
Abstract
Vitamin C (ascorbate) plays important neuroprotective and neuromodulatory roles in the mammalian brain. Astrocytes are crucially involved in brain ascorbate homeostasis and may assist in regenerating extracellular ascorbate from its oxidised forms. Ascorbate accumulated by astrocytes can be released rapidly by a process that is stimulated by the excitatory amino acid, l-glutamate. This process is thought to be neuroprotective against excitotoxicity. Although of potential clinical interest, the mechanism of this stimulated ascorbate-release remains unknown. Here, we report that primary cultures of mouse and rat astrocytes release ascorbate following initial uptake of dehydroascorbate and accumulation of intracellular ascorbate. Ascorbate-release was not due to cellular lysis, as assessed by cellular release of the cytosolic enzyme lactate dehydrogenase, and was stimulated by l-glutamate and l-aspartate, but not the non-excitatory amino acid l-glutamine. This stimulation was due to glutamate-induced cellular swelling, as it was both attenuated by hypertonic and emulated by hypotonic media. Glutamate-stimulated ascorbate-release was also sensitive to inhibitors of volume-sensitive anion channels, suggesting that the latter may provide the conduit for ascorbate efflux. Glutamate-stimulated ascorbate-release was not recapitulated by selective agonists of either ionotropic or group I metabotropic glutamate receptors, but was completely blocked by either of two compounds, TFB-TBOA and UCPH-101, which non-selectively and selectively inhibit the glial Na+-dependent excitatory amino acid transporter, GLAST, respectively. These results suggest that an impairment of astrocytic ascorbate-release may exacerbate neuronal dysfunction in neurodegenerative disorders and acute brain injury in which excitotoxicity and/or GLAST deregulation have been implicated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vitamin C is essential for normal central nervous system (CNS) function in mammals [1, 2]. Importantly, the significance of the vitamin is often overlooked as it is rapidly lost when cells are put into culture [3–5]. Together with the adrenal cortex, pituitary gland, thymus, retina and corpus luteum, the brain contains high concentrations of ascorbate relative to other body tissues [1, 6–8]. Mounting evidence suggests that the reduced form of the vitamin, ascorbate, plays an important neuromodulatory [1, 6, 9] and neuroprotective [10] role during glutamatergic and dopaminergic neurotransmission. Vitamin C in the CNS originates from the blood supply, even in species that are capable of synthesising their own ascorbate, with the majority entering via the sodium-dependent vitamin C transporter isoform 2 (SVCT2) at the choroid plexus [2]. In rats, ascorbate exists in the cerebrospinal fluid at concentrations of 200–400 μM, although in humans the concentration is slightly lower. Ascorbate is actively transported into neurons via SVCT2 [11] where it can accumulate to concentrations of up to 10 mM [12]. As a result of oxidative processes occurring during both normal and disturbed neuronal activity, ascorbate becomes oxidised to dehydroascorbate (DHA) and must be rapidly recycled to avoid an irrevocable loss of the vitamin. However, as DHA is neurotoxic [13], but generally not gliotoxic [14], much of this ascorbate recycling is thought to occur within vicinal astrocytes [15–18], which contain ascorbate concentrations of ~1 mM [12]. Astrocytes are at least as numerous as neurones in the brain [19] and are extremely efficient at importing DHA in a facilitative glucose transporter (GLUT)-dependent manner that probably involves the glial-specific isoform of GLUT1 [7, 15, 17, 20]. DHA that is released by neurones in a possibly GLUT3-dependent manner [7], or is formed by the oxidation of extracellular ascorbate, is thought to be rapidly imported by astrocytes, reduced back to ascorbate, and then be released into the extracellular space; a process that probably supports further neuronal uptake of ascorbate [9, 15, 16, 18]. As a complete turnover of ascorbate, resulting from entry at the choroid plexus, probably occurs over a period of several hours [6], the localised recycling of ascorbate by astrocytes may be vital for its short-term maintenance within the CNS.
l-Glutamate is the major excitatory neurotransmitter in the mammalian CNS. Its release into the synaptic cleft peaks at concentrations of 1–5 mM during neurotransmission [21] and is required to activate ionotropic glutamate receptors (iGluRs), as well as metabotropic glutamate receptors (mGluRs), in the post-synaptic neuron [22]. However, in order to maintain effective signal transduction and avoid the onset of glutamate-induced excitotoxicity, synaptic glutamate must be removed extremely rapidly. Synapses are typically ensheathed by the processes of perisynaptic astrocytes [23]. Under normal circumstances, the removal of synaptic glutamate occurs by the action of sodium-dependent glutamate transporters that are expressed in these astrocytes [24, 25]. Although astrocytes appear to be capable of expressing all five of the known excitatory amino acid transporter (EAAT) isoforms (i.e. termed EAATs 1–5 in humans) [26], the rodent glutamate aspartate transporter (GLAST; orthologue of human EAAT1) and the rodent glutamate transporter 1 (GLT-1; orthologue of human EAAT2) are thought to be the predominantly expressed glutamate transporters in astrocytes [23]. GLAST is the major astrocytic EAAT expressed during CNS development and in the Bergmann glia of the adult brain, while GLT-1 is expressed later in development and primarily in astrocytes localised to the hippocampus, cerebral cortex and striatum [27]. GLAST and/or GLT-1 become reduced in expression and activity in a range of neurodegenerative disorders [24, 25, 28] and during several forms of acute brain injury, and thus may contribute to the excitotoxic aetiology of these disorders (for a review see [29]).
Exposure of both astrocytes [4, 30] and neurones [31] to glutamate is known to trigger the release of ascorbate into the extracellular space, where the ascorbate may help protect neurones against glutamate-induced neuronal dysfunction [10]. The mechanism of glutamate-induced ascorbate-release from astrocytes is unknown, but may involve cell swelling and activation of volume-sensitive osmolyte and anion channels [VSOACs; also known as volume-regulated anion channels (VRACs)] that are permeable to ascorbate [4]. The molecular identities of the plasma membrane conduits involved in VSOAC formation remain to be identified [18, 32].
This study was undertaken to further investigate the mechanism of glutamate-induced release of ascorbate from astrocytes, and to determine the identity of the glutamate receptor/transporter presumably responsible for its mediation. It is shown that the release of ascorbate from primary cultures of rodent astrocytes is stimulated by exposure to submillimolar concentrations of the excitatory amino acids glutamate and aspartate, but not the non-excitatory amino acid glutamine. We extend the previous suggestion [4] that this stimulation may depend on the induction of cell swelling and the consequent activation of VSOACs. Finally, using a pharmacologic approach, we demonstrate for the first time that while the effects of glutamate are not due to the isolated activation of iGluRs, or mGluRs, which can be expressed in astrocytes, GLAST activity appears to be required, at least under the present experimental conditions.
Experimental
Unless otherwise stated, all chemicals were obtained from Sigma-Aldrich (Castle Hill, NSW, Australia) or Merck (Kilsyth, VIC, Australia). The pan-EAAT-specific glutamate inhibitor (2S,3S)-3-[3-[4-(trifluoromethyl)benzoylamino]benzyloxy]aspartate (TFB-TBOA) [33] and the GLT-1-specific inhibitor, dihydrokainic acid (DHK), were purchased from Tocris Bioscience (Ellisville, MO, USA). The novel GLAST-specific inhibitor, “UCPH-101” [25, 34] (originally referred to as “compound 1o” [35]), was obtained as a kind gift from Drs Anders A. Jensen and Lennart Bunch (University of Copenhagen). TFB-TBOA and UCPH-101 were solubilised in dimethyl sulfoxide prior to use, whereas R(+) indanyloxyacetic acid 94 (IAA-94), niflumic acid (NFA) and 1,9 dideoxyforskolin (DDF) required solubilisation in absolute ethanol. N-methyl-d-aspartate (NMDA), (±) α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA), the group 1 mGluR-specific agonist, (S)-3,5-dihydroxyphenylglycine (DHPG) and DHK were solubilised in deionised water.
Spectrophotometric microplate assays were performed on a Benchmark™ Plus microplate spectrophotometer (BioRad, Regents Park Industrial Estate, NSW, Australia) using Nunc 96-well flat-bottomed transparent plates, or Greiner 24-well plates. Orbital mixing of cell suspensions was performed with a MixMate® orbital mixer (Eppendorf South Pacific, North Ryde, NSW, Australia) maintained at 37 °C in a thermoregulated incubator or in a thermoregulated microplate incubator/shaker (Foinoe, Nanjing, Jiangsu, China) set to 37 °C.
Cell Culture
Astrocyte-rich primary cultures were prepared from the brains of newborn Wistar rats and C57BL/6J mice (<24 h old), as previously described [36]. As there may be significant species differences between rat and mouse astrocytes [37], both cultures were employed in some experiments to demonstrate a lack of species specificity in the responses observed. Viable cells were seeded at 3 × 105 cells/well in 24-well culture plates (Greiner Bio-One) and incubated in 90 % (v/v) DMEM (Gibco Invitrogen), 10 % (v/v) foetal bovine serum (Gibco Invitrogen), 20 U/ml penicillin G and 20 μg/ml streptomycin sulphate. Cultures were then maintained at 37 °C in a humidified atmosphere containing 10 % CO2, 90 % air, with the culture medium replaced every seventh day. Cells were used for experiments after 15–21 days of growth in vitro. Immunocytochemical staining using astrocyte markers revealed that ≥90 % of cells were positive for glial fibrillary acidic protein, while >95 % of the cells were positive for vimentin (data not shown). Immediately prior to use, wells in each plate were washed three times with 1.5–2.0 ml of 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES)-buffered saline (HBS; 134 mM NaCl, 5.2 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4 and 20 mM HEPES-Na+, pH 7.2 at 37 °C) that had been pre-warmed to 37 °C .
Ascorbate-Loading of Cells
Ascorbate-loading of cells was typically accomplished by exposing cells to freshly prepared solutions of 200 μM l-DHA dimer for 30 min at 37 °C. Upon hydrolysis of the crystalline l-DHA dimer in aqueous solution, 200 μM DHA dimer should yield 400 μM l-DHA monomer [38]. In order to avoid potential competitive inhibition of GLUT-dependent DHA uptake by d-glucose [39], exposure of astrocytes to DHA was performed in the absence of glucose as described previously [20, 36]. Following DHA-exposure, the extracellular medium was thoroughly removed by aspiration of the supernatant, followed by three successive washes with pre-warmed (37 °C) HBS containing 5 mM d-glucose. This relatively short incubation was well tolerated by the cells as indicated by the absence of a change in LDH release or trypan blue uptake (which were typically <2–3 %) by cultures treated in this way relative to controls (data not shown).
Determination of Intracellular Ascorbate
Intracellular ascorbate levels were determined by either of two procedures. The first was performed exactly as described previously [5], and involves the extraction of ascorbate from cells with 0.1 % saponin in HBS for 30 min/4 °C, followed by the determination of the ascorbate oxidase (AO)-sensitive reduction of ferricyanide to ferrocyanide. The second is a modification of the previous procedure in which, instead of the addition of ferricyanide, ascorbate-containing saponin extracts are combined with an equivalent volume of a freshly made solution containing 25 % (v/v) acetic acid, 1.65 mM FeCl3 and 2.4 mM Ferene-S, resulting in the production of 2 mol Fe2+ per mol ascorbate, with Fe2+ levels detected as their Ferene-S chelate (λ max = 595 nm; ε 595 nm = 35.5 mM−1 cm−1 [40]). In both the methods, ascorbate levels were interpolated from ascorbate standard curves constructed in parallel for each determination. The ascorbate concentrations of the standards were always determined spectrophotometrically (λ max = 265 nm; ε 265 nm = 14.5 mM−1 cm−1). Both the methods provided identical results.
Determination of Ascorbate-Release
Rodent astrocytes typically express SVCT2 under standard culture conditions, but not in vivo [41]. As recently demonstrated for brain capillary endothelial cells in culture [42], the culture-dependent expression of SVCT2 in astrocytes may be related to chronic ascorbate-deficiency under typical culture conditions. The expression of SVCT2 by cells can result in the uptake of a significant proportion of ascorbate that is released by cells under in vitro conditions and can thus confound attempts to measure ascorbate efflux [43]. To circumvent this potential problem, we assessed the release of ascorbate from astrocytes according to a novel modification of a previously described method [5]. Briefly, ascorbate efflux from astrocytes was determined as the AO-sensitive reduction of extracellular ferric citrate to ferrous iron in the presence of the membrane-impermeant Fe(II) chelator, ferene-S. In this way, ascorbate that is released from cells directly reduces extracellular ferric citrate to form ferrous ions that are then rapidly chelated to form an extracellular Fe(II)–ferene-S complex that can be colourimetrically quantified [5].
In order to determine total ascorbate efflux, astrocytes cultured in 24-well plates were washed and overlayed with HBS containing 5 mM d-glucose that additionally contained 200 μM Ferene-S with or without 10 U/ml of AO. Ascorbate-release assays were then initiated by the subsequent addition of a final concentration of 10 μM ferric citrate (1:5 molar ratio), followed immediately by the addition of excitatory (i.e. l-Glu and l-Asp) or non-excitatory (i.e. l-Gln) amino acids to the indicated final concentrations. In experiments with pharmacologic inhibitors of glutamate receptors/transporters or VSOACs, the inhibitors were added to the medium overlying the cells 15 min prior to addition of excitatory amino acids or control medium. This incubation time for inhibitors was chosen: (i) to minimise potential cytotoxicity to cells; (ii) as complete inhibition of glutamate-stimulated ascorbate-release was typically observed; and (iii) as no further inhibition was observed using longer incubation times (e.g. 30 min).
In experiments in which the osmolarity of the medium was changed, the overlying medium was changed immediately prior to addition of Ferene-S with a medium containing the stated osmolarity or iso-osmotic ion replacements. Plates were incubated with gentle orbital agitation for 60 min at 37 °C in the dark. Assays were terminated by the aspiration of the supernatant from each well. Levels of Fe(II)–Ferene-S were then determined according to standard curves constructed with authentic ascorbate for each experiment. Rates of ascorbate efflux-dependent ferric reduction were determined on the basis of the difference between the AO-free and AO-containing wells. This method allows for a highly sensitive determination of ascorbate-release that is additionally not confounded by ascorbate reuptake by astrocytic SVCTs. It is also worth noting here that low micromolar concentrations of cytochalasin B (<10 μM) did not inhibit the apparent rate of ascorbate-release (data not shown). This indicates that the determined ascorbate-release rates were not confounded by reuptake of the low levels (<5 μM) of extracellular DHA that would have been formed upon the reduction of ferric citrate by released ascorbate.
Lactate Dehydrogenase Release
Lactate dehydrogenase (LDH) activity that had been released from astrocyte cultures over the assay period was determined kinetically in 96-well plates according to a modification of a previously described method [44]. Assays were conducted in a final volume of 300 μl using 10 μl of astrocyte supernatant buffered to pH 8.2 at 25 °C with 0.2 M Tris–HCl. LDH that had been released from astrocytes cultures over 60 min in response to various treatments (e.g. exposure to glutamate) was used to oxidise added l-lactate (9.1 mM) to l-pyruvate in the presence of NAD+ (0.5 mM). The NADH formed from this reaction was then detected by measuring the NADH-dependent reduction of 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt (purchased as ‘Cell Proliferation Reagent’ from Boehringer Mannheim) to its water-soluble formazan (λ max = 440 nm) [45]. The production of the formazan was followed spectrophotometrically at 440 nm for periods of 8 min at 25 °C. The rates of formazan reduction were always corrected for the rates of reduction achieved in the absence of exogenously added lactate. The percentage of LDH released into the medium was typically calculated for three separate incubations (mean ± SD) by the following equation: (LDH activity in medium/total LDH activity in medium after cell permeabilisation with 0.1 % saponin for 30 min/4 °C) × 100. Treatment of monolayer cultures with 0.1 % saponin under these conditions causes 100 % cellular lysis [46].
Protein Determination
The protein content of astrocyte cultures was obtained by solubilising cellular protein with 2 % (w/v) sodium dodecyl sulphate and assessing protein concentration using the microplate-adaptation of the bicinchoninic acid assay kit for protein determination (Sigma-Aldrich), according to the manufacturer’s instructions. Bovine serum albumin (fraction V) was employed as a standard.
Statistical Analysis and Curve Fitting
All curve fitting and hypothesis testing were performed in GraphPad Prism® 5.0 (GraphPad Software, San Diego, CA, USA). Dose–response data were modelled by single rectangular hyperbolae in GraphPad Prism® 5.0. Differences between treatments were analysed using either one- or two-way ANOVA with either Bonferroni’s or Dunnett’s post hoc tests of significance using GraphPad Prism® 5.0. Results are typically shown as mean ± SD. Significance levels are given in the figure legends.
Results
The Release of Ascorbate by Astrocytes is Enhanced by l-Glutamate and l-Aspartate, but not l-Glutamine
It has previously been observed that ascorbate-loaded primary astrocyte cultures rapidly release a large fraction of their intracellular ascorbate after exposure to l-glutamate [4, 30]. In these studies, astrocytes were loaded with ascorbate by exposure of cells to either ascorbate [4] or DHA [30]. Although astrocytes typically express SVCTs in culture, there is evidence that these cells do not normally express SVCTs in situ [41]. Therefore, the majority of intra-astrocytic ascorbate in situ probably arises from the uptake and reduction of DHA that is generated extracellularly and/or that is released from neurones [16, 17]. Therefore, to more closely represent this system, astrocyte cultures were pre-loaded with ascorbate by pre-exposure to DHA. Consistent with previous observations [4, 20], mouse and rat astrocytes cultured in the absence of added vitamin C were devoid of intracellular ascorbate (data not shown). The pre-exposure of these astrocytes to 200 μM l-DHA dimer over 30 min resulted in intracellular ascorbate concentrations of 37 ± 5 nmol/mg protein (n = 6; mouse astrocytes) and 35 ± 4 nmol/mg protein (n = 6; rat astrocytes).
Following pre-exposure to DHA, ascorbate-loaded rat and mouse astrocytes demonstrate significant rates of ascorbate-release (Fig. 1a–c). Dose–response analyses indicate that the exposure of ascorbate-loaded mouse (Fig. 1b) and rat (Fig. 1c) astrocytes to synaptically relevant concentrations of glutamate (i.e. 0–1 mM) stimulates the release of ascorbate by these astrocytes. It should be noted that the values shown in Fig. 1b, c have been corrected for basal ascorbate-release rates. The apparent EC50 values for the glutamate-stimulated release of ascorbate are 54 ± 18 μM for mouse astrocytes and 32 ± 20 μM for rat astrocytes. Additionally, the hyperbolic dependence of ascorbate efflux on glutamate concentration cannot be attributed to the exhaustion of intracellular ascorbate as only 47 ± 3.1 % of intracellular ascorbate was released at 1 mM glutamate (Table 1).

Ascorbate-release from astrocytes is stimulated by l-glutamate and l-aspartate, but not l-glutamine. All ascorbate (AA) release experiments were conducted over 1 h. a The release of ascorbate from primary cultures of ascorbate-loaded mouse astrocytes is significantly stimulated by 1 mM glutamate. b, c Dose–response curves for the stimulation of ascorbate-release by glutamate from mouse (b) and rat (c) ascorbate-loaded primary astrocyte cultures. d Dose–response curves for the stimulation of ascorbate-release by aspartate (closed circles) and glutamine (open circles) from primary cultures of ascorbate-loaded mouse astrocytes. Please note the different labellings of the y-axes in (b–d). In panel d, the data have been normalised to the basal rate of ascorbate-release in the absence of amino acids. Data shown are means (±SD) of three experiments. P < 0.001 versus ‘basal’ condition
In support of the notion that the stimulatory effect of glutamate on the release of ascorbate by astrocytes is specific for excitatory amino acids, a similar stimulation was observed with the excitatory amino acid l-aspartate (Fig. 1d, closed circles), but not the structurally similar, non-excitatory amino acid l-glutamine (Fig. 1d, open circles). It is worth noting here that l-glutamine is typically formed intracellularly by astrocytes as a result of l-glutamate uptake and the action of glial glutamine synthetase, and is then released into the extracellular space for subsequent neuronal uptake [22].
Overall these results support previous observations [4, 30] that ascorbate-loaded primary astrocyte cultures are capable of releasing ascorbate when stimulated by glutamate, and further suggest that this response is specific for excitatory, but not for non-excitatory amino acids.
The Release of Ascorbate by Astrocytes is not due to Cellular Lysis
Although glutamate is generally considered to be non-gliotoxic over short periods (<1 h), the sustained exposure (>4 h) of cultured astrocytes to elevated glutamate concentrations may be gliotoxic [40]. To determine the effect of glutamate on the cellular integrity of astrocytes under conditions used in this study, the following experiments were conducted. The cellular release of the cytosolic enzyme LDH is a verified indicator of cellular damage in cultured astrocytes [47, 48]. The levels of LDH released from the cytosol into the extracellular medium by astrocytes were assessed (both in the presence and in the absence of glutamate) as a percentage of the total LDH activity of permeabilised astrocytes. These results were then compared to the percentage of intracellular ascorbate that was released by these cells under the same conditions. From the data presented in Table 1, it is apparent that the low basal release of LDH (1–2 %) by cultured astrocytes remains unchanged upon exposure to 1 mM glutamate for 60 min, whereas the levels of ascorbate released are significantly higher both before (~34 %, P < 0.001) and after (~47 %, P < 0.001) glutamate exposure. Moreover, the levels of glutamate-stimulated release of ascorbate are significantly higher (P < 0.001) than the basal rate of release. These results strongly suggest that both the basal and the glutamate-stimulated releases of ascorbate by cultured astrocytes are not due to a loss of integrity of the plasma membrane.
Glutamate-Stimulated Ascorbate-Release by Astrocytes Requires Cellular Swelling
It is known that the exposure of astrocytes to glutamate induces cellular swelling [49, 50]. This effect appears to be mediated by EAAT-dependent glutamate uptake by astrocytes [49] and/or the mGluR-dependent activation of astrocytic aquaporin 4 [51]. As the glutamate-stimulated swelling of astrocytes has been linked with the astrocytic release of a range of so-called gliotransmitters [49], we next sought to determine whether glutamate-stimulated ascorbate-release by astrocytes was dependent on glutamate-induced cell swelling. It has previously been observed that hyper-osmotic media can inhibit the glutamate-stimulated release of ascorbate by cultured cerebral astrocytes that have been loaded with ascorbate [4]. These results are suggestive of a dependence of glutamate-stimulated ascorbate-release on cellular swelling.
Consistent with the results of the previous study, we have here confirmed that, in comparison to iso-osmotic HBS (310 mOsm), hyper-osmotic media (410 mOsm) containing either excess NaCl (+50 mM) or choline Cl (+50 mM) abolishes the glutamate-stimulated release of ascorbate (Fig. 2a). Importantly, the similarity in inhibition of ascorbate-release by media made hyper-osmotic with either NaCl or choline Cl indicates that the inhibitory effect is in fact mediated by the imposition of hyper-osmolarity rather than some ion-specific (e.g. Na+-specific or choline-specific) inhibitory response.

Glutamate-stimulated ascorbate-release from astrocytes is inhibited by hyper-osmolarity and emulated by hypo-osmolarity. All ascorbate (AA) release experiments were conducted over 1 h with mouse primary astrocyte cultures. Glutamate was always used at a concentration of 1 mM. a The glutamate-stimulated release of ascorbate (expressed as a percentage of the basal release rate) from astrocytes can be inhibited by HBS that has been made hyper-osmotic (410 mOsm) with either NaCl (+50 mM; ‘Glu/NaCl’) or choline chloride (+50 mM; ‘Glu/CCl’). b The glutamate-stimulated release of ascorbate from astrocytes can be emulated by HBS that has been made hypo-osmotic (210 mOsm) by omission of NaCl [−50 mM; ‘Hypo (low Na)’], but not by iso-osmotic HBS made hypo-natraemic by replacement of 50 mM NaCl with 50 mM CCl [‘Iso (low Na)’]. Data shown are means (±SD) of three experiments. ** P < 0.01; *** P < 0.001
To further strengthen the conclusion that glutamate-stimulated ascorbate-release is mediated by cell swelling, we next examined whether the exposure of the cells to hypo-osmotic medium emulated the phenomenon. As shown in Fig. 2b, the exposure of ascorbate-loaded astrocytes to mildly hypo-osmotic medium (210 mOsm), in which 50 mM NaCl had been omitted, significantly stimulated the release of ascorbate. Importantly, this stimulation of ascorbate-release cannot simply be attributed to a lowering of the extracellular [Na+] in the medium, as the iso-osmotic replacement of 50 mM NaCl by 50 mM choline chloride was without effect on ascorbate-release (Fig. 2b). It is also worth noting that exposure of astrocytes to 210 mOsm medium had no effect on LDH release (data not shown), indicating that the stimulatory effect of osmolarity is not attributable to cellular lysis.
Collectively, these data strongly suggest that the ability of glutamate to stimulate ascorbate-release from astrocytes depends on the induction of cell swelling.
Glutamate-Stimulated Ascorbate-Release can be Blocked by Inhibitors of Swelling-Activated Anion Channels
We next sought to examine the release mechanism involved in the mediation of the glutamate-stimulated and hypo-osmolarity-stimulated release of ascorbate by astrocytes. The phenomenon of cell swelling-induced release of small molecules (e.g. glutamate, taurine and d-serine) from astrocytes is well documented [49, 52]. In many cases, the release of such molecules in response to cell swelling can be inhibited by generic anion channel blockers, suggesting the involvement of volume (or swelling)-activated anion channels in the plasma membrane (e.g. VSOACs) [53]. In particular, the glutamate-stimulated release of ascorbate by cultured cerebral astrocytes has previously been observed to be inhibited by putative VSOAC inhibitors, including 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid, 5-nitro-2-(3-phenylpropylamino) benzoic acid and DDF [4]. In order to assess the involvement of VSOACs in the glutamate-stimulated release of ascorbate in the presently described system, we exposed ascorbate-loaded primary astrocyte cultures to DDF and to two other known inhibitors of VSOAC activity: NFA [54, 55] and IAA-94 [53, 55]. Furthermore, we also assessed the effect of these inhibitors on the hypo-osmolarity-stimulated release of ascorbate from these cells.
While DDF inhibits glutamate-stimulated ascorbate-release from cultured astrocytes [4], the effects of NFA and IAA-94 on this process do not appear to have been examined. However, the latter two compounds have been shown to strongly inhibit ATP-induced 14C-ascorbate-release from pig coronary artery endothelial cells [55]. In our experiments, the application of DDF (50 μM), NFA (200 μM) and IAA-94 (200 μM) to ascorbate-loaded astrocytes abolished the stimulation of ascorbate-release that was elicited by 1 mM glutamate (Fig. 3a) and by hypo-osmolarity (210 mOsm; −50 mM NaCl) (Fig. 3b). Although the inhibitory effect of NFA and IAA-94 on ascorbate-release either in the presence of glutamate or following the imposition of hypo-osmolarity exceeded that of DDF (Fig. 3a, b), this may be due to the differing pleiotropic effects of these non-selective inhibitors. Alternatively, as the ascorbate released in the presence of NFA and IAA-94 is significantly less than the relevant vehicle control (Fig. 3a), this suggests that these compounds also inhibit a component of the basal rate of ascorbate-release. Overall, these data suggest that basal and glutamate-stimulated ascorbate efflux from astrocytes may occur by different pathways, with DDF perhaps being more selective for the pathway utilised during glutamate-stimulated ascorbate-release than NFA and IAA-94.

Glutamate-stimulated ascorbate-release from astrocytes can be blocked by inhibitors of VSOACs. All ascorbate (AA) release experiments were conducted over 1 h with mouse primary astrocytes cultures. Glutamate was always used at a concentration of 1 mM. a The glutamate-stimulated release of ascorbate from astrocytes can be abolished by the application of the VSOAC inhibitors DDF (50 μM), NFA (200 μM) and IAA-94 (200 μM), which were added to cells 15 min prior to the addition of glutamate or control medium. b The hypo-osmolarity-stimulated release of ascorbate from astrocytes can be abolished by the same inhibitors as in (a). Data shown are means (±SD) of three experiments. *P < 0.05; **P < 0.01; ***P < 0.001
Moreover, it is also worth noting that as DDF, NFA and IAA-94 inhibit both glutamate-stimulated and hypo-osmolarity-stimulated ascorbate-release to a similar degree, it is unlikely that the inhibitory action of these compounds arises from an inhibition of the interaction of glutamate with the receptor/transporter responsible for mediating glutamate’s stimulatory effect on ascorbate-release (see below).
These data suggest that glutamate-stimulated ascorbate-release by astrocytes is mediated by swelling-activated, ascorbate-permeable anion channels in the plasma membrane (e.g. VSOACs). However, although there are a number of possible protein candidates for VSOAC activity in astrocytes [56], the identities of the molecular entities involved in VSOAC activity remain to be experimentally verified.
Glutamate-Stimulated Ascorbate-Release is not Directly Linked to Activation of NMDA and AMPA iGluRs or Group 1 mGluRs
To assess whether glutamate-stimulated ascorbate-release is dependent on the activation of iGluRs or mGluRs that are known to be expressed in cultured astrocytes, specific agonists for the various receptors were added to the culture medium in place of glutamate. The selective agonists were added at concentrations well above the respective K d and/or EC50 values for the relevant target receptors: NMDA (NMDA receptors: K d = 11–16 μM; [57]), AMPA (AMPA receptors: K d < 3 μM; [58]) and DHPG (group 1 mGluRs: EC50 < 7 μM; [59]).
Incubation of ascorbate-loaded astrocyte cultures with selective agonists of the NMDA [NMDA (50 μM)] or AMPA iGluRs [AMPA (50 μM)], or the group 1 mGluRs, mGluR1 and mGluR5 [DHPG (20 μM)], had no stimulatory effect on ascorbate-release (Fig. 4). Higher concentrations of DHPG (i.e. up to 100 μM) elicited no stimulation of ascorbate-release from ascorbate-replete astrocytes (data not shown). These results strongly suggest that the stimulatory effect of glutamate on ascorbate-release from astrocytes is not due to the isolated activation of NMDA or AMPA iGluRs, or the group 1 mGluRs, which are expressed in cultured astrocytes [60]. Although kainate receptor activation was not directly assessed, clear evidence for a functional expression of these iGluRs in astrocytes is lacking [61]. Additionally, selective agonism of the group II and III mGluRs was not assessed as these mGluRs do not appear to be expressed by rodent astrocytes under standard culture conditions [60].

Glutamate-stimulated ascorbate-release from astrocytes is not due to the isolated activation of iGluRs or mGluRs. All ascorbate (AA) release experiments were conducted over 1 h with rat primary astrocyte cultures. The glutamate (1 mM)-stimulated release of ascorbate from astrocytes cannot be emulated by agonists that are selective for NMDA and AMPA iGluRs or group 1 mGluRs. The agonists used were: NMDA (50 μM), AMPA (50 μM) and DHPG (20 μM). Data shown are means (±SD) of three experiments. ***P < 0.001
Glutamate-Stimulated Ascorbate-Release is Inhibited by GLAST-Specific Inhibitors
The earlier dose–response analysis of glutamate-stimulated ascorbate-release from astrocytes indicates an apparent EC50 value of 30–60 μM for glutamate (cf., Fig. 1b, c). This EC50 range is consistent with the involvement of the constitutively expressed glial EAAT, GLAST (K m for glutamate transport = 77 μM) [24].
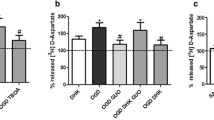
It is well known that undifferentiated cultured rat astrocytes typically express GLAST, but express negligible levels of GLT-1 unless induced by imposed conditions such as incubation with dibutyryl cAMP or co-culture with neurones [62]. Therefore, to more directly assess the relative contribution of GLAST in the mediation of glutamate-stimulated ascorbate-release from rat astrocytes, we employed three glutamate-transport inhibitors [25, 63], with different target specificities. The inhibitors used were: (i) TFB-TBOA (1 μM)—a selective non-transportable inhibitor that targets EAATs 1–5 (K i < 1 μM) without affecting iGluRs or mGluRs [33]; (ii) UCPH-101 (5 μM)—a novel and highly selective non-transportable inhibitor of GLAST (i.e. >400-fold selective for GLAST [K i = 0.66 μM] over GLT-1 and EAAC1 [EAAT3; K i values > 300 μM]) [25, 35]; and (iii) DHK (50 μM)—a selective non-transportable inhibitor of GLT-1 (K i ≈ 30 μM) [25, 63]. It is important to note that as undifferentiated astrocyte cultures such as those used in this study do not typically express detectable levels of GLT-1 [62], the use of DHK is an intended negative control.
Inhibitors were applied to astrocytes 15 min prior to glutamate addition. The application of TFB-TBOA abolished glutamate-stimulated ascorbate-release (Fig. 5a), suggesting the involvement of one or more of EAATs 1–5. The application of UCPH-101 singly or in combination with DHK abolished the stimulatory effect of 1 mM glutamate on ascorbate-release, while DHK alone had no effect (Fig. 5a). Importantly, none of the inhibitors had any significant inhibitory effect on the basal rate of ascorbate-release, with DHK even having a slightly stimulatory effect (P < 0.05; Fig. 5b). These data strongly suggest that in cultured astrocytes from neonatal rats the stimulatory effect of glutamate on ascorbate-release is largely dependent on the glutamate-transport activity of GLAST.

The effect of EAAT inhibitors on glutamate-stimulated ascorbate-release from astrocytes. All ascorbate (AA) release experiments were conducted over 1 h with rat primary astrocyte cultures. a The glutamate (1 mM)-stimulated release of ascorbate from astrocytes is abolished by the pan-EAAT inhibitor, TFB-TBOA (1 μM), the GLAST-specific inhibitor, UCPH-101 (UCPH; 5 μM), but not the GLT-1-specific inhibitor, DHK (50 μM). b The effect of the inhibitors used in (a) on the basal rate of ascorbate-release from astrocytes. Data shown are means (±SD) of three experiments. b ***P < 0.001 versus ‘Glu’ or ‘DHK’ conditions; a *P < 0.05 versus ‘basal’
Discussion
Astrocytes are integrally involved in the regulation of synaptic glutamate concentrations, and as such are responsible for the removal of the majority of neuronally released glutamate during excitatory neurotransmission [23]. The rapid removal of glutamate by astrocytes is crucial for the maintenance of neurotransmitter target-specificity and the minimisation of glutamate-induced excitotoxicity in neurones. It has long been known that extracellular glutamate triggers ascorbate-release within CNS tissue [1, 2, 6]. This release of ascorbate in response to glutamate plays an important neuromodulatory and neuroprotective roles [6, 10]. Mounting evidence suggests that astrocytes are key players in this ascorbate-release [4, 9, 17, 30]. Such considerations are of clinical relevance as they suggest the possibility that a component of the neuropathology presenting in neurodegenerative conditions such as Alzheimer’s, Parkinson’s and Huntington’s diseases (in which glutamate uptake is compromised [24, 28]) may relate to deficits in glutamate-stimulated ascorbate-release from astrocytes. Indeed, a key change that precedes and/or accompanies glutamate excitotoxicity in a diversity of neuropathological conditions is the decreased expression and/or activity of glial glutamate transporters, which may be driven by inflammatory mediators that are released by activated microglia (reviewed in [24]). In vivo support for the notion that a deficit in ascorbate-release may contribute to disease aetiology and/or presentation in Huntington’s disease comes from the observation that R6/2 mice expressing the defective Huntingtin gene have a deficit in striatal ascorbate-release [64] that is co-incident with a decrease in GLT-1 expression [28], and that putative pharmacologic enhancement of GLT-1 expression [65], or reintroduction of ascorbate [66], attenuates the Huntington behavioural phenotype.
In this report, we have explored aspects of the signalling mechanism behind the l-glutamate-stimulated efflux of ascorbate from cultured astrocytes. Our results indicate that this phenomenon is specific for excitatory amino acids (i.e. glutamate and aspartate), as the non-excitatory glutamate analogue, glutamine, is without effect. Using LDH release as a marker of cellular lysis, we demonstrated that neither the basal nor the glutamate-stimulated releases of ascorbate are due to a loss of integrity of the plasma membrane. This result suggests that the release of ascorbate occurs by a specific pathway rather than by non-specific ‘leakage’ from damaged cells.
A Role for Cell Swelling and VSOACs in Glutamate-Stimulated Ascorbate-Release from Astrocytes
We have partially characterised this release pathway by demonstrating a requirement for glutamate-induced cell swelling and the probable involvement of volume-sensitive anion channels (e.g. VSOACs/VRACs) in the plasma membrane.
The induction of astrocyte swelling by glutamate is a well-established phenomenon that is relevant in both physiological and pathophysiological scenarios [49, 51, 52]. Moreover, astrocytes are the major cell type of the brain to show glutamate-induced cell swelling under pathological conditions [50]. The mechanism behind the induction of astrocyte swelling by glutamate is not yet clear, but a recent study suggests a role for the group 1 mGluR-dependent activation of the water channel, aquaporin 4 [51]. A role for EAAT-dependent glutamate uptake has also been suggested [67]. EAAT-dependent glutamate uptake may result in rapid changes in intracellular ions (e.g. Na+) that may trigger directly or indirectly the influx of water. This influx of water results in cellular swelling that leads to the compensatory activation of anion channels in the plasma membrane. These swelling-activated anion channels contribute to the homeostatic process known as regulatory volume decrease (RVD) in which osmotically swollen cells release small anions and osmolytes into the extracellular space to correct their volume [50]. A number of small organic anions have been shown to be released from astrocytes in response to cell swelling, including glutamate, ATP, d-serine [52], taurine [68] and ascorbate [3].
The observations reported here that glutamate-stimulated ascorbate-release from astrocytes can be inhibited by hyper-osmotic and emulated by hypo-osmotic media strongly suggest a requirement cell swelling. Moreover, the experiments reported in this study, in which the effect of several pharmacological VSOAC inhibitors on glutamate- and hypo-osmolarity-stimulated ascorbate efflux was assessed, suggest a likely role for these channels in the ascorbate-release pathway. However, it remains possible that some of the inhibitory effects observed with the VSOAC inhibitors used in this study (i.e. DDF, NFA and IAA-94) are due to inhibition of distinct but nonetheless relevant processes.
First, NFA has been reported to partially inhibit glutamate transport by EAAT4 [69]. Therefore, in principle, some of the inhibition of glutamate-stimulated ascorbate-release by NFA observed in this study could be due to an impairment of EAAT-dependent glutamate uptake. However, the similarity of the inhibition profiles observed for both glutamate- and hypo-osmolarity-stimulated ascorbate-release, suggests instead that the inhibitory action of all three compounds is not directed against EAAT-dependent glutamate uptake, but is instead directed against the release pathway (e.g. VSOACs). Indeed, it was originally reported that although the VSOAC inhibitor NPPB strongly inhibited glutamate-stimulated ascorbate-release, the compound had no effect on glutamate uptake by astrocytes [4]. Thus, although our combined results suggest that it is unlikely, it remains possible that some of the inhibitory effects of DDF, NFA and IAA-94 reported here could be due to an inhibition of EAAT-dependent glutamate uptake.
Second, at least in the case of NFA and IAA-94, there may be some cross-inhibition of connexin hemichannels, which are expressed in cultured astrocytes [53]. This observation is relevant to this results as there is evidence that these hemichannels may be ascorbate permeable [18, 70]. However, connexin hemichannels are typically in a ‘closed’ or non-conductive configuration in the presence of millimolar concentrations of divalent cations (e.g. Ca2+ and Mg2+). Therefore, as the medium used for all experiments in this study contained 1.8 mM Ca2+ and 0.8 mM Mg2+ (i.e. total [metal2+] = 2.6 mM), it is unlikely that the glutamate-stimulated release of ascorbate occurred by a hemichannel-mediated pathway.
Taken together, the results presented here, and previously [3, 4], suggest a probable role for VSOAC-like channels in the mediation of glutamate- and hypo-osmolarity-stimulated ascorbate-releases from cultured rodent astrocytes. However, a definitive assessment of the contribution of such channels to this phenomenon remains to be achieved, pending further identification of the proteins involved.
A Role for GLAST in Regulating Glutamate-Stimulated Ascorbate-Release
In an attempt to determine the identity of the glutamate receptor/transporter responsible for the mediation of these effects by glutamate, we have presented a collection of pharmacological data that exclude the isolated involvement of NMDA and AMPA iGluRs and group 1 mGluRs, while strongly suggesting a requirement for the involvement of the astrocytic EAAT, GLAST. Interestingly, a recent report suggests that glutamate-stimulated ascorbate-release from mixed cultures of cultured chick retinal cells containing neurones growing atop a flat layer of glial cells involves NMDA and non-NMDA receptors but not glutamate transporters [71]. These data suggest that glutamate-dependent ascorbate-release may involve glutamate receptors in some CNS cell types or under conditions that more closely resemble in vivo conditions. However, as these experiments were performed with mixed cultures of neurones and glial cells it is unclear whether the glutamate-stimulated ascorbate-release originated in the neuronal or glial population.
In the experiments presented herein, the abolishment of glutamate-stimulated ascorbate-release by the pan-EAAT inhibitor TFB-TBOA [33] strongly suggests the involvement of one or more of EAATs 1-5. This conclusion is supported by the dose–response data presented for the stimulation of ascorbate-release from astrocytes by glutamate (Fig. 1b, c), which are consistent with the involvement of GLAST. In particular, the data obtained using the novel GLAST-specific inhibitor, UCPH-101 [25, 34, 35], indicate that in the present experimental system GLAST activity is probably entirely responsible for glutamate-stimulated ascorbate-release from cultured neonatal rat astrocytes. These data are to our knowledge the first report of a requirement for GLAST in glutamate-stimulated ascorbate-release from cultured astrocytes.
It is important to note that although the results presented in this report provide strong support for the involvement of GLAST in the mediation of glutamate-stimulated ascorbate-release from cultured neonatal rat astrocytes our results do not rule out the possibility that GLT-1 activity may contribute to this phenomenon under conditions in which GLT-1 is expressed at increased levels (e.g. exposure to dibutyryl cAMP [72], co-culture with neurones [62, 72] or exposure to soluble mediators released by activated microglia (e.g. tumour necrosis factor α) [73]). The determination of GLT-1’s role in this phenomenon is an important next step. Indeed, there is evidence for a direct deficit in ascorbate efflux in Huntington’s disease [64], which is associated with a loss of GLT-1 expression [28]. Moreover, a recent report demonstrates that treatment of R6/2 mice (a mouse model of Huntington’s disease) with the β-lactam, ceftriaxone, which induces GLT-1 expression, reverses the corticostriatal-dependent deficit in ascorbate efflux that occurs in this model [65]. Although, the cell types responsible for the increased ascorbate efflux in ceftriaxone-treated R6/2 mice were not determined, the role of astrocytes is likely [65]. Moreover, it will be important to determine whether there is still some involvement of iGluRs or mGluRs in modulating GLAST-dependent glutamate-stimulated ascorbate-release.
A further potential caveat is that although the GLAST inhibitor, UCPH-101, is more than 400-fold selective for GLAST over GLT-1 and EAAC1 (EAAT3), it has not been characterised against EAATs 4 and 5 [35]. However, significant structural differences between subtypes of the groupings of EAATs 1-3 and EAATs 4/5 suggest that it is unlikely that a compound such as UCPH-101 that exhibits such a high selectivity for one subtype of the former group would have any significant cross-reactivity with any subtype of the latter group [35].
In conclusion, the results presented in this study provide evidence that the glutamate-stimulated release of ascorbate from cultured rodent astrocytes occurs by a mechanism that is dependent on: (i) glutamate-induced cell swelling and the activation of VSOAC-like channels, and (ii) the activity of glial EAATs, and in particular GLAST. This mechanism may be of clinical relevance to neurodegenerative diseases in which the glial EAATs become deregulated.
Abbreviations
- AMPA:
-
(±) α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
- AO:
-
Ascorbate oxidase
- DDF:
-
1,9 dideoxyforskolin
- DHA:
-
Dehydroascorbate
- DHK:
-
Dihydrokanic acid
- DHPG:
-
(S)-3,5-dihydroxyphenylglycine
- EAAT:
-
Excitatory amino acid transporter
- GLAST:
-
Glutamate asparate transporter
- GLT-1:
-
Glutamate transporter 1
- GLUT:
-
Facilitative glucose transporter
- IAA-94:
-
R(+) indanyloxyacetic acid
- iGluR:
-
Ionotropic glutamate receptor
- mGluR:
-
Metabotropic glutamate receptor
- NFA:
-
Niflumic acid
- NMDA:
-
N-methyl-d-aspartate
- TFB-TBOA:
-
(2S,3S)-3-[3-[4-(trifluoromethyl)benzoylamino]benzyloxy]aspartate
- SVCT2:
-
Sodium-dependent vitamin C transporter, isoform 2
- VRAC:
-
Volume-regulated anion channel
- VSOAC:
-
Volume-sensitive osmolyte and anion channel
References
Grünewald, R. A. (1993). Ascorbic acid in the brain. Brain Research Brain Research Reviews, 18, 123–133.
Harrison, F. E., & May, J. M. (2009). Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radical Biology and Medicine, 46, 719–730.
Siushansian, R., Dixon, S. J., & Wilson, J. X. (1996). Osmotic swelling stimulates ascorbate efflux from cerebral astrocytes. Journal of Neurochemistry, 66, 1227–1233.
Wilson, J. X., Peters, C. E., Sitar, S. M., Daoust, P., & Gelb, A. W. (2000). Glutamate stimulates ascorbate transport by astrocytes. Brain Research, 858, 61–66.
Lane, D. J. R., & Lawen, A. (2008). Non-transferrin iron reduction and uptake are regulated by transmembrane ascorbate cycling in K562 cells. Journal of Biological Chemistry, 283, 12701–12708.
Rebec, G. V., & Pierce, R. C. (1994). A vitamin as neuromodulator: Ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Progress in Neurobiology, 43, 537–565.
Hediger, M. A. (2002). New view at C. Nature Medicine, 8, 445–446.
Du, J., Cullen, J. J., & Buettner, G. R. (2012). Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochimica et Biophysica Acta. Epub ahead of print. doi:10.1016/j.bbcan.2012.06.003
Castro, M. A., Beltrán, F. A., Brauchi, S., & Concha, I. I. (2009). A metabolic switch in brain: Glucose and lactate metabolism modulation by ascorbic acid. Journal of Neurochemistry, 110, 423–440.
Rice, M. E. (2000). Ascorbate regulation and its neuroprotective role in the brain. Trends in Neurosciences, 23, 209–216.
Qiu, S., Li, L., Weeber, E. J., & May, J. M. (2007). Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. Journal of Neuroscience Research, 85, 1046–1056.
Rice, M. E., & Russo-Menna, I. (1998). Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience, 82, 1213–1223.
Song, J. H., Shin, S. H., & Ross, G. M. (2001). Oxidative stress induced by ascorbate causes neuronal damage in an in vitro system. Brain Research, 895, 66–72.
Kim, E. J., Park, Y. G., Baik, E. J., Jung, S. J., Won, R., Nahm, T. S., et al. (2005). Dehydroascorbic acid prevents oxidative cell death through a glutathione pathway in primary astrocytes. Journal of Neuroscience Research, 79, 670–679.
Wilson, J. X. (1997). Antioxidant defense of the brain: A role for astrocytes. Canadian Journal of Physiology and Pharmacology, 75, 1149–1163.
Swanson, R. A., Ying, W., & Kauppinen, T. M. (2004). Astrocyte influences on ischemic neuronal death. Current Molecular Medicine, 4, 193–205.
Astuya, A., Caprile, T., Castro, M., Salazar, K., García Mde, L., Reinicke, K., et al. (2005). Vitamin C uptake and recycling among normal and tumor cells from the central nervous system. Journal of Neuroscience Research, 79, 146–156.
Lane, D. J. R., & Lawen, A. (2009). Ascorbate and plasma membrane electron transport—Enzymes vs efflux. Free Radical Biology and Medicine, 47, 485–495.
Hilgetag, C. C., & Barbas, H. (2009). Are there ten times more glia than neurons in the brain? Brain Structure and Function, 213, 365–366.
Lane, D. J. R., Robinson, S. R., Czerwinska, H., & Lawen, A. (2010). A role for Na+/H+ exchangers and intracellular pH in regulating vitamin C-driven electron transport across the plasma membrane. Biochemical Journal, 428, 191–200.
Clements, J. D. (1996). Transmitter timecourse in the synaptic cleft: Its role in central synaptic function. Trends in Neurosciences, 19, 163–171.
Walton, H. S., & Dodd, P. R. (2007). Glutamate-glutamine cycling in Alzheimer’s disease. Neurochemistry International, 50, 1052–1066.
Marcaggi, P., & Attwell, D. (2004). Role of glial amino acid transporters in synaptic transmission and brain energetics. Glia, 47, 217–225.
Tilleux, S., & Hermans, E. (2007). Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. Journal of Neuroscience Research, 85, 2059–2070.
Bunch, L., Erichsen, M. N., & Jensen, A. A. (2009). Excitatory amino acid transporters as potential drug targets. Expert Opinion on Therapeutic Targets, 13, 719–731.
Liang, J., Takeuchi, H., Doi, Y., Kawanokuchi, J., Sonobe, Y., Jin, S., et al. (2008). Excitatory amino acid transporter expression by astrocytes is neuroprotective against microglial excitotoxicity. Brain Research, 1210, 11–19.
Danbolt, N. C. (2001). Glutamate uptake. Progress in Neurobiology, 65, 1–105.
Behrens, P. F., Franz, P., Woodman, B., Lindenberg, K. S., & Landwehrmeyer, G. B. (2002). Impaired glutamate transport and glutamate-glutamine cycling: Downstream effects of the Huntington mutation. Brain, 125, 1908–1922.
Sheldon, A. L., & Robinson, M. B. (2007). The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochemistry International, 51, 333–355.
Wilson, J. X., & Dragan, M. (2005). Sepsis inhibits recycling and glutamate-stimulated export of ascorbate by astrocytes. Free Radical Biology and Medicine, 39, 990–998.
May, J. M., Li, L., Hayslett, K., & Qux, Z.-c. (2006). Ascorbate transport and recycling by SH-SY5Y neuroblastoma cells: Response to glutamate toxicity. Neurochemical Research, 31, 785–794.
Davies, A. R. L., Belsey, M. J., & Kozlowski, R. Z. (2004). Volume-sensitive organic osmolyte/anion channels in cancer: Novel approaches to studying channel modulation employing proteomics technologies. Annals of the New York Academy of Sciences, 1028, 38–55.
Shimamoto, K., Sakai, R., Takaoka, K., Yumoto, N., Nakajima, T., Amara, S. G., et al. (2004). Characterization of novel L-threo-β-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Molecular Pharmacology, 65, 1008–1015.
Erichsen, M. N., Huynh, T. H. V., Abrahamsen, B., Bastlund, J. F., Bundgaard, C., Monrad, O., et al. (2010). Structure-activity relationship study of first selective inhibitor of excitatory amino acid transporter subtype 1: 2-amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (UCPH-101). Journal of Medicinal Chemistry, 53, 7180–7191.
Jensen, A. A., Erichsen, M. N., Nielsen, C. W., Stensbøl, T. B., Kehler, J., & Bunch, L. (2009). Discovery of the first selective inhibitor of excitatory amino acid transporter subtype 1. Journal of Medicinal Chemistry, 52, 912–915.
Lane, D. J. R., Robinson, S. R., Czerwinska, H., Bishop, G. M., & Lawen, A. (2010). Two routes of iron accumulation in astrocytes: Ascorbate-dependent ferrous iron uptake via the divalent metal transporter (DMT1) plus an independent route for ferric iron. Biochemical Journal, 432, 123–132.
Puschmann, T. B., Dixon, K. J., & Turnley, A. M. (2010). Species differences in reactivity of mouse and rat astrocytes in vitro. Neurosignals, 18, 152–163.
Deutsch, J. C. (2000). Dehydroascorbic acid. Journal of Chromatography A, 881, 299–307.
Siushansian, R., Tao, L., Dixon, S. J., & Wilson, J. X. (1997). Cerebral astrocytes transport ascorbic acid and dehydroascorbic acid through distinct mechanisms regulated by cyclic AMP. Journal of Neurochemistry, 68, 2378–2385.
Lane, D. J. R., & Lawen, A. (2008). A highly sensitive colorimetric microplate ferrocyanide assay applied to ascorbate-stimulated transplasma membrane ferricyanide reduction and mitochondrial succinate oxidation. Analytical Biochemistry, 373, 287–295.
Berger, U. V., & Hediger, M. A. (2000). The vitamin C transporter SVCT2 is expressed by astrocytes in culture but not in situ. NeuroReport, 11, 1395–1399.
Qiao, H., & May, J. M. (2008). Development of ascorbate transporters in brain cortical capillary endothelial cells in culture. Brain Research, 1208, 79–86.
May, J. M., & Z-c, Qu. (2009). Ascorbic acid efflux and re-uptake in endothelial cells: Maintenance of intracellular ascorbate. Molecular and Cellular Biochemistry, 325, 79–88.
Korzeniewski, C., & Callewaert, D. M. (1983). An enzyme-release assay for natural cytotoxicity. Journal of Immunological Methods, 64, 313–320.
Berridge, M. V., Herst, P. M., & Tan, A. S. (2005). Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnology Annual Review, 11, 127–152.
Wassler, M., Jonasson, I., Persson, R., & Fries, E. (1987). Differential permeabilzation of membranes by saponin treatment of isolated rat hepatocytes. Release of secretory proteins. Biochemical Journal, 247, 407–415.
Chen, C.-J., Liao, S.-L., & Kuo, J.-S. (2000). Gliotoxic action of glutamate on cultured astrocytes. Journal of Neurochemistry, 75, 1557–1565.
Dringen, R., Kussmaul, L., & Hamprecht, B. (1998). Detoxification of exogenous hydrogen peroxide and organic hydroperoxides by cultured astroglial cells assessed by microtiter plate assay. Brain Research Protocols, 2, 223–228.
Hansson, E., Muyderman, H., Leonova, J., Allansson, L., Sinclair, J., Blomstrand, F., et al. (2000). Astroglia and glutamate in physiology and pathology: Aspects on glutamate transport, glutamate-induced cell swelling and gap-junction communication. Neurochemistry International, 37, 317–329.
Mulligan, S. J., & MacVicar, B. A. (2006) VRACs CARVe a path for novel mechanisms of communication in the CNS. Science’s STKE, 2006, pe42.
Gunnarson, E., Zelenina, M., Axehult, G., Song, Y., Bondar, A., Krieger, P., et al. (2008). Identification of a molecular target for glutamate regulation of astrocyte water permeability. Glia, 56, 587–596.
Hamilton, N. B., & Attwell, D. (2010). Do astrocytes really exocytose neurotransmitters? Nature Reviews Neuroscience, 11, 227–238.
Ye, Z.-C., Oberheim, N., Kettenmann, H., & Ransom, B. R. (2009). Pharmacological “cross-inhibition” of connexin hemichannels and swelling activated anion channels. Glia, 57, 258–269.
Sorota, S. (1994). Pharmacologic properties of the swelling-induced chloride current of dog atrial myocytes. Journal of Cardiovascular Electrophysiology, 5, 1006–1016.
Davis, K. A., Samson, S. E., Best, K., Mallhi, K. K., Szewczyk, M., Wilson, J. X., et al. (2006). Ca2+-mediated ascorbate release from coronary artery endothelial cells. British Journal of Pharmacology, 147, 131–139.
Parkerson, K. A., & Sontheimer, H. (2004). Biophysical and pharmacological characterization of hypotonically activated chloride currents in cortical astrocytes. Glia, 46, 419–436.
Patneau, D. K., & Mayer, M. L. (1990). Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-d-aspartate and quisqualate receptors. Journal of Neuroscience, 10, 2385–2399.
Honoré, T., Lauridsen, J., & Krogsgaard-Larsen, P. (1982). The binding of [3H]AMPA, a structural analogue of glutamic acid, to rat brain membranes. Journal of Neurochemistry, 38, 173–178.
Wiśniewski, K., & Car, H. (2002). (S)-3,5-DHPG: A review. CNS Drug Reviews, 8, 101–116.
Peavy, R. D., & Conn, P. J. (1998). Phosphorylation of mitogen-activated protein kinase in cultured rat cortical glia by stimulation of metabotropic glutamate receptors. Journal of Neurochemistry, 71, 603–612.
Seifert, G., & Steinhäuser, C. (2001). Ionotropic glutamate receptors in astrocytes. Progress in Brain Research, 132, 287–299.
Swanson, R. A., Liu, J., Miller, J. W., Rothstein, J. D., Farrell, K., Stein, B. A., et al. (1997). Neuronal regulation of glutamate transporter subtype expression in astrocytes. Journal of Neuroscience, 17, 932–940.
Bridges, R. J., & Esslinger, C. S. (2005). The excitatory amino acid transporters: Pharmacological insights on substrate and inhibitor specificity of the EAAT subtypes. Pharmacology & Therapeutics, 107, 271–285.
Rebec, G. V., Barton, S. J., & Ennis, M. D. (2002). Dysregulation of ascorbate release in the striatum of behaving mice expressing the Huntington’s disease gene. Journal of Neuroscience, 22, RC202.
Miller, B. R., Dorner, J. L., Bunner, K. D., Gaither, T. W., Klein, E. L., Barton, S. J., et al. (2012). Up-regulation of GLT1 reverses the deficit in cortically evoked striatal ascorbate efflux in the R6/2 mouse model of Huntington’s disease. Journal of Neurochemistry, 121, 629–638.
Rebec, G. V., Barton, S. J., Marseilles, A. M., & Collins, K. (2003). Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. NeuroReport, 14, 1263–1265.
Han, B. C., Koh, S. B., Lee, E. Y., & Seong, Y. H. (2004). Regional difference of glutamate-induced swelling in cultured rat brain astrocytes. Life Sciences, 76, 573–583.
Kimelberg, H. K., Goderie, S. K., Higman, S., Pang, S., & Waniewski, R. A. (1990). Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. Journal of Neuroscience, 10, 1583–1591.
Poulsen, M. V., & Vandenberg, R. J. (2001). Niflumic acid modulates uncoupled substrate-gated conductances in the human glutamate transporter EAAT4. Journal of Physiology, 534, 159–167.
Ahmad, S., & Evans, W. H. (2002). Post-translational integration and oligomerization of connexin 26 in plasma membranes and evidence of formation of membrane pores: Implications for the assembly of gap junctions. Biochemical Journal, 365, 693–699.
Portugal, C. C., Miya, V. S., da Costa Calaza, K., Santos, R. A. M., & Paes-de-Carvalho, R. (2009). Glutamate receptors modulate sodium-dependent and calcium-independent vitamin C bidirectional transport in cultured avian retinal cells. Journal of Neurochemistry, 108, 507–520.
Schlag, B. D., Vondrasek, J. R., Munir, M., Kalandadze, A., Zelenaia, O. A., Rothstein, J. D., et al. (1998). Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Molecular Pharmacology, 53, 355–369.
Tilleux, S., Gorsaud, S., & Hermans, E. (2009). Selective up-regulation of GLT-1 in cultured astrocytes exposed to soluble mediators released by activated microglia. Neurochemistry International, 55, 35–40.
Acknowledgments
We are thankful to Drs Lennart Bunch and Anders A. Jensen (University of Copenhagen) for the generous gift of UCPH-101, and to A/Prof. Stephen Robinson and Ms. Hania Czerwinska (Monash University) for the generous supply of astrocyte cultures and for helpful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
D. J. R. Lane and A. Lawen contributed equally as senior authors
Rights and permissions
About this article
Cite this article
Lane, D.J.R., Lawen, A. The Glutamate Aspartate Transporter (GLAST) Mediates l-Glutamate-Stimulated Ascorbate-Release Via Swelling-Activated Anion Channels in Cultured Neonatal Rodent Astrocytes. Cell Biochem Biophys 65, 107–119 (2013). https://doi.org/10.1007/s12013-012-9404-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-012-9404-8