
Abstract
HspB3, an as yet uncharacterized sHsp, is present in muscle, brain, heart, and in fetal tissues. A point mutation correlates with the development of axonal motor neuropathy. We purified recombinant human HspB3. Circular dichroism studies indicate that it exhibits β-sheet structure. Gel filtration and sedimentation velocity experiments show that HspB3 exhibits polydisperse populations with predominantly trimeric species. HspB3 exhibits molecular chaperone-like activity in preventing the heat-induced aggregation of alcohol dehydrogenase (ADH). It exhibits moderate chaperone-like activity towards heat-induced aggregation of citrate synthase. However, it does not prevent the DTT-induced aggregation of insulin, indicating that it exhibits target protein-dependent molecular chaperone-like activity. Unlike other sHsps, it has a very short C-terminal extension. Fusion of the C-terminal extension of αB-crystallin results in altered tertiary and quaternary structure, and increase in polydispersity of the chimeric protein, HspB3αB-CT. The chimeric protein shows comparable chaperone-like activity towards heat-induced aggregation of ADH and citrate synthase. However, it shows enhanced activity towards DTT-induced aggregation of insulin. Our study, for the first time, provides the structural and chaperone functional characterization of HspB3 and also sheds light on the role of the C-terminal extension of sHsps.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Small heat shock proteins (sHSPs) play important roles in stress tolerance, prevention of aggregation of target proteins, degradation of cellular proteins as well as in regulation of other cellular functions such as cell cycle, apoptosis, differentiation, signal transduction and in maintaining cytoskeletal integrity [1–7]. Based on sequence homology, ten mammalian sHsps have been identified so far [8, 9]. Of these, only Hsp27, αA-crystallin, αB-crystallin, Hsp20, and Hsp22 have been characterized for their structural and chaperone functional aspects. Many point mutations have been identified in members of the mammalian sHsp family that cause cataracts, several neuropathies and myopathies [10].
Muscle cells express many sHsps [11]; however, the reason for the recruitment of so many sHsps is not known. Moreover, the individual functions of these sHsps are not completely understood. The monomeric molecular masses of sHSPs range from 12 to 43 kDa [1–3]. Some members (Hsp27, α-crystallins) form large oligomeric assemblies ranging from 150 to 800 kDa [1–3], whereas other sHsps such as HspB2, HspB6 exhibit small oligomeric populations and HspB8 exists as monomer or dimer [12–15]. A hallmark feature of the sHSPs is the presence of a conserved sequence of 80–100 amino acids, referred to as the “α-crystallin domain” [16]. However, the flanking N-terminal domains and C-terminal regions (called “C-terminal extension”) of sHsps vary both in length and sequence [16].
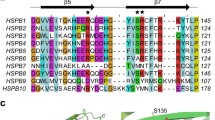
HspB3 is expressed in adult smooth muscle, brain and heart as well as in several other fetal tissues [17, 18]. A point mutation, R7S, in HspB3 correlates with development of axonal motor neuropathy [19]. However, the structural and functional aspects of HspB3 are not known so far. In order to address these aspects, in this study we have cloned its coding sequence in pET21a vector, expressed the protein in Escherichia coli and purified it to homogeneity. We have performed biophysical characterization for its structural aspects and investigated its molecular chaperone property. A striking feature of HspB3 is the almost complete absence of the C-terminal extension [17; see Fig. 1a]. The C-terminal extension in sHsps has many polar and charged residues, and is believed to play a solubilizing role in keeping the chaperone-target protein complexes in solution [20]. The C-terminal extension also has a conserved IXI/V motif [16] that makes inter-subunit contacts with the hydrophobic groove in the β-sandwich of the α-crystallin domain [21, 22] and has a propensity to engage in alternate inter-subunit contacts [23]. These interactions are important in determining the oligomeric size and chaperone activity of sHsps [24, 25]. The C-terminal extension encompassing the IXI motif in αB-crystallin, in addition to other regions, has been shown to interact with microtubules and desmin [26, 27]. Mutations in the IXI motif of plant and prokaryotic sHsps or truncations spanning this region were shown to dissociate the oligomeric structure of the proteins and result in loss of chaperone-like activity [28, 29]. Intrigued by the lack of a C-terminal extension in HspB3, we have also investigated the effect of fusing the C-terminal extension of the well-studied sHSP, αB-crystallin to HspB3 on its structural and chaperone functional aspects.

a Sequence alignment of human HspB3 with other human sHSPs. The sequence alignment shows the N-terminal domain, the conserved α-crystallin domain and the variable C-terminal extension. The C-terminal extension of HspB3 is replaced by that of αB-crystallin (HspB5) to create the chimeric protein HspB3αB-CT. The C-terminal extensions of HspB3 and αB-crystallin (HspB5) are shown in the box. b SDS-PAGE showing marker (lane 1), purified proteins HspB3 (lane 2), and HspB3αB-CT (lane 3)
Materials and Methods
Cloning of Human HspB3 and the Chimeric Protein, HspB3αB-CT
Since HspB3 is an intronless gene (Genbank accession: NM_006308.2 GI: 306966173), we have isolated genomic DNA from IMR32 human neuroblastoma cells to clone HspB3. IMR32 cells were harvested and lysed in 10 mM Tris–HCl buffer, pH 7.5 containing 10 mM EDTA, 150 mM NaCl, 0.5 % SDS, 200 μg/ml of proteinase K. Aqueous layer containing genomic DNA was separated upon extraction with phenol:chloroform:isoamyl alcohol (25:24:1), followed by isopropanol precipitation. HspB3 gene was PCR-amplified from the genomic DNA using the forward primer 5′-ACTCATATGGCAAAAATCATTTTGAGGCAC-3′ (NdeI site underlined) and reverse primer 5′-ATAGAATTCGATACGATGTCACTTAGTCCC-3′ (EcoRI site underlined). The PCR amplicon was cloned into pET21a vector (Novagen, Madison, WI, USA) using the restriction sites present in the primers and the vector. The sequence of the HspB3 pET21a construct was verified and found to be identical to that reported earlier [17].
The DNA sequence corresponding to amino acids 1–145 of HspB3 was amplified using the forward primer 5′-ACTCATATGGACATCGCCATCCACCACCC-3′ and reverse primer 5′-GGGATTTGCCTTCTCGGGGTGGTGAGAGTCCAGTGTCAAACC-3′. The DNA fragment corresponding to the C-terminal extension (amino acids 150–175) of αB-crystallin (HspB5) was amplified using forward primer 5′-CCACCCCGAGAAGGCAAATCCC-3′ and reverse primer 5′-ATAGAATTCTCACTTAGTCCCAACTGGATC-3′ from an αB-crystallin clone, already present in the laboratory as template. Amplicons were gel purified and allowed to extend via overlapping regions, followed by PCR using extreme end forward and reverse primer, thereby yielding the amplicon for the chimeric protein. The amplicon was further cloned into the pET21a vector and sequenced for validation.
Expression and Purification of Recombinant Human HspB3 and HspB3αB-CT
Escherichia coli strain BL21DE3 (Novagen, Madison, WI, USA) was transformed with pET21a expression vector containing the human HspB3 coding sequence. Protein expression profile was investigated at three temperatures (37, 30, and 20 °C) and at various isopropyl β-d-thiogalactoside (IPTG) concentrations 1, 0.75, 0.5, and 0.25 mM and a combination of both—low temperature and low IPTG concentrations. It was found that under all the conditions of temperature and IPTG concentrations, HspB3 was expressed in the insoluble fraction.
For purification of HspB3, the transformed cells were cultured in Luria–Bertani medium at 37 °C in a rotary shaker at 250 rpm. Protein expression was induced with 1 mM IPTG. Cells were harvested after 4 h of induction and lysed by 40 min incubation in buffer A (20 mM phosphate buffer, pH 7.4, containing 100 mM NaCl, 2 mM DTT), containing 150 μg/ml lysozyme and 1 mM PMSF on ice, followed by sonication. HspB3 partitioned into the insoluble inclusion body. After centrifugation, the inclusion body was washed with 20 mM phosphate buffer, pH 7.4, containing 100 mM NaCl and 0.05 % Triton X-100. It was further washed twice with buffer A to remove Triton X-100. The inclusion body was then solubilized in buffer A containing 6 M urea. The sample in 6 M urea was first diluted twofold with buffer A (to 3 M urea) and then fivefold from the sample in 3 M urea (to 0.6 M urea), followed by dialysis. The refolded protein thus obtained yielded a clear solution and was homogeneous as judged by SDS-polyacrylamide gel electrophoresis. We further subjected this sample to gel filtration on a Sephacryl S-300 column (90 × 1 cm2 diameter) to remove trace contamination, if any. Fractions containing the protein were pooled, concentrated by ultrafiltration and stored at 4 °C for further experiments. A similar procedure was followed for the purification of HspB3αB-CT.
Fluorescence Studies
Fluorescence measurements were carried out on a Hitachi F-4500 fluorescence spectrophotometer. For intrinsic fluorescence studies, a 0.2 mg/ml solution of HspB3 or HspB3αB-CT in buffer A was used. Intrinsic tryptophan fluorescence spectra were recorded with the excitation wavelength set at 295 nm and the excitation and emission band passes set at 2.5 nm. In bis-ANS-binding studies, the hydrophobic probe, bis-ANS, was used at a final concentration of 10 μM. The samples were excited at 390 nm and emission spectra were recorded with the excitation and emission band passes set at 2.5 nm. All spectra were recorded in the corrected spectrum mode.
Tryptophan Fluorescence Quenching Studies
KI quenching to study the accessibility of the lone tryptophan of HspB3 and HspB3αB-CT were performed using a Hitachi F-4500 fluorescence spectrophotometer with the excitation wavelength set at 295 nm and fluorescence emission spectra were scanned from 300 to 400 nm. Fluorescence quenching titration was performed at room temperature by sequentially adding aliquots of concentrated KI solution (7 M) to a 0.2 mg/ml HspB3 solution. Sodium thiosulphate was added to the KI stock solution to prevent I −3 formation.
The fluorescence quenching data in the presence of KI were analyzed by fitting to the Stern–Volmer equation:
where F 0 and F are fluorescence intensities in the absence or presence of KI, respectively, K SV is the Stern–Volmer quenching constant, and Q is the KI concentration. Percentage accessibility of tryptophan to the quencher was calculated using the Lehrer plot.
Circular Dichroism (CD) Spectroscopy
CD spectra of HspB3 or HspB3αB-CT were recorded on a Jasco J-815 spectropolarimeter. Near-UV CD spectra were recorded at protein concentration of 1 mg/ml in buffer A in a 1 cm path length cuvette, and far-UV CD spectra were recorded with a 0.2 mg/ml sample of protein in a 0.1 cm path length cuvette. Each spectrum is the average of four accumulations.
Gel Filtration Chromatography
The quaternary structure of HspB3 and HspB3αB-CT was investigated by gel filtration chromatography. Human HspB3 or HspB3αB-CT (200 μl of 1 mg/ml solution) in buffer A was loaded on to a Superose-6 HR 10/30 FPLC column from Amersham Biosciences previously equilibrated with buffer A. The proteins were eluted with the same buffer with a flow rate of 0.5 ml/min. Molecular mass standards thyroglobulin (669 kDa), ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), bovine serum albumin (67 kDa), ovalbumin (45 kDa), chymotrypsinogen A (25 kDa), and ribonuclease A (13.7 kDa) were used for calibration.
Sedimentation Velocity Measurements
Sedimentation velocity measurements were performed using an Optima XL-I analytical ultracentrifuge (Beckman Coulter, Fullerton, CA, USA). HspB3 and HspB3αB-CT protein samples at 1 mg/ml in phosphate buffer pH 7.4 were subjected to centrifugation at 40,000 rpm at 20 °C, using An50Ti rotor. The sedimentation coefficient S 20,w and molecular mass of the protein was calculated using the program SEDFIT [30], which uses nonlinear regression fitting of the sedimenting boundary profile with Lamm equation,
which describes the concentration distribution c(r, t) of a species with sedimentation coefficient s and diffusion coefficient D in a sector-shaped volume and in the centrifugal field ω2 r.
Chaperone-Like Activity
The chaperone-like activity of HspB3 or HspB3αB-CT was investigated against the heat-induced aggregation of yeast alcohol dehydrogenase (ADH) and citrate synthase (CS), as well as the DTT-induced aggregation of insulin. The thermal aggregation of yeast ADH (100 μg/ml) in the absence or the presence of different concentrations of HspB3 was monitored at 48 °C in 50 mM phosphate buffer, pH 7.2, containing 100 mM NaCl. Thermal aggregation of CS (at 43.5 °C) was monitored in 20 mM HEPES–KOH buffer, pH 7.4. The buffer containing different concentrations of HspB3 was incubated at 43.5 °C for 4 min, before the addition of CS at a final concentration of 25 μg/ml. Aggregation was monitored by measuring light scattering at right angles in a Hitachi 4000 fluorescence spectrophotometer. The excitation and emission wavelengths were set at 465 nm and excitation and emission band passes were set at 3 nm. The chaperone-like activity of HspB3 and HspB3αB-CT was also investigated against aggregation of CS at 48 °C.
DTT-induced aggregation of insulin was monitored in 10 mM phosphate buffer, pH 7.2, containing 100 mM NaCl at 37 °C in the absence or presence of human HspB3. The buffer containing required concentrations of HspB3 (to obtain different target protein to HspB3 ratios) was incubated for 4 min with constant stirring in a cuvette at 37 °C, using a thermostatically maintained Julabo water bath. Insulin was added at a final concentration of 0.2 mg/ml. Aggregation of insulin was initiated by the addition of 1 M DTT to a final concentration of 20 mM. Aggregation was monitored by measuring light scattering as described above. Percentage protection was calculated using the formula: (I t−I t+chaperone)/I t × 100, where I t is the intensity of light scattering of the target protein at the end of the assay and I t+chaperone is the intensity of light scattering of the target protein in the presence of chaperone at the end of the assay.
Results
Cloning, Expression and Purification of HspB3 and HspB3αB-CT
We have cloned human HspB3 gene in pET21a and expressed the recombinant protein in E. coli BL21DE3. HspB3 was purified from inclusion bodies as described in “Materials and Methods”. HspB3 refolded thus was essentially homogeneous as judged by electrophoresis on a 12 % SDS-polyacrylamide gel (Fig. 1b). Since HspB3 lacks a prominent C-terminal extension, we cloned and expressed the chimeric protein, HspB3αB-CT that lacks 5 amino acids from the C-terminus of HspB3 but has the C-terminal extension of αB-crystallin fused. C-terminal extensions, due to the presence of large number of polar residues, are considered to perform a solubilising role [20]. Despite having the C-terminal extension of αB-crystallin, the chimeric protein also partitioned to the inclusion bodies and was refolded and purified using the procedure described for the wild type protein.
Structural Characterization of HspB3 and HspB3αB-CT
We have studied the secondary structure of the proteins by far-UV CD spectroscopy. The far-UV CD spectrum of HspB3 exhibits a minimum at 218 nm and another at 208 nm (Fig. 2a). Analysis of the far-UV CD spectrum using the CDNN program [31] indicates that HspB3 has ~29.6 % antiparallel and 5.4 % parallel β-sheet, 20.5 % β-turns, 10.8 % α-helix and ~35.9 % random coil. Thus, the CD study indicates significant β-sheet structure for the protein as in the case of other sHsps such as α-crystallins. HspB3αB-CT exhibits an almost similar far-UV CD spectrum as the wild type protein, except for a slight difference in the ellipticity in the 210–220 nm region (Fig. 2a). CDNN analysis of the far-UV CD spectrum of HspB3αB-CT also reflects the similarity in the structure and yields 30.6 % antiparallel and 5.4 % parallel β-sheet, 20.5 % β-turns, 10.3 % α-helix, and 35.9 % random coil.

CD spectroscopy of HspB3 and HspB3αB-CT. a Far-UV CD spectrum of HspB3 (curve a) and HspB3αB-CT (curve b). b Near-UV CD spectrum of HspB3 (curve a) and HspB3αB-CT (curve b). [θ]mrm represents the mean residue mass ellipticity
We have studied the tertiary structure of the proteins by near-UV CD and fluorescence spectroscopy. The near-UV CD spectrum of HspB3 (Fig. 2b) exhibits peaks in the 270–290 nm region corresponding to tryptophan and tyrosine residues and in the 255–265 nm region corresponding to phenylalanine residues. HspB3 has a single-tryptophan residue in its sequence located at the 93rd position. The significant chiral structure shown by the near-UV CD spectrum of HspB3 indicates tight packing of the side chains of the aromatic amino acids in the tertiary structure of HspB3. The near-UV CD spectrum of HspB3αB-CT shows significant difference from that of the wild type protein (Fig. 2b), indicating differences in the tertiary structural packing upon fusing the C-terminal extension of αB-crystallin to HspB3.
Figure 3a shows the fluorescence emission spectrum of HspB3 and HspB3αB-CT upon excitation at 295 nm. The spectra exhibit emission maximum at ~344.6 and 343.8 nm, respectively, indicating that the sole tryptophan residue in both proteins is in a polar environment. This was further confirmed by quenching studies using the dynamic quencher, KI. Figure 3b shows the Stern–Volmer plot of the quenching of tryptophan fluorescence by the polar quencher molecule, KI. The fractional accessibilities of the sole Trp residue of HspB3 and HspB3αB-CT to the quencher molecules obtained from the Lehrer plot (modified Stern–Volmer plot) are 1.009 and 0.83, respectively, indicating that Trp is almost completely accessible to the polar environment in HspB3, while that in HspB3αB-CT is in polar environment, but its accessibility to the quencher is slightly decreased compared to that of HspB3.

Intrinsic tryptophan fluorescence and surface hydrophobicity of HspB3 and HspB3αB-CT. a Intrinsic tryptophan fluorescence of 0.2 mg/ml sample of HspB3 (curve a) and HspB3αB-CT (curve b) in 20 mM phosphate buffer (pH 7.4) containing 100 mM NaCl. b Modified Stern–Volmer plot of the quenching of tryptophan fluorescence of HspB3 (dark circle) and HspB3αB-CT (open circle) by the polar quencher molecule, KI. Fluorescence spectra were recorded by setting the excitation wavelength at 295 nm and the excitation and emission band passes at 2.5 nm. c Fluorescence spectra of the hydrophobic fluorescent probe, bis-ANS in the presence of the well-characterized sHsp, αB-crystallin (curve a), HspB3 (curve b), HspB3αB-CT (curve c) and in buffer alone (curve d). The excitation wavelength was 390 nm and excitation and emission band passes were set at 2.5 nm
Bis-ANS Binding Studies
Hydrophobic interactions between the chaperone and target protein play an important role in the binding of the chaperone to the target protein. Molecular chaperones bind to the non-native states of target proteins and prevent their aggregation [32]. We have used bis-ANS, a hydrophobic probe, to probe the hydrophobic surfaces of HspB3. Upon binding to the hydrophobic surfaces of a protein, the fluorescence intensity of bis-ANS increases several-fold accompanied by a blue shift in the emission maximum [33]. An increase in the fluorescence intensity of bis-ANS accompanied by a blue shift in its emission maximum to ~502 nm could be seen when bound to HspB3, suggesting that HspB3 has exposed hydrophobic surfaces (Fig. 3c). A comparison of the bis-ANS binding to HspB3 with that to another sHSP, αB-crystallin, shows that the extent of surface hydrophobicity of HspB3 is lower than that of αB-crystallin (Fig. 3c). The emission maximum of bis-ANS upon binding to HspB3αB-CT was also found to be 502 nm; however, the fluorescence intensity of bis-ANS bound to HspB3αB-CT is slightly less compared to that bound to HspB3 (Fig. 3c).
Gel Filtration Studies
sHSPs are known to have monomeric molecular masses ranging from 12 to 43 kDa, but some members of the family form large oligomeric assemblies with molecular masses ranging from 150 to 1,000 kDa. We have investigated the quaternary structure of HspB3 by Superose-6 gel filtration chromatography. The elution pattern of HspB3 from the Superose-6 column exhibits a peak corresponding to a molecular mass of ~70 kDa, suggesting that HspB3 may predominantly exist as a tetramer (Fig. 4a). It may be noted that a small fraction of higher molecular mass species are also observed, but a distinct peak is not seen in the chromatogram (Fig. 4a). We have used samples of HspB3 at different protein concentrations (1, 0.5, and 0.25 mg/ml) and found that the overall profiles do not differ significantly (data not shown).

Quaternary structure studies of HspB3 and HspB3αB-CT. a Gel filtration chromatography of HspB3 (curve a) and HspB3αB-CT (curve b) on a Superose 6 HR gel filtration column. The elution positions of molecular mass standards: 1 thyroglobulin, 2 ferritin, 3 catalase, 4 aldolase, 5 albumin, 6 ovalbumin, 7 chymotrypsinogen A, and 8 ribonuclease A, are shown by down arrows. The y-axis (absorbance at 280 nm) is normalized to highlight the differences in the polydispersity of the wild type and chimeric HspB3. b Distribution of sedimentation coefficients of HspB3 (open circles) and HspB3αB-CT (closed squares). The molecular masses of the species determined from the sedimentation velocity data by solving Lamm’s equation (see “Materials and Methods” for details) are also indicated. Numbers on the peak represent the molecular weight in kDa of that species
Sedimentation velocity measurement study (Fig. 4b) shows that HspB3 exhibits polydispersed populations with the major population exhibiting a molecular mass of 57 kDa. Considering the sequence-based theoretical molecular mass of the subunit as 17 kDa, the ratio of the molecular mass obtained by the sedimentation velocity method to the theoretical subunit molecular mass is calculated to be 3.35. This result suggests that the major population of HspB3 is trimeric. Thus, there seems to be a discrepancy in the oligomeric state of HspB3 revealed by gel filtration and sedimentation velocity measurements. Since sedimentation velocity measurements yield relatively more precise determination of molecular mass, we presume that HspB3 predominantly forms trimeric population. However, we cannot completely rule out the existence of some tetrameric population which is not resolved in the technique used.
HspB3αB-CT exhibits relatively more polydispersed species. In gel filtration chromatography (Fig. 4a), it yielded a peak corresponding to a dimeric species (35.3 kDa); in addition it also exhibited peaks corresponding to peak molecular masses of 152 and 668 kDa. Sedimentation velocity measurement reveals a major population having a molecular mass of 36 kDa, indicating a dimeric species and other species of various indicated molecular masses (Fig. 4b). The apparent differences in the relative amounts of different species reflected by gel filtration chromatography and sedimentation velocity measurements could be due to the intrinsic differences in the resolution of these two different techniques.
Chaperone-Like Activity of HspB3 and HspB3αB-CT
We used heat-induced aggregation of yeast ADH and CS as well as the DTT-induced aggregation of insulin as model systems to assay chaperone-like activity. Yeast ADH aggregates at a temperature of 48 °C. Figure 5a shows the effect of HspB3 on the heat-induced aggregation of yeast ADH. At a target protein to chaperone ratio of 1:1 (w/w), HspB3 offers ~78 % protection; on the other hand HspB3αB-CT offers slightly lesser protection (~60 %) at the same target protein to chaperone ratio. A comparison of the chaperone-like activity of HspB3 and HspB3αB-CT as a function of their concentration shows that at all the ratios, HspB3 exhibits higher chaperone-like activity than HspB3αB-CT (Fig. 5b); however, at a target protein to chaperone ratio of 1:1 (w/w), the extent of difference in their chaperone-like activities is much less.

Chaperone-like activity of HspB3 and HspB3αB-CT. a Chaperone-like activity of HspB3 against heat-induced aggregation of yeast ADH at 48 °C. Heat-induced aggregation of yeast ADH (0.1 mg/ml) alone (curve 1), in the presence of 0.1 mg/ml of HspB3αB-CT (curve 2) and in the presence of 0.1 mg/ml HspB3 (curve 3). b The percentage protection of thermal aggregation of ADH by HspB3 (hashed bar) and HspB3αB-CT (closed bar) at various target protein to chaperone weight ratios. c Chaperone-like activity of HspB3 against heat-induced aggregation of citrate synthase (CS) at 43.5 °C. Aggregation of CS (25 μg/ml) alone (curve 1) and in the presence of the chaperones: at weight ratio of 1:1 CS to HspB3αB-CT (curve 2), 1:1 CS to HspB3 (curve 3), 1:8 CS to HspB3αB-CT (curve 4), and 1:8 CS to HspB3 (curve 5). d The percentage protection of thermal aggregation of CS by HspB3 (hashed bar) and HspB3αB-CT (closed bar) at various target protein to chaperone weight ratios. e Chaperone-like activity of Human HspB3 towards DTT-induced aggregation of insulin. Curve 1 aggregation profile of insulin (0.2 mg/ml) alone; curve 2 in the presence of 1:1 ratio of insulin to HspB3, curve 3 in the presence of 1:1 and curve 4 in the presence of 1:2 ratios of insulin to HspB3αB-CT (w/w). f Chaperone-like activity of HspB3 and HspB3αB-CT towards DTT-induced aggregation of insulin represented by hashed and closed bars, respectively, at insulin to chaperone protein ratio of 1:1 and 1:2. It is to be noted that the light scattering values of the sample of insulin in the presence of HspB3 eventually reaches higher than that of the control (insulin alone; see e). Since there is no prevention of aggregation, we considered the percentage protection in this case as zero
Interestingly, a very high concentration of HspB3 was required to prevent the heat-induced aggregation of another protein, CS. At a target protein to chaperone ratio of 1:1 (w/w), HspB3 delayed the aggregation of CS. However, it eventually aggregated to almost the same extent as CS alone, thus offering little or no protection (Fig. 5c). At 1:1 ratio (w/w) of CS to HspB3αB-CT, the aggregation kinetics of CS was similar to that in the presence of HspB3. At a target protein to chaperone ratio of 1:4 (w/w), HspB3 prevented the aggregation of CS to an extent of ~44 %, whereas HspB3αB-CT exhibited only 21 % chaperone-like activity, indicating that the chimeric protein has lesser chaperone-like activity than the wild type protein (Fig. 5d). At a higher concentration of chaperone (target protein to chaperone ratio of 1:8, w/w), the difference between the chaperone-like activities of HspB3 and the chimeric protein decreased and were 80 and 72 %, respectively (Fig. 5C). It is interesting to note that ~80 % protection was offered by HspB3 against CS at a target protein to chaperone ratio of 1:8 (w/w), while a comparable extent of protection was offered against ADH at a target protein to chaperone ratio of 1: 0.5 (w/w) (Fig. 5b, d).
HspB3 failed to prevent the aggregation of DTT-induced aggregation of insulin at a 1:1 (w/w) ratio (Fig. 5e), as well as 1:2 (w/w) ratio of insulin to HspB3 (data not shown). In fact, the aggregation of insulin in the presence of HspB3 was higher than that of insulin alone, suggesting that HspB3 co-precipitated out with insulin. On the other hand, HspB3αB-CT offered ~31 % protection at a 1:1 ratio (Fig. 5e). At a 1:2 ratio (w/w) of target protein to chaperone, the chimeric protein prevented the DTT-induced aggregation of insulin to an extent ~75 % (Fig. 5f). We have also performed gel filtration chromatography and collected the peaks corresponding to the dimeric population and the higher oligomeric populations of the chimeric protein separately and investigated their chaperone property against CS and insulin. The dimeric and other oligomeric species showed 69 and 70 % protection, respectively, against CS aggregation at a weight ratio of 1:8 of CS to the chimeric protein, while they showed 24 and 23 % protection, respectively, against insulin aggregation at a weight ratio of 1:1 of insulin to the chimeric protein. Thus, the dimeric and higher oligomeric populations of the chimeric protein do not significantly differ in their chaperone activity towards these target proteins.
Thus, our results show that HspB3 does not prevent the aggregation of insulin significantly, while it prevents the aggregation of ADH efficiently and prevents the aggregation of CS only moderately. In order to investigate if the temperature difference at which ADH and CS aggregation assays are carried out is responsible for the observed differences in the chaperone activity, we have performed the chaperone assay at 48 °C (aggregation temperature for ADH) using the temperature-dependent aggregation model of CS. The percentage protection obtained for HspB3 and HspB3αB-CT at a 1:1 weight ratio of CS to the chaperone was 9 and 5, respectively, which are comparable to those obtained at the experimental temperature of 43.5 °C (Fig. 5d). Thus, these results clearly show that HspB3 exhibits target protein-dependent chaperone-like activity.
Discussion
As mentioned earlier, HspB3 is expressed in adult smooth muscle, brain and heart as well as in several other fetal tissues [17, 18]. A point mutation, R7S, in HspB3 results in axonal motor neuropathy [19]. Though HspB3 seems to have an important biological role, its structural and functional aspects have not been addressed so far.
Sequence-based comparison of HspB3 with other members of the sHsp family reveals many interesting variations. It has been found to be the most deviating member of the sHSP family. Firstly, it has an exceptionally short “C-terminal extension”. The C-terminal extension in sHsps has many polar and charged residues, and is believed to play a solubilizing role in keeping the chaperones or the chaperone-target protein complexes in solution [20, 34]. Swapping the C-terminal extension between αA- and αB-crystallin results in altered oligomeric size and modulation of chaperone function [35]. It has been shown that deletion of 17 amino acids from the C-terminal end of αA-crystallin results in the formation of large insoluble aggregates and appreciable decrease in chaperone activity [36]. Notably, the C-terminal extensions in sHsps have a conserved IXI/V motif, which provides important subunit contacts and plays a role in oligomerization. In αA- and αB-crystallin this region makes alternative interactions involving inter-subunit interactions [23]. Mutation of the motif to GXG in both αA- and αB-crystallin results in the formation of larger oligomers with enhanced chaperone property [23]. Solid state NMR and SAXS studies on αB-crystallin show that the IXI motif is involved in interdimeric and intermolecular interactions, which play an important role in the determination of oligomeric size and chaperone activity [24]. The NMR study revealed intermolecular interactions between the IXI motif (I159-P160-I161) and the substrate binding groove (β4 and β8 strands) which is released upon decrease in pH, potentially leading to chaperone activation [24]. Studies on another sHsp, StHsp14.0 showed that hydrophobicity and size of amino acids in IXI/V motif are responsible not only for assembly of the oligomer but also for the maintenance of β-sheet rich secondary structure and hydrophobicity [25].
Interestingly, as opposed to the observation in the case of mammalian sHsps described above, mutations in IXI motif of plant and prokaryotic sHsps or truncations spanning this region were shown to dissociate the oligomeric structure of the proteins and result in loss of chaperone-like activity [28, 29]. Thus, the IXI motif, and hence the C-terminal extension, may have different types of interactions, modulations of oligomerization and chaperone function in different sHsps. The understanding on the exact details on the structural aspects and functional outcome, therefore, needs further studies. It needs to be reiterated that HspB3 lacks this motif. Secondly, HspB3 also shows deviation in the N-terminal domain. The N-terminal domain of sHsps is important in the formation of higher order oligomers [37, 38]. HspB3 has very poor conservation in a stretch of sequence in the N-terminal domain (SRLFDQFFG motif) which is otherwise well conserved among the other mammalian members. This motif is important in higher order oligomerization [39]. It is, therefore, intriguing as to whether HspB3 can form oligomeric assembly and exhibit chaperone property like other known members of the sHsp family. In this context, our present study provides information, for the first time, on the structural and chaperone properties of HspB3.
Our study shows that HspB3 exhibits β-sheet conformation as exhibited by many members of the sHSP family. However, there are distinct differences in terms of quaternary structure and the chaperone property. Despite lacking a proper C-terminal extension and exhibiting poor sequence conservation in the N-terminal domain, HspB3 forms polydispersed oligomeric species with a major population of trimer, indicating the possibility of alternative subunit interaction sites. The well-conserved “α-crystallin” domain is known to make contacts in the dimeric interface [24]. Thus, it appears that potentially the α-crystallin domain and some parts of the N-terminal domain of HspB3 interact to form the oligomeric assembly.
Our study shows that HspB3 exhibits chaperone-like activity; however, it is target protein-dependent. Whereas it shows fairly good activity against temperature-induced aggregation of yeast ADH, it prevents the temperature-induced aggregation of CS only at very high target protein to HspB3 ratios and does not prevent the DTT-induced aggregation of insulin. Several studies have shown that the C-terminal extension of sHsps is important for keeping the chaperone-target protein complex in solution [34], and truncation of the C-terminal leads to a drastic reduction or loss of chaperone-like activity [36]; the fact that HspB3 has a very short C-terminal extension may explain the moderate chaperone-like activity of HspB3. Various factors such as oligomeric size, surface hydrophobicity and subunit exchange are believed to affect the chaperoning ability of sHsps. Based on our studies, we conclude that HspB3 forms a major population of trimer and exhibits a target protein-dependent chaperone-like activity. This property of HspB3 appears to be different from that of other sHsps, such as Hsp27, αA- and αB-crystallin. Thus, there seem to be two broad classes among the mammalian sHsps: one class includes those like Hsp27 (HspB1), αA-crystallin (HspB4), and αB-crystallin (HspB5) that exhibit general chaperone property in preventing the aggregation of large number of target proteins and the other includes those that exhibit observable target protein-dependence in their chaperone property (such as HspB3 as revealed by this study). Interestingly, an earlier study of Sugiyama et al. [11] has shown that five members of the sHsp family present in the muscle cells form two types of mutually exclusive oligomers/complexes: one comprises Hsp27, αB-crystallin and p20 (Hsp20 or HspB6) and another comprises HspB2 and HspB3 with apparent molecular mass of 150 kDa. A study of den Engelsman et al. [40] shows that co-expression of HspB2 and HspB3 in E. coli and subsequent purification yielded a series of hetero-oligomers consisting of 4, 8, 12, 16, 20, and 24 subunits with HspB2:HspB3 subunit ratio of 3:1; the complex exhibited poor chaperone property. A recent study from our laboratory [12] showed that HspB2 exhibits concentration-dependent oligomerization property and exhibits target protein-dependent chaperone property. However, as opposed to HspB3 (present study), HspB2 prevents the aggregation of insulin significantly, while both of them prevent the aggregation of ADH very effectively. Thus, it appears that HspB3 or HspB2 may form distinct oligomerization pattern and functionally distinct species compared to other sHsps. The sequence and structural features that determine specific oligomerization patterns or differences among sHsps and their importance in the functional modulations are not yet completely understood. Therefore, characterization of individual sHsps for their structural and chaperone properties would be useful. In this context understanding and further designing experiments to address specific sequence and structural features of sHsps that determine the formation of oligomeric assemblies and their functional significance are important.
Since HspB3 lacks a C-terminal extension, it would be of interest to address the effect of fusion of a C-terminal extension to HspB3. The chimeric protein, HspB3αB-CT, having the C-terminal extension of αB-crystallin shows a predominantly dimeric population; however, it also includes some proportions of higher order oligomeric populations. The fact that the trimeric populations are diminished in the chimeric protein indicate the dominant role of the C-terminal extension in dictating subunit assembly of HspB3. It is possible that the potential inter-subunit contacts by the C-terminal extension in dimer exclude other inter-subunit contacts necessary for trimeric assembly. Conceivably, the C-terminal extension also affects the chaperone property of the system. The chimeric protein exhibits chaperone property towards DTT-induced aggregation of insulin, an activity that HspB3 lacks. Our study indicates that the C-terminal extension plays an important role in the prevention of aggregation of insulin. Interestingly, similar results of modulation of chaperone activity by C-terminal extension have been reported in the case of mouse Hsp25 and Xenopus Hsp30C [41, 42]. There are two possible mechanisms by which C-terminal extension exerts it effect on the chaperone property. It may either improve the solubilization of the complex due to its preponderance of charged residues or may exhibit direct interaction with the target protein, or both. For instance, it has been shown that the region 155 PERTIPITRE 164 of the C-terminal extension of αB-crystallin directly interacts with desmin [26] and microtubules [27].
Thus, our study for the first time describes the structural and chaperone functional properties of the sHSP, HspB3. It demonstrates that though it shows certain properties that are characteristic of sHSPs, such as significant β-sheet structure and chaperone activity, it differs from them in terms of its small oligomeric structure and target protein-dependent chaperone activity. Our study further reinforces the important role of C-terminal extensions in sHsps in structural and chaperone properties. Thus, this study reports a candidate of the mammalian sHsp family that exhibits target protein-dependent chaperone-like activity. The partial structural and chaperone functional differences exhibited by individual sHsps, in addition to their shared properties, may be important for the shared as well as diverse functional role of sHsps in cell survival and stress tolerance, which may explain why cell expresses many sHsps differentially and temporally.
References
Van Montfort, R., Slingsby, C., & Vierling, E. (2001). Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Advances in Protein Chemistry, 59, 105–156.
Haslbeck, M., Franzmann, T., Weinfurtner, D., & Buchner, J. (2005). Some like it hot: The structure and function of small heat-shock proteins. Nature Structural & Molecular Biology, 12, 842–846.
Narberhaus, F. (2002). Alpha-crystallin-type heat shock proteins: Socializing minichaperones in the context of a multichaperone network. Microbiology Molecular Biology Reviews, 66, 64–93.
Bai, F., Xi, J., Higashikubo, R., & Andley, U. P. (2004). A comparative analysis of alphaA- and alphaB-crystallin expression during the cell cycle in primary mouse lens epithelial cultures. Experimental Eye Research, 79, 795–805.
Ito, H., Kamei, K., Iwamoto, I., Inaguma, Y., & Kato, K. (2001). Regulation of the levels of small heat-shock proteins during differentiation of C2C12 cells. Experimental Cell Research, 266, 213–221.
Kamradt, M. C., Lu, M., Werner, M. E., Kwan, T., Chen, F., Strohecker, A., et al. (2005). The small heat shock protein alpha B-crystallin is a novel inhibitor of TRAIL-induced apoptosis that suppresses the activation of caspase-3. Journal of Biological Chemistry, 280, 11059–11066.
Charette, S. J., Lavoie, J. N., Lambert, H., & Landry, J. (2000). Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Molecular and Cellular Biology, 20, 7602–7612.
Kappé, G., Franck, E., Verschuure, P., Boelens, W. C., Leunissen, J. A. M., & de Jong, W. W. (2003). The human genome encodes 10 α-crystallin-related small heat shock proteins: HspB1-10. Cell Stress and Chaperones, 8, 53–61.
Fontaine, J. M., Rest, J. S., Welsh, M. J., & Benndorf, R. (2003). The sperm outer dense fiber protein is the 10th member of the superfamily of mammalian small stress proteins. Cell Stress and Chaperones, 8, 62–69.
Benndorf, R., & Welsh, M. J. (2004). Shocking degeneration. Nature Genetics, 36, 547–548.
Sugiyama, Y., Suzuki, A., Kishikawa, M., Akutsu, R., Hirose, T., Waye, M. M., et al. (2000). Muscle develops a specific form of small heat shock protein complex composed of MKBP/HSPB2 and HSPB3 during myogenic differentiation. Journal of Biological Chemistry, 275, 1095–1104.
Prabhu, S., Raman, B., Ramakrishna, T., & Rao, C. M. (2012). HspB2/myotonic dystrophy protein kinase binding protein (MKBP) as a novel molecular chaperone: Structural and functional aspects. PLoS ONE, 7, e29810.
Bukach, O. V., Seit-Nebi, A. S., Marston, S. B., & Gusev, N. B. (2004). Some properties of human small heat shock protein Hsp20 (HspB6). European Journal of Biochemistry, 271, 291–302.
Kim, M. V., Seit-Nebi, A. S., Marston, S. B., & Gusev, N. B. (2004). Some properties of human small heat shock protein Hsp22 (H11 or HspB8). Biochemical and Biophysical Research Communications, 315, 796–801.
Chowdary, T. K., Raman, B., Ramakrishna, T., & Rao, C. M. (2004). Mammalian Hsp22 is a heat-inducible small heat-shock protein with chaperone-like activity. Biochemical Journal, 381, 379–387.
de Jong, W. W., Caspers, G. J., & Leunissen, J. A. (1998). Genealogy of the alpha-crystallin—small heat-shock protein superfamily. International Journal of Biological Macromolecules, 22, 151–162.
Boelens, W. C., Van Boekel, M. A., & de Jong, W. W. (1998). HspB3, the most deviating of the six known human small heat shock proteins. Biochimica et Biophysica Acta, 1388, 513–516.
Molyneaux, B. J., Arlotta, P., Fame, R. M., MacDonald, J. L., MacQuarrie, K. L., & Macklis, J. D. (2009). Novel subtype-specific genes identify distinct subpopulations of callosal projection neurons. Journal of Neuroscience, 29, 12343–12354.
Kolb, S. J., Snyder, P. J., Poi, E. J., Renard, E. A., Bartlett, A., Gu, S., et al. (2010). Mutant small heat shock protein B3 causes motor neuropathy: Utility of a candidate gene approach. Neurology, 74, 502–506.
Smulders, R. H. P. H., Carver, J. A., Lindner, R. A., van Boekel, M. A., Bloemendal, H., & de Jong, W. W. (1996). Immobilization of the C-terminal extension of bovine alphaA-crystallin reduces chaperone-like activity. Journal of Biological Chemistry, 271, 29060–29066.
Van Montfort, R. L., Basha, E., Friedrich, K. L., Slingsby, C., & Vierling, E. (2001). Crystal structure and assembly of a eukaryotic small heat shock protein. Nature Structural Biology, 8, 1025–1030.
Kim, K. K., Kim, R., & Kim, S. H. (1998). Crystal structure of a small heat-shock protein. Nature, 394, 595–599.
Pasta, S. Y., Raman, B., Ramakrishna, T., & Rao, C. M. (2004). The IXI/V motif in the C-terminal extension of alpha-crystallins: Alternative interactions and oligomeric assemblies. Molecular Vision, 10, 655–662.
Jehle, S., Rajagopal, P., Bardiaux, B., Markovic, S., Kühne, R., Stout, J. R., et al. (2010). Solid-state NMR and SAXS studies provide a structural basis for the activation of alphaB-crystallin oligomers. Nature Structural & Molecular Biology, 17, 1037–1042.
Saji, H., Iizuka, R., Yoshida, T., Abe, T., Kidokoro, S., Ishii, N., et al. (2008). Role of the IXI/V motif in oligomer assembly and function of StHsp14.0, a small heat shock protein from the acidothermophilic archaeon, Sulfolobus tokodaii strain 7. Proteins, 71, 771–782.
Houck, S. A., Landsbury, A., Clark, J. I., & Quinlan, R. A. (2011). Multiple sites in αB-crystallin modulate its interactions with desmin filaments assembled in vitro. PLoS ONE, 6, e25859.
Ghosh, J. G., Houck, S. A., & Clark, J. I. (2007). Interactive domains in the molecular chaperone human aB crystallin modulate microtubule assembly and disassembly. PLoS ONE, 2, e498.
Studer, S., Obrist, M., Lentze, N., & Narberhaus, F. (2002). A critical motif for oligomerization and chaperone activity of bacterial alpha-heat shock proteins. European Journal of Biochemistry, 269, 3578–3586.
Kirschner, M., Winkelhaus, S., Thierfelder, J. M., & Nover, L. (2000). Transient expression and heat-stress-induced co-aggregation of endogenous and heterologous small heat-stress proteins in tobacco protoplasts. Plant Journal, 24, 397–411.
Schuck, P. (2000). Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophysical Journal, 78, 1606–1619.
Böhm, G., Muhr, R., & Jaenicke, R. (1992). Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Engineering, 5, 191–195.
Raman, B., Siva Kumar, L. V., Ramakrishna, T., & Rao, C. M. (2001). Redox-regulated chaperone function and conformational changes of Escherichia coli Hsp33. FEBS Letters, 489, 19–24.
Musci, G., Metz, G. D., Tsunematsu, H., & Berliner, L. J. (1985). 4,4′-Bis[8-(phenylamino)naphthalene-1-sulfonate] binding to human thrombins: A sensitive exo site fluorescent affinity probe. Biochemistry, 24, 2034–2039.
Leroux, M. R., Melki, R., Gordon, B., Batelier, G., & Candido, E. P. (1997). Structure-function studies on small heat shock protein oligomeric assembly and interaction with unfolded polypeptides. Journal of Biological Chemistry, 272, 24646–24656.
Pasta, S. Y., Raman, B., Ramakrishna, T., & Rao, C. M. (2002). Role of the C-terminal extensions of α-crystallins: Swapping the C-terminal extension of αA-crystallin to αB-crystallin results in enhanced chaperone activity. Journal of Biological Chemistry, 277, 45821–45828.
Andley, U. P., Mathur, S., Griest, T. A., & Petrash, J. M. (1996). Cloning, expression, and chaperone-like activity of human alphaA-crystallin. Journal of Biological Chemistry, 271, 31973–31980.
Merck, K. B., De Haard-Hoekman, W. A., Oude Essink, B. B., Bloemendal, H., & de Jong, W. W. (1992). Expression and aggregation of recombinant alpha A-crystallin and its two domains. Biochimica et Biophysica Acta, 1130, 267–276.
Feil, I. K., Malfois, M., Hendle, J., van Der Zandt, H., & Svergun, D. I. (2001). A novel quaternary structure of the dimeric alpha-crystallin domain with chaperone-like activity. Journal of Biological Chemistry, 276, 12024–12029.
Pasta, S. Y., Raman, B., Ramakrishna, T., & Rao, C. M. (2003). Role of the conserved SRLFDQFFG region of alpha-crystallin, a small heat shock protein: Effect on oligomeric size, subunit exchange, and chaperone-like activity. Journal of Biological Chemistry, 278, 51159–51166.
den Engelsman, J., Boros, S., Dankers, P. Y. M., Kamps, B., Egberts, W. T. V., Böde, C. S., et al. (2009). The small heat-shock proteins HSPB2 and HSPB3 form well-defined hetero oligomers in a unique 3 to 1 subunit ratio. Journal of Molecular Biology, 393, 1022–1032.
Lindner, R. A., Carver, J. A., Ehrnsperger, M., Buchner, J., Esposito, G., Behlke, J., et al. (2000). Mouse Hsp25, a small shock protein: The role of its C-terminal extension in oligomerization and chaperone action. European Journal of Biochemistry, 267, 1923–1932.
Fernando, P., & Heikkila, J. J. (2000). Functional characterization of Xenopus small heat shock protein, Hsp30C: The carboxyl end is required for stability and chaperone activity. Cell Stress and Chaperones, 5, 148–159.
Acknowledgments
A. Asthana acknowledges senior research fellowship from CSIR, New Delhi. Ch. M. Rao acknowledges the J. C. Bose National Fellowship of Department of Science and Technology, New Delhi.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Asthana, A., Raman, B., Ramakrishna, T. et al. Structural Aspects and Chaperone Activity of Human HspB3: Role of the “C-Terminal Extension”. Cell Biochem Biophys 64, 61–72 (2012). https://doi.org/10.1007/s12013-012-9366-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-012-9366-x