HDL, LDL, triglycerides, fasting glucose, 2-h glucose, fasting insulin, glycosylated haemoglobin, sodium, potassium, bicarbonate, chloride, urea, creatinine, glomerular filtration rate, CD4 count, CD8 count, CD45 count and CD3 count. The physical measurements of weight, height, abdominal circumference, waist circumference and patient history were conducted by the cardiologist (Dr. S. Khan). The inclusion criteria for CAD patients were: Indian ancestry and unrelated, adults aged <45 years, and stable CAD confirmed at angiography. The exclusion criteria for controls included an acute coronary syndrome/revascularisation procedure in the preceding 3 months, chronic renal or liver disease, malignancy and known inflammatory or infectious disease.
DNA Extraction
Genomic DNA was extracted from the whole blood sample of each patient and control, according to the method described by Sambrook et al. [26]. Cells were transferred to 600 μl lysis buffer (0.5 % sodium dodecyl sulphate (SDS), 150 mM NaCl, 10 mM ethylenediaminetetra-acetic acid (EDTA), 10 mM Tris–HCl (pH 8.0)). To this, RNase A (100 μg/ml; DNase-free) was added to the solution and incubated (37 °C, 1 h). Proteinase K (200 μg/ml) was then added and incubated (3 h, 50 °C). Protein contaminants were then precipitated by adding 5 mM 0.1 % potassium acetate before centrifugation (5,000×g; 15 min). Supernatants containing genomic DNA were transferred to fresh tubes and extracted with 100 % isopropanol on ice, and thereafter washed with 70 % ethanol. DNA samples were dissolved in 10 mM Tris and 0.1 mM EDTA (pH 7.4, 4 °C). DNA concentration was determined using the Nanodrop2000 spectrophotometer, and all samples were standardised to a concentration of 10 ng/μl.
Genotyping
An optimised polymerase chain reaction (PCR) was used to obtain the highest specificity and yield of the 147 bp PCR product. This was achieved by amplification of the genomic DNA using 40 pmol of each primer (Forward Primer: 5′-CATGGGTTGTGTCAGTGTCAGAGCT-3′; Reverse Primer: 5′-TGCCTTCTGTCTCCAGTCTTCCAA-3′) [16]. A no-template sample was run with the positive samples as a quality control measure against PCR contamination.
The 30 μl reaction consisted of 200 μM of each dNTP, 2.5 mM MgCl2, 1× Green GoTaq Flexi buffer, 0.2U GoTaq DNA polymerase (Promega) and 30 ng genomic DNA template.
PCR was performed under the following cycling conditions: 94 °C for 10 min (initial denaturation), followed by 30 cycles of 94 °C for 30 s, 65 °C for 30 s (annealing) and 72 °C for 7 min (final extension).
PCR products were electrophoresed on agarose gel (1.8 %, 0.5 mg/ml ethidium bromide) and visualised using the Uvitech image documentation system (Uvitech Alliance 2.7).
PCR–RFLP was used to determine the miR-146a rs2910164 genotypes. 15 μl of each PCR product was subjected to restriction by 1.5 μl (10u/μl) Sac I and 2 μl 10× Buffer-Sac I (Fermentas). Overnight restriction occurred at 37 °C, and thereafter restriction products were electrophoresed on agarose gel (3 %, 0.5 mg/ml ethidium bromide) and visualised as was the PCR product.
Presence of the wild-type G-allele resulted in no cleavage of the PCR product. The variant C-allele yielded two fragments of 122 and 25 bp. The homozygous genotype yielded three bands of 147, 122 and 25 bp. Restriction products were run alongside a DNA ladder for accurate reading of fragment sizes, thus enabling correct analysis of genotypes.
RNA Extraction
Total RNA was isolated from peripheral blood mononuclear cells (PBMC) from each miR-146a rs2910164 genotype in CAD patients and controls. RNA was isolated with Trizol reagent following an in-house protocol. Briefly, Trizol (300 μl) was added to PBMCs and frozen for 1 h at −80 °C. Thereafter, chloroform (100 μl) was added and centrifuged at (12,000×g, 15 min, 4 °C). Isopropanol (250 μl) was added to the aqueous phase and once again subjected to freezing (1 h, −80 °C). This was followed by centrifuging (12,000×g, 20 min, 4 °C), and then washing with 500 μl cold ethanol (75 %). Finally, samples were centrifuged at 7,400×g for 15 min (4 °C). Ethanol was removed and RNA pellets were resuspended in 15 μl nuclease-free water.
RNA was quantified using the Nanodrop2000 spectrophotometer. The ratio of absorbance at 260 and 280 nm were used to assess the purity of RNA, and samples were run on 2 % agarose gel to check the integrity of the RNA. All RNA samples were standardised to a concentration of 100 ng/μl.
Total RNA was used to prepare cDNA using the miScript II RT kit (miScript Reverse Transcriptase Mix, 10× miScript Nucleics Mix, 5× miScript HiSpec Buffer; Qiagen, Catalog No. 218161), according to the manufacturer’s instructions.
Quantitative PCR
miR-146a expression was analysed using the miScript SYBR Green PCR kit (Qiagen, Catalog No. 218073), according to the manufacturer’s instructions. The miR-146a expression assay was run in triplicates. Human U6 was used as the internal control.
The comparative threshold cycle (C t) method was used to assess the relative changes in expression. 2−ΔΔCt represents the fold change in miR-146a expression between sample groups. A no-template control was run with the positive samples to assess the overall specificity of the reaction [27].
Western Blot Analysis
Cytobuster™ reagent (Novagen) supplemented with protease and phosphatase inhibitors (Roche, 05892791001 and 04906837001, respectively) were used for protein isolation. Cytobuster (200 μl) was added to PBMCs (4 °C, 10 min) and centrifuged (180×g; 4 °C, 10 min) to obtain a crude protein extract. Protein samples were quantified using the bicinchoninic acid assay and standardized to 0.5 mg/ml. Protein samples were prepared with laemlii buffer (0.5 M Tris (pH 6.8), glycerol, 10 % sodium dodecyl sulphide (SDS), 0.5 % bromophenol blue, β-mercaptoethanol) and denatured at 95 °C.
Samples (25 μl) were electrophoresed on 7.5 % SDS-polyacrylamide gel electrophoresis gels, and thereafter transferred to nitrocellulose membranes using the Trans-Blot® Turbo™ Transfer system (BIORAD). Membranes were blocked with 5 % bovine serum albumin (BSA) in Tris buffer saline (20 mM Tris–HCl (pH 7.4), 500 mM NaCl and 0.01 % Tween 20 (TBST)) for 1 h, and incubated with primary antibody (IRAK-1; Cell Signalling Technology 4769, TRAF-6; Cell Signalling Technology 8347, NF-κB; ab17742 and β-actin; ab8226, 1:1000) in 1 % BSA in TBST at 4 °C overnight. Membranes were then washed (TBST, 4 × 15 min) and treated with secondary antibody (Anti-rabbit IgG, HRP-linked; Cell Signalling Technology 4769, 1:2000) for 1 h at room temperature. Membranes were washed again (TBST, 4 × 15 min) and protein expression was detected by the LumiGLO® chemiluminescent substrate system (KPL) with the Alliance 2.7 Image Documentation System (UViTech). Protein bands were standardised against β-actin, and analysed with the Advanced Image Analysis software (UViTech v12.14). The results were expressed as mean relative band intensity (RBI).
Statistical Analysis
The Hardy–Weinberg equilibrium was used to test for deviation of allele/genotype frequency. All other statistical analysis was performed with Graphpad prism software (version 5.0). Allele and genotype frequencies were calculated using the Fisher’s exact and Chi square tests, respectively. The comparison of miR-146a expression between CAD patients and controls were done by a nonparametric t test. All other calculations were done by performing a one-way ANOVA.
Results are expressed as mean ± standard error. A p value less than 0.05 was considered statistically significant.
Results
Amongst the clinical parameters assessed between the groups, body mass index (BMI), hypertension, diabetes, triglycerides, fasting glucose and HbA1c was higher in patients than controls (Table 1, published data).
The genotype distribution complied with the Hardy–Weinberg equilibrium in patients and controls (Chi square p = 0.7356 and p = 0.8501, respectively). No significant difference was observed in distribution of the miR-146a genotypes between the patients and controls (p = 0.9164, OR = 1.025, 95 % CI 0.6782–1.550; Table 1). The rare C allele was more frequent in CAD patients (69) than controls (64).
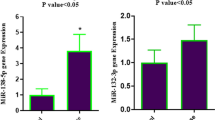
Relative to the controls, CAD patients exhibited a 2.58 fold increase in miRNA-146a expression (p = 0.3939) (Fig. 1). CAD patients with the miR-146a rs2910164 CC genotype expressed significantly higher levels of miR-146a (6.25-fold increase compared to controls and CAD patients with the reference GG genotype) *** p < 0.0001 (Fig. 2).

Fold change in miR-146a expression levels in CAD patients relative to controls

miR-146a expression in CAD patients and controls with different genotypes *** p < 0.0001
Patients with the CC genotype expressed significantly lower levels of IRAK-1 (0.38 ± 0.02; p = 0.0072; compared to GC, Fig. 3) and TRAF-6 (0.44 ± 0.02; p = 0.0146; compared to GC and GG, Fig. 4). These patients also expressed the lowest levels of NF-κB compared to the other genotypes, although this did not reach statistical significance (7.18 ± 0.32; p = 0.4843, Fig. 5).

IRAK-1 expression in CAD patients (** p = 0.0072) and controls

TRAF-6 expression in CAD patients (* p = 0.0146) and controls (# p = 0.0247)

NF-κB expression in CAD patients and controls
CAD patients and controls with the CC genotype presented with the lowest mean hsCRP levels compared to the other genotypes (6.53 ± 1.47 and 4.23 ± 1.17 mg/l, respectively). However this did not reach statistical significance (p = 0.7989 for CAD patients and p = 0.9683 for controls, Tables 2 and 3; Fig. 6).

The expression of IRAK-1, TRAF-6 and NF-κB in a CAD patients compared to b Controls
Discussion
The present study investigated the possible association between the miR-146a rs2910164 and miR-146a expression in young South African Indian CAD patients.
CAD patients with the miR-146a rs2910164 CC genotype expressed significantly higher levels of miR-146a (6.25-fold increase compared to controls and CAD patients with the reference GG genotype; p < 0.0001) and significantly lower levels of IRAK-1 (0.38 ± 0.04; p = 0.0072; compared to GC) and TRAF-6 (0.44 ± 0.05; p = 0.0146; compared to GC and GG). Furthermore, these patients also expressed the lowest levels of NF-κB and hsCRP compared to the other genotypes, although this did not reach statistical significance. We propose a possible mechanism for the observed phenomenon.
IRAK-1 and TRAF-6 are both involved in the MyD88-dependent branch of the TLR signalling pathway [19, 28]. MyD88 is important for the activation of inflammatory cytokines triggered by all TLRs. Upon stimulation, MyD88 recruits IRAK-1 to the receptor. IRAK-1 is phosphorylated and complexed with TRAF-6, and together they dissociate from the receptor. A series of further interactions lead to phosphorylation of IκB, thereby releasing the transcription factor NF-κB into the nucleus and allowing the induction of inflammatory genes [28].
IRAK-1 and TRAF-6 are known targets of miR-146a [18]. The promoter region of miR-146a contains several binding sites for the transcription factor NF-κB, and the induced expression of miR-146a occurs in a NF-κB-dependent manner [7].
miR-146a inhibits IRAK-1 and TRAF-6 expression through negative feedback regulation (Fig. 7) [7, 29]. Therefore, higher levels of miR-146a levels lead to increased inhibition of IRAK-1 and TRAF-6, causing decreased levels of pro-inflammatory cytokines such as IL-1, IL-6, IL-8, tumour necrosis factor-α (TNF-α) [29]. These cytokines are known regulators of CRP [30], and a decrease in their levels consequently reduce CRP levels.

The role of miRNA-146a in the inflammatory response through a negative feedback mechanism
Decreased hsCRP levels observed in CAD patients with the CC genotype may be as a result of them expressing significantly higher levels of miR-146a, increased binding to and inhibition of IRAK-1 and TRAF-6.
In contrast to our study, a previous investigation of the miR-146a rs2910164 in hepatocellular carcinoma by Xu et al. [9] observed an increased production of miR-146a associated with the GG genotype. In 2008, Shen et al. observed an increase in miR-146a expression in breast/ovarian cancer patients with the variant C allele [31]. However, neither of these studies investigated the target molecules of miR-146a and downstream effects of dysregulated miR-146a. This is the first study to investigate the miR-146a rs2910164 and its influence on miR-146a and miR-146a target molecule expression in young South African Indian CAD patients.
Since high levels of pro-inflammatory cytokines and CRP contribute to the chronic inflammation seen in CAD, the CC genotype serves a protective function by increasing miR-146a levels and reducing inflammation. Our study suggests miR-146a as an important modulator of the inflammatory response. Based on these findings, miR-146a could serve as a potential target in therapeutic interventions towards lowering inflammation and alleviating the complications in CAD.
Study limitations include sample size, lack of exclusion criteria on BMI, hypertension, diabetes and effects of medication. The involvement of additional SNPs and/or regulatory molecules which are yet to be discovered may be a confounding factor in our study. A follow up study with a larger patient population would provide further clarification.
References
Hung, P. S., Chang, K. W., Kao, S. Y., Chu, T. H., Liu, C. J., & Lin, S. C. (2012). Association between the rs2910164 polymorphism in pre-mir-146a and oral carcinoma progression. Oral Oncology, 48, 404–408.
Takahashi, Y., Satoh, M., Minami, Y., Tabuchi, T., Itoh, T., & Nakamura, M. (2010). Expression of miR-146a/b is associated with the Toll-like receptor 4 signal in coronary artery disease: Effect of renin-angiotensin system blockade and statins on miRNA-146a/b and Toll-like receptor 4 levels. Clinical Science, 119, 395–405.
Zorc, M., Jevsinek Skok, D., Godnic, I., Calin, G. A., Horvat, S., Jiang, Z., et al. (2012). Catalog of microRNA seed polymorphisms in vertebrates. PLoS ONE, 7(1), e30737.
Ellwanger, D. C., Buttner, F. A., Mewes, H. W., & Stumpflen, V. (2011). The sufficient minimal set of miRNA seed types. Bioinformatics, 27(10), 1346–1350.
Hu, Z., Liang, J., Wang, Z., Tian, T., Zhou, X., Chen, J., et al. (2008). Common genetic variants in pre-microRNAs were associated with increased risk of breast cancer in Chinese women. Human Mutation, 30(1), 79–84.
Mittal, R. D., Gangwar, R., George, G. P., Mittal, T., & Kapoor, R. (2011). Investigative role of pre-microRNAs in bladder cancer patients: A case-control study in North India. DNA and Cell Biology, 30(6), 401–406.
Li, L., Chen, X., & Li, Y. (2010). MicroRNA-146a and human disease. Scandinavian Journal of Immunology, 71, 227–231.
Yang, B., Chen, J., Li, Y., Zhang, J., Li, D., Huang, Z., et al. (2012). Association of polymorphisms in pre-miRNA with inflammatory biomarkers in rheumatoid arthritis in the Chinese Han population. Human Immunology, 73, 101–106.
Xu, T., Zhu, Y., Wei, Q., Yuan, Y., Zhou, F., Ge, Y., et al. (2008). A functional polymorphism in the miR-146a gene is associated with the risk for hepatocellular carcinoma. Carcinogenesis, 29(11), 2126–2131.
Li, D., Wang, T., Song, X., Qucuo, M., Yang, B., Zhang, J., et al. (2011). Genetic study of two single nucleotide polymorphisms within corresponding microRNAs and susceptibility to tuberculosis in a Chinese Tibetan and Han population. Human Immunology, 72, 598–602.
Raitoharju, E., Lyytikainen, L. P., Levula, M., Oksala, N., Mennander, A., Tarkka, M., et al. (2011). miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis, 219, 211–217.
Xu, J., Zhao, J., Evan, G., Xiao, C., Cheng, Y., & Xiao, J. (2012). Circulating microRNAs: Novel biomarkers for cardiovascular diseases. Journal of Molecular Medicine, 90, 865–875.
Jamaluddin, M. S., Weakly, S. M., Zhang, L., Kougias, P., Lin, P. H., Yao, Q., et al. (2011). miRNAs: Roles and clinical applications in vascular disease. Expert Review of Molecular Diagnostics, 11(1), 79–89.
Zilahi, E., Tarr, T., Papp, G., Griger, Z., Sipka, S., & Zeher, M. (2011). Increased microRNA-146a/b, TRAF6 gene and decreased IRAK1 gene expressions in the peripheral mononuclear cells of patients with Sjögren’s syndrome. Immunology Letters, 141, 165–168.
Jazdzewski, K., Liyanarachchi, S., Swierniak, M., Pachucki, J., Ringel, M. D., Jarzab, B., et al. (2009). Polymorphic mature microRNAs from passenger strand of pre-miR-146a contribute to thyroid cancer. Proceedings of the National Academy of Sciences, 106(5), 1502–1505.
Chatzikyriakidou, A., Voulgari, P. V., Georgiou, I., & Drosos, A. A. (2010). A polymorphism in the 3′-UTR of interleukin-1 receptor-associated kinase (IRAK1), a target gene of miR-146a, is associated with rheumatoid arthritis susceptibility. Joint Bone Spine, 77(5), 411–413.
Yue, C., Wang, M., Ding, B., Wang, W., Fu, S., Zhou, D., et al. (2011). Polymorphism of the pre-miR-146a is associated with risk of cervical cancer in a Chinese population. Gynecologic Oncology, 122, 33–37.
Curtale, G., Citarella, F., Carissimi, C., Goldoni, M., Carucci, N., Fulci, V., et al. (2010). An emerging player in the adaptive immune response: MicroRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood, 115(2), 265–273.
Taganov, K. D., Boldin, M. P., Chang, K. J., & Baltimore, D. (2006). NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proceedings of the National Academy of Sciences, 103(33), 12481–12486.
Mohan, V., Venkatraman, J. V., & Pradeepa, R. (2010). Epidemiology of cardiovascular disease in type 2 diabetes: The Indian scenario. Journal of Diabetes Science and Technology, 4(1), 158–170.
Murray, C. J., & Lopez, A. D. (1997). Alternative projections of mortality and disability by cause 1990–2020: Global burden of disease study. The Lancet, 349, 1498–1504.
Reddy, K. S., & Yusuf, S. (1998). Emerging epidemic of cardiovascular disease in developing countries. Circulation, 97(6), 596–601.
Seedat, Y. K. (2005). Review: Diabetes mellitus in South African Indians. The British Journal of Diabetes & Vascular Disease, 5(5), 249–251.
Sharma, M., & Ganguly, N. (2005). Premature coronary artery disease in Indians and its associated risk factors. Vascular Health and Risk Management, 1(3), 217–225.
Norman, R., Bradshaw, D., Schneider, M., Pieterse, D., & Groenwald, P. (2006). Revised burden of disease estimates for the comparative risk factor assessment, South Africa 2000. Cape Town: Medical Research Council.
Sambrook, J., & Russell, D. W. (2001). Rapid isolation of mammalian DNA in molecular cloning: A laboratory manual (3rd ed.). New York: Cold Spring Harbor Laboratory Press.
Livak, K. J., & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods, 25(4), 402–408.
Takeda, K., & Akira, S. (2004). TLR signalling pathways. Seminars in Immunology, 16, 3–9.
Pauley, K. M., Satoh, M., Chan, A. L., Bubb, M. R., Reeves, W. H., & Chan, E. K. (2008). Upregulated miR-146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Research & Therapy, 10(4), 101.
Hirschfield, G. M., & Pepys, M. B. (2003). C-reactive protein and cardiovascular disease: New insights from an old molecule. QJM, 96(11), 793–807.
Shen, J., Ambrosone, C. B., DiCioccio, R., Odunsi, K., Lele, S. B., & Zhao, H. (2008). A functional polymorphism in the miR-146a gene and age of familial breast/ovarian cancer diagnosis. Carcinogenesis, 29(10), 1963–1966.
Acknowledgments
This work was supported by the National Research Foundation and College of Health Science Scholarships.
Conflict of interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ramkaran, P., Khan, S., Phulukdaree, A. et al. miR-146a Polymorphism Influences Levels of miR-146a, IRAK-1, and TRAF-6 in Young Patients with Coronary Artery Disease. Cell Biochem Biophys 68, 259–266 (2014). https://doi.org/10.1007/s12013-013-9704-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-013-9704-7