
Abstract
Hypertension is one of the most prevalent cardiovascular disorders worldwide, affecting 1.13 billion people, or 14% of the global population. Hypertension is the single biggest risk factor for cerebrovascular dysfunction. According to the American Heart Association, high blood pressure (BP), especially in middle-aged individuals (~ 40 to 60 years old), is associated with an increased risk of dementia, later in life. Alzheimer’s disease and cerebrovascular disease are the two leading causes of dementia, accounting for around 80% of the total cases and usually combining mixed pathologies from both. Little is known regarding how hypertension affects cognitive function, so the impact of its treatment on cognitive impairment has been difficult to assess. The brain renin-angiotensin system (RAS) is essential for BP regulation and overactivity of this system has been established to precede the development and maintenance of hypertension. Angiotensin II (Ang-II), the main peptide within this system, induces vasoconstriction and impairs neuro-vascular coupling by acting on brain Ang-II type 1 receptors (AT1R). In this review, we systemically analyzed the association between RAS and biological mechanisms of cognitive impairment, from the perspective of AT1R located in the central nervous system. Additionally, the possible contribution of brain AT1R to global cognition decline in COVID-19 cases will be discussed as well.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Decline in certain cognitive abilities is part of the process of normal aging, it includes reduced speed of acquiring new information and more difficulties in extracting recent memory. During aging, with some pathological conditions, the cognitive function could further decline, thus develop into mild cognitive impairment or dementia. On one hand, mild cognitive impairment is usually triggered during the early stages of dementia, and shares the same risk factors, including aging, genetic causes, and cardiovascular diseases. On the other, dementia is defined as a brain disease that manifests long-term and gradual loss of cognitive abilities, the symptoms of which are much more severe, and with no ideal treatment in most cases. The number of patients suffering from cognitive impairment is estimated to triplicate by 2050 as a result of aging of the world population and lack of disease-modifying treatments [1,2,3,4].
The most common type of dementia is Alzheimer’s disease (AD), whose etiology is actually still unclear. It has been believed, since quite a long time, that AD is caused by abnormal deposits of protein in brain, such as amyloid β (Aβ) and Tau, which are toxic to the neurons located in cognition-functioning areas. This well-known theory has been recently challenged more than ever, due to the unsatisfactory outcomes of the medications targeting amyloid in clinical trials [5]. One third of elderly people who have amyloid deposits in their brains are reported to have intact cognitive function [6]. In some cases, the impairment of neuronal function itself, has been considered as a possible etiology of AD [5]. Cerebrovascular dysfunction is another leading cause of dementia, involving vascular contribution to cognitive impairment and dementia. Together with AD, they account for around 80% of the total cases and usually combine mixed pathologies from each other [1]. In the last 2 decades, numerous studies have linked cardio- and cerebro-vascular risk factors to AD [7]. As a result, the roles of hypertension, diabetes, and dyslipidemia in the progression of AD have drawn more attention than ever. Notably, therapeutic strategies that target hypertension have been found to exhibit protective effects on cognitive function and ability to decrease the incidents of AD [8]. Commonly used anti-hypertensive drugs include thiazide diuretics, calcium channel blockers (CCB), angiotensin-converting enzyme inhibitors (ACEI), angiotensin-II (Ang-II) receptor blockers (ARB), and beta blockers (BB). Among all categories, drugs that directly target the renin-angiotensin system (RAS), such as ACEI and ARB, have shown more consistent and promising ability to preserve cognitive function, and to reduce AD-associated pathological alterations in both preclinical and clinical trials [9,10,11].
The RAS regulates fluid and electrolyte balance, as well as systemic vascular resistance. Beyond its many roles in maintaining homeostasis, the RAS is an essential regulator of blood pressure (BP) [12]. Upon decrease in renal blood flow, renin is generated by hydrolysis of its precursor-prorenin, then directly released into the circulation. In the presence of renin, angiotensinogen is converted into angiotensin I (Ang-I), and subsequently into Ang-II by the action of angiotensin-converting enzyme (ACE). Ang-II is a potent vasoconstrictive peptide and the main effector of the RAS, acting through its dominant receptor, Ang-II type 1 receptor (AT1R). Overactivation of the RAS has been proved to be involved in the development of multiple cardiovascular diseases and metabolic disorders, such as hypertension, heart failure, atherosclerosis, obesity, and diabetes [13, 14], many of which have been identified as risk factors for cognitive impairment. It has been shown that Ang-II impairs neuro-vascular coupling and cognitive function independently of its pressor response, highlighting the deleterious effect of RAS overactivation on memory [15]. Neuro-vascular coupling, also known as functional hyperemia, represents alterations in local perfusion that take place in response to changes in neuronal activity, thereby functioning as the basic unit of cerebral blood flow regulation. Compromised neuro-vascular coupling is also embedded in AD, where activated RAS is involved too [16]. A pathological study based of AD patients’ brains showed significant immuno-reactivities for ACE and Ang-II surrounding the vasculature in the cerebral cortex [17]. In addition to vessels, immuno-reactivities of ACE and Ang-II were also found to be higher in AD patients’ cortical neurons, suggesting that RAS may have a direct impact on neuronal functions as well.
Thereby in this review, the possible roles of brain AT1R, especially those neuronal residents, in the development of cognitive impairment will be summarized and discussed. In addition, recent related updates, including possible contribution to global cognition decline in Coronavirus Disease 2019 (COVID-19) cases will be discussed.
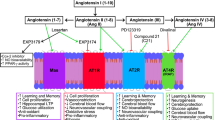
The Science of Angiotensin Receptors in Cognitive Impairment
In addition to the systemic RAS, there is also believed to have tissue specific expression and regulation of the RAS. Locally expressed RAS components have been found within multiple organs, including the kidneys, adrenal glands, adipose tissue, cardiovascular system, as well as brain, and have a variety of physiological or pathophysiological functions in association or independently of the systemic RAS [18]. Although it is still a controversial topic, there is growing body of evidence demonstrating the existence and pivotal functions of brain-specific RAS, which contains brain-locally expressed angiotensinogen, renin, (Pro)renin receptor, ACE, angiotensin-converting enzyme type 2 (ACE2)/angiotensin-(1-7) [Ang-(1-7)]/Mas axis, and most members of angiotensin receptors [19]. So far, three types of angiotensin receptors have been reported, and they are type 1, 2, and 4, with Ang-II or Ang-IV as their ligands. Notably, it has to be clarified that rodents express two AT1R subtypes, AT1aR and AT1bR; and within the brain, neurons are positive for both subtypes, while glia only contains AT1bR [20]. In brain vasculature, AT1R is responsible for the impairment of endothelial function and neuro-vascular coupling [15, 21, 22], through increasing oxidative stress. Similar to ACE, AT1R is also expressed in areas that are critical to learning and formation of memory [23], however, their direct role in the development of cognitive impairment is still unclear and quite debatable. As to the type 2 receptors, their expression is more plentiful in fetal tissues, and the ratio of AT2R and AT1R drops dramatically after birth, according to related autoradiographic studies [24]. Because AT2R and AT1R are coupled with different types of G protein, for AT2R is inhibitory G proteins (Gi), their effects are quite opposite (as shown in Fig. 1). In general, AT2R-mediated actions counter most of the effects induced by AT1R activation, including anti-proliferation, anti-inflammation, and vasodilation [25]. Therefore, AT2R-mediated signaling is considered to be neuroprotective. Selectively knocking down AT2R leads to impairments in spatial memory and exploratory behavior [26], while direct stimulation of AT2R by C21, effectively prevents post stroke cognitive impairment in aged animals [27]. However, considering that the affinity of Ang-II to AT1R is significantly higher, the effects of Ang-II/AT2R-mediated signaling are often masked, especially when the expression of AT1R is up-regulated under certain circumstances, such as cerebral ischemic injury and hypertension [28, 29]. In mice with AD-related mutations of human amyloid precursor protein (APP), C21 showed no significant improvement in memory, suggesting that AT2R might not be an ideal therapeutic target for APP-related cognitive and cerebrovascular deficits [30]. As to “AT4R”, it is actually considered as a binding site for Ang-IV and its existence is still controversial. There are recent studies reporting that Ang-IV/AT4R signaling is able to rescue cerebrovascular dysfunction and cognitive impairment in AD models, suggesting a potential new target for treatment of cognitive decline [31, 32]. Interestingly, in a study simultaneously targeting AT4R and AT1R, divalinal (selective AT4R blocker) countered losartan’s ability to rescue spatial learning and memory in APP mutated mice, suggesting that the beneficial effects of ARB on cognitive function could be resulting from the unmasked Ang-IV/AT4R signaling [31, 33].

The science of renin-angiotensin system in hypertension and cognitive impairment. Through different signaling pathways, Ang-II leads to specific effects on blood pressure (BP) regulation and hypertension-associated cognitive impairment. In general, Ang-II/AT1R pathway induces deleterious impact on cognitive function, promoting cognitive impairment, while the others exhibit protective effects at different levels
Regulatory RAS, the Yin of Y-Ang-II
In addition to its classic arm, the RAS is also composed of compensatory mechanisms preventing it from overactivation. By removing one amino acid, ACE2 transforms Ang-II into Ang-(1-7), thus turning the vasoconstrictor into a vasodilator peptide. As a major component of the regulatory RAS, Ang-(1-7) counters most of the effects of Ang-II, leading to nitric oxide (NO) release, reduction of oxidative stress, and preservation of baroreflex and autonomic balance. More recently, Ang-(1-7) has been found to protect against brain damage in related neurological disorders, such as brain ischemia, traumatic brain injury, and AD, with remarkable inhibition of microglial activation, reduction of inflammatory response, and protection of neurons from apoptosis [34,35,36]. Importantly, in the case of AD, the enzymatic activity of ACE2 in brain was found to be markedly reduced and associated with increases in Aβ and tau pathology. Unlike cortical ACE2, the activity of ACE2 in the cerebrospinal fluid of AD patients was increased, suggesting the reduction of tissue-bound ACE2 could be result from ectodomain shedding [37, 38]. The mechanism of ACE2 impairment during AD is still unknown, besides ubiquitination, the activity of membrane-bound ACE2 can also be decreased via a disintegrin and metalloprotease 17 (ADAM17)-mediated shedding. Within the central nervous system (CNS), ACE2 protein is mostly observed in the cell body of neurons. Our previous work shows that the enzymatic activity of ACE2 in cortical neurons is significantly decreased by the treatment with high glutamate concentrations, suggesting that excitotoxicity could impair the compensatory activity of ACE2 [39]. This glutamate-induced reduction of ACE2 activity was then demonstrated to be mediated through ADAM17, which reacts to oxidative stress and p38-mitogen-activated protein kinase (MAPK) pathway. Together with the Ca2+ influx via ionotropic glutamate receptors, all of those factors are within the typical signaling flows of excitotoxicity during AD.
Possible Roles of AT1R in Etiology of Cognitive Impairment
Ang-II/AT1R-mediated signaling impairs cognitive function independently of its pressor effect. Outside of the brain nuclei involved in cardiovascular regulation, AT1R is also expressed in various cell types within regions responsible for learning and memory, such as amygdala, hippocampus, and multiple cortical areas [23, 40,41,42]. Previous pre-clinical studies have demonstrated a significant neuroprotective effect of ARB on neuronal injury, decline of neurological performance, and cognitive loss [43]. Regarding Ang-II/AT1R signaling, there are several mechanisms should be considered in the etiology of cognitive impairment (Fig. 2).

Schematic illustration of how brain AT1R promotes neurodegeneration and cognitive impairment. Neurodegeneration and vascular dysfunction are not independent of each other. For one thing, they share similar signaling pathways, as shown in the yellow bubble. For another, during hypertension, vascular dysfunction could be a key trigger for neurodegeneration, while changed acetylcholine (Ach) tone and neuronal activity would further compromise the functional hyperemia Possible roles of AT1R in etiology of cognitive impairment
AT1R and Oxidative Stress
Increased oxidative stress in brain has been considered as an early feature of AD, and contributes to its pathogenesis [44]. Via AT1R, Ang-II evokes reactive oxygen species (ROS) production by NADPH oxidase (NOX), mitochondrial dysfunction, and reduction of endogenous antioxidant enzymes. That is to say, when Ang-II/AT1R signaling is upregulated during AD progression, it can increase oxidative stress and further deteriorate related pathological alterations in brain. In Tg2576 mice, NOX2-dependent pathways and ROS production in perivascular macrophages (PVM) were found to contribute to AD-associated cerebrovascular dysfunction [16]. In another study from the same group, the authors identified that AT1R located in PVM are responsible for increased production of ROS [15], thus promoting cerebrovascular dysfunction and cognitive impairment in mice with Ang-II-induced hypertension. In vascular adventitial fibroblasts, Ang-II decreased both the expression and activity of catalase, and further investigation revealed that Ang-II/AT1R-induced activation of ERK1/2 (extracellular-signal regulated kinase) pathway is involved in the downregulation of this endogenous antioxidant enzyme [45]. Beyond the vessels, in Neuro2A neuronal cells, Ang-II was also reported to downregulate the expression of Nrf2 (nuclear factor erythroid 2-related factor 2), a key regulator of the endogenous anti-oxidative response [46]. Reduced Nrf2 protein level is also associated with AD, as recent studies have shown significant therapeutic influence of Nrf2 activators on AD pathology both in vitro and in vivo [47]. Ang-II-mediated signaling pathway has been shown to be related to mitochondrial dysfunction and mitochondria-produced ROS in the neurons of rostral ventrolateral medulla (RVLM), by damaging the electron transport chain complexes [48]. In SH-SY5Y neuronal cells, Ang-II treatment was also found to induce aberrant mitochondrial fission, through protein kinase C (PKC) δ activation [49]. Moreover, Ang-II/AT1R signaling can affect NO production and bioavailability through mechanisms involving ACE2/Ang-(1-7) and AT2R, which have been partially discussed in the previous section. Notably, in the CNS, Ang-II induces both ACE2 internalization and ADAM17-mediated ACE2 shedding via AT1R; therefore, compromising the compensatory activity of ACE2 and promoting RAS overactivation [29, 50]. In such case, AT1R-mediated signaling in brain could facilitate cognitive impairment and deteriorate AD-related pathology by exacerbating oxidative stress that triggers neurodegeneration and neuronal apoptosis. According to a recent study, NO deficit in cerebral endothelial cells can reduce neuronal calpain nitrosylation and lead to tau phosphorylation by activating cyclin-dependent kinase 5 [51]. This might explain how central Ang-II administration induced Alzheimer-like tau phosphorylation in normal rat brains [52].
AT1R and Neuroinflammation
A growing body of literature has proposed brain inflammation as a driving force or causative factor of neuro-degradation [53]. In the brain of AD and vascular dementia, cytokines and inflammatory mediators, such as tumor necrosis factor α (TNFα), interleukin (IL)-1α, IL-1β, IL-6, cyclooxygenase (COX)-2, and nuclear factor (NF)-κB, are among the genes that are markedly upregulated [54]. Meanwhile, gliosis, represented by massively activated/increased astrocytes and microglia, is considered to be one of the hallmarks of AD progression [55]. Ang-II has been known to evoke pro-inflammatory responses in various brain cell types, especially in the vasculature and glial cells. In brain vessels, Ang-II promotes inflammation not only through activation of NF-κB-mediated genes, but also by suppression of peroxisome proliferator-activated receptors (PPAR) [56]. Therefore, Ang-II/AT1R signaling is able to stimulate NLRP3 inflammasome activation and increase the activity of caspase-1 and -3, thus promoting the release of pro-inflammatory cytokines [57]. Taking advantage of a novel transgenic mouse model, we previously showed that neuronal AT1R plays a vital role in the development of neuro-inflammation, by demonstrating its regulation of ADAM17 activation [29]. Besides ACE2, various inflammatory cytokines, chemokines, and adhesion molecules are the substrates of ADAM17, including TNFα, ICAM-1 (intercellular adhesion molecule 1), VCAM-1 (vascular adhesion molecule 1), and CX3CL1 (also known as Fractalkine) [58]. In deoxycorticosterone acetate (DOCA)-salt hypertension, the activity of ADAM17 is solely determined by AT1R-mediated signaling, which involves ROS and downstream MAPK signaling pathway (both ERK1/2 and p38-MAPK). In the same study, evidence of neuronal AT1R in modulating neuron-microglia communication was also provided. In FACS-sorted hypothalamic cells, DOCA-salt treatment significantly and selectively up-regulated the mRNA level of TNFα in non-neuron/non-astrocyte cells, while such alteration was not observed in mice with neuronal AT1R deficiency [29]. ADAM17 is considered as a player in the modulation of neuron-microglia communication, through shedding of ectodomains of ACE2 and CX3CL1. As mentioned earlier, the enzymatic activity of ACE2 is critical to the endogenous production of Ang-(1-7), which has direct inhibitory effect on microglia activation and related pro-inflammatory responses [59]. In addition, ADAM17 also cleaves the membrane-bound CX3CL1, a ‘turn off’ signal from neuron to microglia [60, 61], and increase the release of TNFα and IL-1β from microglia through soluble CX3CL1.
Does microglia express AT1R? The answer is controversial. It has been reported that microglial AT1R can cross-talk with TLR4 (Toll-like receptor 4) and facilitate ROS production in the hypothalamic paraventricular nucleus (PVN) [62]. However, there is no evidence so far showing that Ang-II activates microglia through directly acting on microglial AT1R, further investigations are certainly required to understand the role of microglial Ang-II/AT1R-induced pro-inflammatory response. As to astrocytes, the role of their AT1R is also underdeveloped. It is known that astrocytes actively participate in synaptic transmission and plasticity due to their essential role in glutamate turnover [63]. Recently, the role of astrocytes in CNS inflammation has been stepping into the spotlight. Multiple pro-inflammatory pathways have been associated with reactive astrocytes within the brain, including both cytokines and damage-associated molecular patterns [64]. In normal conditions, astrocytes express very few AT1R, while these receptors can be markedly up-regulated upon Ang-II stimulation [65, 66]. However, the specific role of AT1R expressed on astrocytes in the progression of CNS inflammation is still a question waiting for its answer. In a mouse model of multiple sclerosis, Ang-II/AT1R signaling is found to affect astrocytes by inducing cytokines release, such as tumor growth factor β (TGFβ) [67]. In the brains of hypertensive rats, treatments with ARB attenuated the hypertension-related astrocyte activation and neuroinflammation, by activating Wnt/β-catenin signaling [68]. But without manipulations specific to AT1R on astrocytes, it is still unclear whether Ang-II/AT1R signaling is responsible for the activation of astrocytes during pathological states. Selective ablation of astrocytic AT1R was found to attenuate sympatho-excitation as well, in a heart failure mouse model induced by myocardial infarction [65]. It was reported earlier that Ang-II inhibited glutamate transporter function via astrocytic AT1R, thereby increasing extracellular glutamate levels, which could support the activation of pre-sympathetic glutamatergic neurons and sympatho-excitation [69]. Later on, we identified the indispensable role of neuronal AT1R to the activation of pre-sympathetic glutamatergic neurons. Using selective deletion of AT1R on glutamatergic neurons, kidney-related pre-sympathetic glutamatergic neurons were no longer able to depolarize upon Ang-II stimulation, when compared to neurons with an intact neuronal AT1R signaling pathway [28].
AT1R and Neurodegeneration
Immuno-reactivity for ACE and Ang-II was found to be higher in cortical neurons of AD patients, suggesting that the RAS might support cognitive impairment directly through neuronal mechanisms [17]. Acute application of Ang-II can excite hippocampal CA1 neurons by abolishing incoming GABAergic inhibition [70]. However, this is considered as an anxiolytic effect, and exhibits no ability to enhance memory or reverse memory deficits. In rodents’ hippocampal slices, the administration of Ang-II was found to impair the induction of LTP (long-term potentiation) in the dentate gyrus, which is critical to spatial memory (or memory discrimination), and this effect can be blocked by losartan, an AT1R antagonist [42, 71]. In addition, Ang-II/AT1R signaling also suppresses LTP induction and LTD (long-term depression) stabilization in the lateral nucleus of the amygdala from rat brain slices [72, 73]. Both LTP and LTD are associated with the activity of synaptic transmission, in which the involvement of Ang-II/AT1R signaling highlights its impact on synaptic plasticity and cognitive functions. A more recent study reported that hippocampal p38-MAPK pathway is critical to the inhibition of LTP in Ang-II-dependent hypertensive mice [74], indicating again that the AT1R can impinge hippocampal neuronal function and memory through its downstream signaling.
Phosphatidylinositol-4,5-bisphosphate (PIP2), an important membrane phosphatidylinositol lipid, plays a critical role in maintaining normal synaptic transmission [75]. Blocking PIP2 recruitment can induce a significant inhibition in LTP, and mimic aging-related memory loss [76]. Notably, a significant reduction of PIP2 level in cortex has been associated with AD progression [77]. Phosphatidylinositol 4-kinase (PI4K) is one of the key enzymes for PIP2 bio-synthesis, and its activity was also found to be decreased, by ~ 50%, in the cortex of AD patients [78]. However, PI4K activity might not be significantly affected by aging, as supported by an enzymatic study in old rats [79], suggesting that reduced PI4K activity could be a hallmark for neurodegenerative disorders. It has been well-known that PIP2 is a second messenger downstream of Gq protein-coupled receptors, including AT1R. In cardiomyocytes, the activity of PI4K can be upregulated by Ang-II/AT1R through PKCs [80], therefore the cellular content of PIP2 increased following Ang-II treatment. However, the boost of PIP2 re-synthesis could be limited, and more investigation is necessary to determine how chronic brain RAS activation could affect the activity of PI4K as well as PIP2 turnover in neurons.
As mentioned earlier, AT1R and AT2R exhibit opposite effects from many perspectives. Unlike AT2R, AT1R activation supports cell proliferation and tissue growth [81]. Therefore, it is not surprising that Ang-II treatment can induce expression of brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family of growth factors [82, 83]. High salt diet induces the activation of vasopressin neurons and hypertension via BDNF, while removal of BDNF significantly attenuates Ang-II-induced hypertension [84, 85]. From this perspective, it looks like Ang-II/AT1R signaling could improve neuronal function through up-regulating BDNF. However, these findings relate to BDNF within regions that are predominantly responsible for cardiovascular regulation, but BDNF levels in cortical and hippocampal areas are actually downregulated in hypertension [86], suggesting that neurogenesis in those regions could be passively affected by brain RAS overactivation.
In addition to BDNF, Ang-II/AT1R signaling is also involved in other mechanisms that contribute to neurodegenerative alterations. In the etiology of AD, one of the theories is that impairment of cholinergic signaling in the brain underlies the development of AD [87]. Lesion of cholinergic neurons or inhibiting cholinergic pathways, lead to impaired neuro-vascular coupling, due to the reduced neuronal activity (demonstrated by the lower gamma oscillations of local field potential), indicating the critical role of neuro-activation in neuro-vascular coupling and the importance of acetylcholine (ACh) tone [88]. Both synthesis and release of ACh are markedly reduced during AD, and replenishing ACh levels pharmacologically show therapeutic benefits on AD-related cognition decline [88]. In both human temporal cortex and rat entorhinal cortex, Ang-II was found to significantly decrease the release of ACh, and antagonizing its binding sites was able to abolish this inhibitory effect, indicating that brain RAS activation has a negative impact on cholinergic neurons [89, 90]. Interestingly, ADAM17 can be a double-edge sword in the progression of AD. On one hand, ADAM17 cleaves ACE2, TNFα, and CX3CL1, promoting the development of neuroinflammation. On the contrary, ADAM17 is an α-secretase, which degrades APP into soluble APP α fragment, helping to ameliorate neurotoxic Aβ deposition [91]. Since the muscarinic type 1 receptor is thought to lead the beneficial effect of ADAM17 [92, 93], this good side of ADAM17 could be masked when ACh tone is low, while Ang-II level is high.
AT1R and Apoptosis
Although anti-proliferation is one of the roles of AT2R-mediated signaling, it is AT1R that participate in Ang-II-induced neuronal cell death. Ang-II was found to support the development of Parkinson’s disease by triggering apoptosis of dopaminergic neurons [94]. It was further revealed that the processes of autophagy and apoptosis induced by Ang-II treatment were the consequences of AT1R activation [95]. Enhanced oxidative stress through NOXs is thought to be one of the mechanisms underlying Ang-II/AT1R-induced neuronal apoptosis. Oxidative stress is able to damage all components within a living cell, including lipids, proteins, and DNA, and it is considered as a major contributor to neuronal loss during AD [44]. In hippocampal neural stem cells, Ang-II induces ROS production, which can be blocked by pre-treatment of losartan. ROS then activates AMPK (AMP‐activated protein kinase)-mediated apoptosis and autophagy signaling, eventually causing neuronal death [96]. Moreover, Ang-II/AT1R signaling was found to trigger apoptosis in dopaminergic neurons by undermining mitochondria permeability, therefore, this mitochondria-dependent apoptotic pathway is considered as a separate mechanism from the ROS-induced apoptosis [97]. In addition to oxidative stress, enhanced pro-inflammatory response also supports the apoptotic effect of Ang-II on neurons. For example, increased release of pro-inflammatory cytokines, such as TNFα, activate NF-κb-mediated NLRP3 inflammasome, thus leading to the process of neuronal apoptosis [98, 99]. In INS-1 islet β-cells, Ang-II has been demonstrated to induce apoptosis by activating NLRP3 inflammasome via AT1R [57]. Blocking AT1R via ARB has been shown to provide an anti-apoptotic effect in various disease models and related cell types, and this could be one of many factors that support the therapeutic use of ARB in treating neurodegenerative alterations in AD.
AT1R and BBB Disruption
The blood–brain barrier (BBB) is a tissue matrix located within the endothelium of cerebral capillaries and choroids plexus, controlling substances exchange between the CNS and periphery. Due to its protective role, the integrity of BBB is critical to the maintenance of a highly regulated brain internal homeostatic process. Compromised integrity of the BBB facilitates the entry of neurotoxic substances into the brain, and is often associated with increased inflammatory and immune responses [100]. In patients with early-stage AD, magnetic resonance imaging has showed global BBB leakage, which is associated with cognitive decline, highlighting the necessity of BBB integrity to normal cognitive function [101]. In a rat model of postoperative cognitive dysfunction, the laparotomy surgery was shown to increase Ang-II release in the hippocampus and impair BBB integrity via Ang-II/AT1R-mediated signaling pathways [102]. While in spontaneously hypertensive rats, normalization of brain Ang-II levels could decrease BBB leakage and improve its function within the autonomic brain areas [103]. Treatment with Ang-II was found to increase the permeability of cultured BBB micro-vessel endothelial cells in a time-dependent manner, and this Ang-II-induced leakage could be prevented by telmisartan [104], demonstrating the contributory role of Ang-II/AT1R signaling in the disruption of BBB integrity. A recent study reported that specific knockdown of cerebral endothelial AT1R completely prevented BBB disruption in the Ang-II-induced hypertension model, indicating that AT1R-mediated signaling in endothelial cells could initiate the BBB opening during the development of hypertension [22]. Moreover, HMGB-1 (high mobility group box 1), a possible downstream effector of Ang-II/AT1R signaling, was found to be involved in BBB disruption as well. An in vitro study has revealed that HMGB-1 induced a significant and concentration-dependent increase of dextran leak-out from the BBB and pre-treatment with anti-HMGB-1 antibody exhibited positive effect on keeping the integrity of BBB [105]. Additionally, a marked increase of serum HMGB-1 was observed in AD patients and positively correlated with the patients’ Aβ levels [105], suggesting that HMGB-1-mediated BBB disruption could be considered as a biomarker for AD progression. Due to the damage induced by Ang-II/AT1R signaling, the BBB disruption initiated by hypertension might contribute to the development of AD in late-life of hypertensive patients, and this could be one of the reasons why ARB exhibit the potential to reduce AD risk.
β-Arrestin-Biased AT1R Signaling
After binding to AT1R, Ang-II actually triggers multiple intracellular signaling pathways, in which the classic one is predominant due the excessive activation of Gαq/11. Among the additional mechanisms of Ang-II/AT1R signaling, AT1R-mediated β-arrestin recruitment, with its subsequent desensitization and internalization of AT1R, has gained significant attentions due to its potential as remedy for pathological manifestations involving an over-activated RAS [106]. β-Arrestins are ubiquitous scaffolding proteins that induce receptor desensitization, internalization, and other intracellular signaling mechanisms, so the β-arrestin-biased AT1R signaling is able to counter most of the classic effects of Ang-II. Between the two subtypes of β-arrestin identified so far, β-arrestin 2, instead of β-arrestin 1, is the mediator of the Gαq/11-independent responses during AT1R activation [107]. Ligands able to selectively stimulate a specific pathway over the others are called biased agonists, and biased activation of AT1R can be triggered by stretch or its biased agonists. So far, several biased agonists of AT1R, most of which are Ang-II analogs, have been synthesized, such as [Sar1, Ile4, Ile8]-Ang-II and TRV027 [106, 108]. TRV027 ([Sar1, D-Ala8]-Ang-II) serves as a β-arrestin-biased ligand of AT1R and antagonizes the classic Gαq/11 pathway of Ang-II/AT1R. We have previously reported the beneficial effects of central administration of TRV027 on hypertension, autonomic, and vascular function, as well as its ACE2-preserving ability in neurons [108], suggesting a potential of AT1R-biased agonists, especially TRV027, in treating hypertension-associated cognitive impairment and the vascular component of AD.
Sex Differences
The level of AT1R expression is upregulated throughout the developmental stage, and should exhibit no sex-related preference, according to the nature of AT1R gene. However, sex-specific modulation of BP, along with activity of the RAS, have been reported in various hypertension models. For example, mean arterial pressure rose faster in wildtype male than female mice after systemic Ang-II infusion, and this sex difference was found to be attenuated in mice with ACE2 deficiency [109]. Accordingly, it is possible that AT1R-mediated signaling can be affected by the compensatory activity of ACE2. However, despite the fact that ACE2 gene is located on the X chromosome, it is still controversial whether females have more ACE2 expression or higher activity [110]. Moreover, using global ACE2 knockout mice, we detected higher mRNA levels of ADAM17 and Mas1 in the hypothalamus, but no significant change in AT1R mRNA (unpublished data), suggesting that AT1R might not be significantly affected by ACE2 expression under normal circumstances. Interestingly, lower mRNA expression of AT1R and AT1R to AT2R ratio were detected in the aorta and mesenteric vessels of female mice, compared to males. Instead of ACE2, estrogen and its related signaling were considered to be a possible explanation [111]. Whether there is any difference in neuronal AT1R abundance between males and females is still unclear, despite that expression of AT1R in the vasculature does show sex differences. Prior to menopause, females are protected by their sex hormones, such as estrogen and the compensatory activity of ACE2. However, with aging and the loss of these protections, females are found to be more vulnerable to cardiovascular diseases, as well as to cognitive impairment. Since mid-life BP is considered to be critical to normal cognitive performance in late-life, it is important for women who are undergoing menopause to maintain adequate estrogen levels for protection from hypertensive stimuli.
An update on ARB in Clinical Trials
The association between hypertension and age-related cognitive impairment has now been accepted, and more recently, hypertension has also been linked to the development of AD [112]. Multiple clinical trials and meta-analysis have been performed to study whether anti-hypertensive medications show a beneficial effect on either treating dementia, including AD, or delaying its onset in the elderly [8,9,10]. Although ARB showed promising data in rodent models and in vitro, it is still controversial whether ARB have the ability to decrease AD risk in human, while CCB, ACEI, and diuretics were found to reach significant lower risk ratio, compared to 1 [8]. A recent study reported that treatment of losartan alone was not able to restore the functional hyperemia in hypertensive mice, and the treatment effect of losartan was found to be weaker than amlodipine, a CCB drug, closely aligning with clinical observations [113]. Possible reasons have been considered for this discrepancy between clinical trials and experimental models, with regards to the therapeutic potential of ARB in dementia. Due to the possible difficulties of ARB getting into the brain (related data are contradictory), there might not be enough amount of the active molecules reaching the neuro-vascular unit, where neuronal and peri-vascular AT1R can be targeted; or cardiovascular nuclei, such as PVN and RVLM, where RAS promotes sympatho-excitation via neuronal AT1R. As to the 3 other types of anti-hypertensive drugs, which were shown to significantly reduce AD incidents in the meta-analysis, they are thought to work effectively in the periphery as well, through reducing blood volume and disrupting the bio-synthesis of Ang-II in the circulation, unlike the ARB.
Possible Contribution of AT1R to COVID-19-Related Cognitive Dysfunction
As the major cell entry receptor for SARS-CoV-2, ACE2 has gained much attention since December 2019. As mentioned earlier in this review, ACE2 is a key player in the regulatory axis of the RAS. At the beginning of the pandemic, there was a debate regarding the role of RAS in both infection and progression of COVID-19, and whether its overactivation could prevent or promote SARS-CoV-2 infection. As more patients were diagnosed and hospitalized, an agreement has been reached, showing that RAS overactivation, or loss of ACE2 compensatory activity, would lead to more severe symptoms of infection, difficulties in recovery from the illness, and importantly worse outcomes [114, 115]. About 30–50% of the people hospitalized due to COVID-19 have hypertension and those hypertensive patients have a two-fold higher chance of dying from COVID-19 [116]. Treatments that target RAS, especially AT1R, have already been used to prevent infection-related tissue damage [117]. Notably, it has been suggested that the entry of SARS-CoV-2 into the cells via ACE2 binding is actually AT1R dependent [118]. In vitro, blocking AT1R with losartan or β-arrestin-biased AT1R ligand, TRV027, fully prevented the internalization of ACE2 activated by the binding of S1-spike protein [118]. In fact, TRV027 is currently under clinical trial to determine the role of RAS overactivation in COVID-19 infection, and recently announced data from 30 patients of the proof-of-concept study (COVRAS). The results showed that TRV027 was well-tolerated and provided initial evidence of its potential to improve biomarker and clinical endpoints regarding the disease severity and progression of COVID-19 (https://www.trevena.com/investors/press-releases/detail/279/trevena-announces-results-of-trv027-proof-of-concept-study).
Beyond serious respiratory and cardiovascular symptoms, negative influence of SARS-CoV-2 infection has also been reported in the CNS [119]. One of the most common, important, and definitely infection-related symptoms in COVID-19 patients is the loss of smell and taste. With the continuous mutation of the virus, as well as more and more people being vaccinated, the symptoms of the COVID-19 are becoming lighter and less severe, nevertheless, loss of smell and taste is still being reported in most infected people, as shown in the CDC data. Some would even suggest that loss of smell and taste better predicts the disease than other well-known symptoms such as fever and cough [120]. Although the underlying mechanism is still not clear, it has been suggested that loss of olfactory receptor neurons and brain infiltration of the olfactory centers could be considered to explain the dysfunction of smell [121], implying that SARS-CoV-2 could infect the CNS through a “quiet” manner, instead of causing major inflammation. At the beginning of the pandemic, we have identified the expression of ACE2 protein in neurons of the human brain, highlighting the potential of SARS-CoV-2 for neuro-invasion [122]. In addition to olfactory dysfunction, it has also been found that patients with recent SARS-CoV-2 infection could experience global cognition decline, associated with memory, attention, and executive function, particularly verbal fluency [123]. Indeed, SARS-CoV-2 is able to invade human neurons in vivo. Using human brain organoids, mice expressing human ACE2, and autopsies from deceased COVID-19 patients, the neuro-invasive capacity of SARS-CoV-2 has been detected and further studied [124]. It has also been shown that the infection by SARS-CoV-2 of brain neurons in humans is: (1) ACE2 dependent, (2) would induce metabolic changes of the cell, and (3) would lead to local vascular alteration. Therefore, RAS in the brain, especially vascular and neuronal AT1R, could be actively involved in the development of COVID-19-related cognitive impairment. Moreover, following the neuro-invasion and replication of the virus, compromised ACE2 compensatory activity resulting from the virus-associated internalization and the RAS overactivation, would be in favor of neuro-toxicity and neuro-inflammation, which could further contribute to the development of cognitive dysfunction in the COVID-19 patients (Fig. 3).

Possible contributions of brain AT1R in COVID-19-related global cognition decline. SARS-CoV-2 is neuro-invasive and this might lead to the onset of COVID-19-related global cognition decline. The entry of SARS-CoV-2 into the cells via ACE2 binding requires AT1R-mediated signaling, which has already been considered as a therapeutic target for treatment of COVID-19
Perspectives
The negative impact of RAS overactivation on cognitive impairment has now been accepted. Some argued that this was due to its pressor effect, and some suggested otherwise. In addition to the cognition-related neural circuits, neuro-vascular coupling, which is initiated by evoked neurons, is important in cognitive behavior as well. Therefore, the processes of learning and memory rely on neurons with normal activity and regulated activation, and a growing body of people start to consider that treating or reversing neurodegeneration is the key for the AD remedy. Accordingly, brain AT1R, especially those located in neurons, and associated signaling pathways, should also be considered as potential therapeutic targets. This could open a new avenue for a better understanding and treatment of cognitive impairment.
References
Benjamin, E. J., Blaha, M. J., Chiuve, S. E., Cushman, M., Das, S. R., Deo, R., de Ferranti, S. D., Floyd, J., Fornage, M., Gillespie, C., Isasi, C. R., Jimenez, M. C., Jordan, L. C., Judd, S. E., LacklanSd, D., Lichtman, J. H., Lisabeth, L., Liu, S., Longenecker, C. T., … Stroke Statistics, S. Heart disease and stroke statistics—2017 update: A report from the American Heart Association. Circulation, 135(10), e146–e603. https://doi.org/10.1161/CIR.0000000000000485
Merkler, A. E., & Iadecola, C. (2017). Rollercoaster blood pressure: An Alzheimer disease risk factor? Circulation, 136(6), 526–528. https://doi.org/10.1161/CIRCULATIONAHA.117.029618
Alzheimer’s, A. (2016). 2016 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, 12(4), 459–509. https://doi.org/10.1016/j.jalz.2016.03.001
Lennon, M. J., Makkar, S. R., Crawford, J. D., & Sachdev, P. S. (2019). Midlife hypertension and Alzheimer’s disease: A systematic review and meta-analysis. Journal of Alzheimer’s Disease (JAD), 71(1), 307–316. https://doi.org/10.3233/JAD-190474
Cummings, J., Lee, G., Ritter, A., Sabbagh, M., & Zhong, K. (2020). Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s & Dementia, 6(1), e12050. https://doi.org/10.1002/trc2.12050
Huang, S. Y., Zhu, J. X., Shen, X. N., Xu, W., Ma, Y. H., Li, H. Q., Dong, Q., Tan, L., & Yu, J. T. (2020). Prevalence of the preclinical stages of Alzheimer’s disease in cognitively intact older adults: The CABLE study. Journal of Alzheimer’s Disease (JAD), 75(2), 483–492. https://doi.org/10.3233/JAD-200059
Cechetto, D. F., Hachinski, V., & Whitehead, S. N. (2008). Vascular risk factors and Alzheimer’s disease. Expert Review of Neurotherapeutics, 8(5), 743–750. https://doi.org/10.1586/14737175.8.5.743
Ou, Y. N., Tan, C. C., Shen, X. N., Xu, W., Hou, X. H., Dong, Q., Tan, L., & Yu, J. T. (2020). Blood pressure and risks of cognitive impairment and dementia: A systematic review and meta-analysis of 209 prospective studies. Hypertension, 76(1), 217–225. https://doi.org/10.1161/HYPERTENSIONAHA.120.14993
Mishra, N., Mohan, D., Fuad, S., Basavanagowda, D. M., Alrashid, Z. A., Kaur, A., Rathod, B., Nosher, S., & Heindl, S. E. (2020). The association between hypertension and cognitive impairment, and the role of antihypertensive medications: A literature review. Cureus, 12(12), e12035. https://doi.org/10.7759/cureus.12035
Yang, W., Luo, H., Ma, Y., Si, S., & Zhao, H. (2021). Effects of antihypertensive drugs on cognitive function in elderly patients with hypertension: A review. Aging and Disease, 12(3), 841–851. https://doi.org/10.14336/AD.2020.1111
Lebouvier, T., Chen, Y., Duriez, P., Pasquier, F., & Bordet, R. (2020). Antihypertensive agents in Alzheimer’s disease: Beyond vascular protection. Expert Review of Neurotherapeutics, 20(2), 175–187. https://doi.org/10.1080/14737175.2020.1708195
Atlas, S. A. (2007). The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. Journal of Managed Care Pharmacy (JMCP), 13(8 Suppl B), 9–20.
Boustany, C. M., Bharadwaj, K., Daugherty, A., Brown, D. R., Randall, D. C., & Cassis, L. A. (2004). Activation of the systemic and adipose renin-angiotensin system in rats with diet-induced obesity and hypertension. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 287(4), R943–R949. https://doi.org/10.1152/ajpregu.00265.2004
Warnholtz, A., Nickenig, G., Schulz, E., Macharzina, R., Brasen, J. H., Skatchkov, M., Heitzer, T., Stasch, J. P., Griendling, K. K., Harrison, D. G., Bohm, M., Meinertz, T., & Munzel, T. (1999). Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: Evidence for involvement of the renin-angiotensin system. Circulation, 99(15), 2027–2033. https://doi.org/10.1161/01.cir.99.15.2027
Faraco, G., Sugiyama, Y., Lane, D., Garcia-Bonilla, L., Chang, H., Santisteban, M. M., Racchumi, G., Murphy, M., Van Rooijen, N., Anrather, J., & Iadecola, C. (2016). Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. The Journal of Clinical Investigation, 126(12), 4674–4689. https://doi.org/10.1172/JCI86950
Park, L., Uekawa, K., Garcia-Bonilla, L., Koizumi, K., Murphy, M., Pistik, R., Younkin, L., Younkin, S., Zhou, P., Carlson, G., Anrather, J., & Iadecola, C. (2017). Brain perivascular macrophages initiate the neurovascular dysfunction of Alzheimer abeta peptides. Circulation Research, 121(3), 258–269. https://doi.org/10.1161/CIRCRESAHA.117.311054
Savaskan, E., Hock, C., Olivieri, G., Bruttel, S., Rosenberg, C., Hulette, C., & Muller-Spahn, F. (2001). Cortical alterations of angiotensin converting enzyme, angiotensin II and AT1 receptor in Alzheimer’s dementia. Neurobiology of aging, 22(4), 541–546.
Nehme, A., Zouein, F. A., Zayeri, Z. D., & Zibara, K. (2019). An update on the tissue renin angiotensin system and its role in physiology and pathology. Journal of Cardiovascular Development and Disease. https://doi.org/10.3390/jcdd6020014
Nakagawa, P., & Sigmund, C. D. (2017). How Is the Brain Renin-Angiotensin System Regulated? Hypertension, 70(1), 10–18. https://doi.org/10.1161/HYPERTENSIONAHA.117.08550
Johren, O., Inagami, T., & Saavedra, J. M. (1995). AT1A, AT1B, and AT2 angiotensin II receptor subtype gene expression in rat brain. NeuroReport, 6(18), 2549–2552. https://doi.org/10.1097/00001756-199512150-00024
De Silva, T. M., Modrick, M. L., Grobe, J. L., & Faraci, F. M. (2021). Activation of the central renin-angiotensin system causes local cerebrovascular dysfunction. Stroke, 52(7), 2404–2413. https://doi.org/10.1161/STROKEAHA.121.034984
Santisteban, M. M., Ahn, S. J., Lane, D., Faraco, G., Garcia-Bonilla, L., Racchumi, G., Poon, C., Schaeffer, S., Segarra, S. G., Korbelin, J., Anrather, J., & Iadecola, C. (2020). Endothelium-macrophage crosstalk mediates blood-brain barrier dysfunction in hypertension. Hypertension, 76(3), 795–807. https://doi.org/10.1161/HYPERTENSIONAHA.120.15581
Parrish, J. N., Bertholomey, M. L., Pang, H. W., Speth, R. C., & Torregrossa, M. M. (2019). Estradiol modulation of the renin-angiotensin system and the regulation of fear extinction. Translational Psychiatry, 9(1), 36. https://doi.org/10.1038/s41398-019-0374-0
Gao, J., Chao, J., Parbhu, K. J., Yu, L., Xiao, L., Gao, F., & Gao, L. (2012). Ontogeny of angiotensin type 2 and type 1 receptor expression in mice. Journal of the Renin-Angiotensin-Aldosterone System (JRAAS), 13(3), 341–352. https://doi.org/10.1177/1470320312443720
Gao, L., & Zucker, I. H. (2011). AT2 receptor signaling and sympathetic regulation. Current Opinion in Pharmacology, 11(2), 124–130. https://doi.org/10.1016/j.coph.2010.11.004
Maul, B., von Bohlen und Halbach, O., Becker, A., Sterner-Kock, A., Voigt, J. P., Siems, W. E., Grecksch, G., & Walther, T. (2008). Impaired spatial memory and altered dendritic spine morphology in angiotensin II type 2 receptor-deficient mice. Journal of Molecular Medicine, 86(5), 563–571.
Ahmed, H. A., Ishrat, T., Pillai, B., Bunting, K. M., Vazdarjanova, A., Waller, J. L., Ergul, A., & Fagan, S. C. (2019). Angiotensin receptor (AT2R) agonist C21 prevents cognitive decline after permanent stroke in aged animals-A randomized double- blind pre-clinical study. Behavioural Brain Research, 359, 560–569. https://doi.org/10.1016/j.bbr.2018.10.010
Xu, J., Molinas, A. J. R., Mukerjee, S., Morgan, D. A., Rahmouni, K., Zsombok, A., & Lazartigues, E. (2019). Activation of ADAM17 (a disintegrin and metalloprotease 17) on glutamatergic neurons selectively promotes sympathoexcitation. Hypertension, 73(6), 1266–1274. https://doi.org/10.1161/HYPERTENSIONAHA.119.12832
Xu, J., Sriramula, S., Xia, H., Moreno-Walton, L., Culicchia, F., Domenig, O., Poglitsch, M., & Lazartigues, E. (2017). Clinical relevance and role of neuronal AT1 receptors in ADAM17-mediated ACE2 shedding in neurogenic hypertension. Circulation Research, 121(1), 43–55. https://doi.org/10.1161/CIRCRESAHA.116.310509
Royea, J., Lacalle-Aurioles, M., Trigiani, L. J., Fermigier, A., & Hamel, E. (2020). AT2R’s (Angiotensin II type 2 receptor’s) role in cognitive and cerebrovascular deficits in a mouse model of Alzheimer disease. Hypertension, 75(6), 1464–1474. https://doi.org/10.1161/HYPERTENSIONAHA.119.14431
Royea, J., Zhang, L., Tong, X. K., & Hamel, E. (2017). Angiotensin IV receptors mediate the cognitive and cerebrovascular benefits of losartan in a mouse model of Alzheimer’s disease. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 37(22), 5562–5573. https://doi.org/10.1523/JNEUROSCI.0329-17.2017
Royea, J., Martinot, P., & Hamel, E. (2020). Memory and cerebrovascular deficits recovered following angiotensin IV intervention in a mouse model of Alzheimer’s disease. Neurobiology of Disease, 134, 104644. https://doi.org/10.1016/j.nbd.2019.104644
Wayner, M. J., Armstrong, D. L., Phelix, C. F., Wright, J. W., & Harding, J. W. (2001). Angiotensin IV enhances LTP in rat dentate gyrus in vivo. Peptides, 22(9), 1403–1414. https://doi.org/10.1016/s0196-9781(01)00475-2
Zheng, J., Li, G., Chen, S., Bihl, J., Buck, J., Zhu, Y., Xia, H., Lazartigues, E., Chen, Y., & Olson, J. E. (2014). Activation of the ACE2/Ang-(1-7)/Mas pathway reduces oxygen-glucose deprivation-induced tissue swelling, ROS production, and cell death in mouse brain with angiotensin II overproduction. Neuroscience, 273, 39–51. https://doi.org/10.1016/j.neuroscience.2014.04.060
Jiang, T., Gao, L., Lu, J., & Zhang, Y. D. (2013). ACE2-Ang-(1-7)-Mas axis in brain: A potential target for prevention and treatment of ischemic stroke. Current Neuropharmacology, 11(2), 209–217. https://doi.org/10.2174/1570159X11311020007
Xu, P., Sriramula, S., & Lazartigues, E. (2011). ACE2/ANG-(1–7)/Mas pathway in the brain: the axis of good. American Journal of Physiology Regulatory, Integrative and Comparative Physiology, 300(4), 804–817. https://doi.org/10.1152/ajpregu.00222.2010
Kehoe, P. G., Al Mulhim, N., Zetterberg, H., Blennow, K., & Miners, J. S. (2019). Cerebrospinal fluid changes in the renin-angiotensin system in Alzheimer’s disease. Journal of Alzheimer’s Disease (JAD), 72(2), 525–535. https://doi.org/10.3233/JAD-190721
Kehoe, P. G., Wong, S., Al Mulhim, N., Palmer, L. E., & Miners, J. S. (2016). Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-beta and tau pathology. Alzheimer’s Research & Therapy, 8(1), 50. https://doi.org/10.1186/s13195-016-0217-7
Xu, J., Sriramula, S., & Lazartigues, E. (2018). Excessive glutamate stimulation impairs ACE2 activity through ADAM17-mediated shedding in cultured cortical neurons. Cellular and Molecular Neurobiology. https://doi.org/10.1007/s10571-018-0591-8
Huang, Z., Ohno, N., Terada, N., Saitoh, Y., Chen, J., & Ohno, S. (2013). Immunohistochemical detection of angiotensin II receptors in mouse cerebellum and adrenal gland using “in vivo cryotechnique.” Histochemistry and Cell Biology, 140(4), 477–490. https://doi.org/10.1007/s00418-013-1084-y
Basmadjian, O. M., Occhieppo, V. B., Marchese, N. A., Silvero, C. M., Becerra, M. C., Baiardi, G., & Bregonzio, C. (2021). Amphetamine induces oxidative stress, glial activation and transient angiogenesis in prefrontal cortex via AT1-R. Frontiers in Pharmacology, 12, 647747. https://doi.org/10.3389/fphar.2021.647747
Denny, J. B., Polan-Curtain, J., Wayner, M. J., & Armstrong, D. L. (1991). Angiotensin II blocks hippocampal long-term potentiation. Brain Research, 567(2), 321–324. https://doi.org/10.1016/0006-8993(91)90812-A
Villapol, S., & Saavedra, J. M. (2015). Neuroprotective effects of angiotensin receptor blockers. American Journal of Hypertension, 28(3), 289–299. https://doi.org/10.1093/ajh/hpu197
Nunomura, A., Castellani, R. J., Zhu, X., Moreira, P. I., Perry, G., & Smith, M. A. (2006). Involvement of oxidative stress in Alzheimer disease. Journal of neuropathology and Experimental Neurology, 65(7), 631–641. https://doi.org/10.1097/01.jnen.0000228136.58062.bf
Yang, W., Zhang, J., Wang, H., Gao, P., Singh, M., Shen, K., & Fang, N. (2011). Angiotensin II downregulates catalase expression and activity in vascular adventitial fibroblasts through an AT1R/ERK1/2-dependent pathway. Molecular and Cellular Biochemistry, 358(1–2), 21–29. https://doi.org/10.1007/s11010-011-0915-1
Ma, A., Gao, L., Wafi, A. M., Yu, L., Rudebush, T., Zhou, W., & Zucker, I. H. (2020). Overexpression of central ACE2 (angiotensin-converting enzyme 2) attenuates the pressor response to chronic central infusion of Ang II (Angiotensin II): A potential role for Nrf2 (nuclear factor [erythroid-derived 2]-like 2). Hypertension, 76(5), 1514–1525. https://doi.org/10.1161/HYPERTENSIONAHA.120.15681
Uruno, A., Matsumaru, D., Ryoke, R., Saito, R., Kadoguchi, S., Saigusa, D., Saito, T., Saido, T. C., Kawashima, R., & Yamamoto, M. (2020). Nrf2 suppresses oxidative stress and inflammation in app knock—in Alzheimer’s disease model mice. Molecular and Cellular Biology. https://doi.org/10.1128/MCB.00467-19
Chan, S. H., Tai, M. H., Li, C. Y., & Chan, J. Y. (2006). Reduction in molecular synthesis or enzyme activity of superoxide dismutases and catalase contributes to oxidative stress and neurogenic hypertension in spontaneously hypertensive rats. Free Radical Biology & Medicine, 40(11), 2028–2039. https://doi.org/10.1016/j.freeradbiomed.2006.01.032
Qi, X., Disatnik, M. H., Shen, N., Sobel, R. A., & Mochly-Rosen, D. (2011). Aberrant mitochondrial fission in neurons induced by protein kinase C{delta} under oxidative stress conditions in vivo. Molecular Biology of the Cell, 22(2), 256–265. https://doi.org/10.1091/mbc.E10-06-0551
Deshotels, M. R., Xia, H., Sriramula, S., Lazartigues, E., & Filipeanu, C. M. (2014). Angiotensin II mediates angiotensin converting enzyme type 2 internalization and degradation through an angiotensin II type I receptor-dependent mechanism. Hypertension, 64(6), 1368–1375. https://doi.org/10.1161/HYPERTENSIONAHA.114.03743
Faraco, G., Hochrainer, K., Segarra, S. G., Schaeffer, S., Santisteban, M. M., Menon, A., Jiang, H., Holtzman, D. M., Anrather, J., & Iadecola, C. (2019). Dietary salt promotes cognitive impairment through tau phosphorylation. Nature, 574(7780), 686–690. https://doi.org/10.1038/s41586-019-1688-z
Tian, M., Zhu, D., Xie, W., & Shi, J. (2012). Central angiotensin II-induced Alzheimer-like tau phosphorylation in normal rat brains. FEBS Letters, 586(20), 3737–3745. https://doi.org/10.1016/j.febslet.2012.09.004
Block, M. L., & Hong, J. S. (2005). Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Progress in neurobiology, 76(2), 77–98. https://doi.org/10.1016/j.pneurobio.2005.06.004
Corriveau, R. A., Bosetti, F., Emr, M., Gladman, J. T., Koenig, J. I., Moy, C. S., Pahigiannis, K., Waddy, S. P., & Koroshetz, W. (2016). The science of vascular contributions to cognitive impairment and dementia (VCID): A framework for advancing research priorities in the cerebrovascular biology of cognitive decline. Cellular and Molecular Neurobiology, 36(2), 281–288. https://doi.org/10.1007/s10571-016-0334-7
Cai, Z. Y., Yan, Y., & Chen, R. (2010). Minocycline reduces astrocytic reactivation and neuroinflammation in the hippocampus of a vascular cognitive impairment rat model. Neuroscience Bulletin, 26(1), 28–36. https://doi.org/10.1007/s12264-010-0818-2
Tham, D. M., Martin-McNulty, B., Wang, Y. X., Wilson, D. W., Vergona, R., Sullivan, M. E., Dole, W., & Rutledge, J. C. (2002). Angiotensin II is associated with activation of NF-kappaB-mediated genes and downregulation of PPARs. Physiological Genomics, 11(1), 21–30. https://doi.org/10.1152/physiolgenomics.00062.2002
Wang, J., Feng, Y., Huo, H., Zhang, X., Yue, J., Zhang, W., Yan, Z., & Jiao, X. (2019). NLRP3 inflammasome mediates angiotensin II-induced islet beta cell apoptosis. Acta Biochimica et Biophysica Sinica, 51(5), 501–508. https://doi.org/10.1093/abbs/gmz032
Xu, J., Mukerjee, S., Silva-Alves, C., Carvalho-Galvão, A., Cruz, J., Balarini, C., Braga, V., Lazartigues, E., França-Silva, M., & d. S. (2016). A disintegrin and metalloprotease 17 in the cardiovascular and central nervous systems. Frontiers in Physiology. https://doi.org/10.3389/fphys.2016.00469
Liu, M., Shi, P., & Sumners, C. (2016). Direct anti-inflammatory effects of angiotensin-(1-7) on microglia. Journal of Neurochemistry, 136(1), 163–171. https://doi.org/10.1111/jnc.13386
Sheridan, G. K., & Murphy, K. J. (2013). Neuron-glia crosstalk in health and disease: Fractalkine and CX3CR1 take centre stage. Open Biology, 3(12), 130181. https://doi.org/10.1098/rsob.130181
Garton, K. J., Gough, P. J., Blobel, C. P., Murphy, G., Greaves, D. R., Dempsey, P. J., & Raines, E. W. (2001). Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). Journal of Biological Chemistry, 276(41), 37993–38001. https://doi.org/10.1074/jbc.M106434200
Biancardi, V. C., Stranahan, A. M., Krause, E. G., de Kloet, A. D., & Stern, J. E. (2016). Cross talk between AT1 receptors and Toll-like receptor 4 in microglia contributes to angiotensin II-derived ROS production in the hypothalamic paraventricular nucleus. American Journal of Physiology. Heart and Circulatory Physiology, 310(3), H404–H415. https://doi.org/10.1152/ajpheart.00247.2015
Perea, G., & Araque, A. (2007). Astrocytes potentiate transmitter release at single hippocampal synapses. Science, 317(5841), 1083–1086. https://doi.org/10.1126/science.1144640
Giovannoni, F., & Quintana, F. J. (2020). The Role of Astrocytes in CNS Inflammation. Trends in Immunology, 41(9), 805–819. https://doi.org/10.1016/j.it.2020.07.007
Isegawa, K., Hirooka, Y., Katsuki, M., Kishi, T., & Sunagawa, K. (2014). Angiotensin II type 1 receptor expression in astrocytes is upregulated leading to increased mortality in mice with myocardial infarction-induced heart failure. American Journal of Physiology—Heart and Circulatory Physiology, 307(10), H1448-1455. https://doi.org/10.1152/ajpheart.00462.2014
Li, S., Zhou, C., Zhu, Y., Chao, Z., Sheng, Z., Zhang, Y., & Zhao, Y. (2021). Ferrostatin-1 alleviates angiotensin II (Ang II)-induced inflammation and ferroptosis in astrocytes. International Immunopharmacology, 90, 107179. https://doi.org/10.1016/j.intimp.2020.107179
Lanz, T. V., Ding, Z., Ho, P. P., Luo, J., Agrawal, A. N., Srinagesh, H., Axtell, R., Zhang, H., Platten, M., Wyss-Coray, T., & Steinman, L. (2010). Angiotensin II sustains brain inflammation in mice via TGF-beta. The Journal of Clinical Investigation, 120(8), 2782–2794. https://doi.org/10.1172/JCI41709
Bhat, S. A., Goel, R., Shukla, S., Shukla, R., & Hanif, K. (2018). Angiotensin receptor blockade by inhibiting glial activation promotes hippocampal neurogenesis via activation of Wnt/beta-catenin signaling in hypertension. Molecular Neurobiology, 55(6), 5282–5298. https://doi.org/10.1007/s12035-017-0754-5
Stern, J. E., Son, S., Biancardi, V. C., Zheng, H., Sharma, N., & Patel, K. P. (2016). Astrocytes contribute to angiotensin II stimulation of hypothalamic neuronal activity and sympathetic outflow. Hypertension, 68(6), 1483–1493. https://doi.org/10.1161/HYPERTENSIONAHA.116.07747
Hadjiivanova, C. H., & Georgiev, V. (1998). In vitro effect of angiotensin II on GABA release in rat hippocampus. Neuropeptides, 32(5), 431–434. https://doi.org/10.1016/s0143-4179(98)90067-1
Wayner, M. J., Armstrong, D. L., Polan-Curtain, J. L., & Denny, J. B. (1993). Role of angiotensin II and AT1 receptors in hippocampal LTP. Pharmacology, Biochemistry, and Behavior, 45(2), 455–464. https://doi.org/10.1016/0091-3057(93)90265-U
Tchekalarova, J., & Albrecht, D. (2007). Angiotensin II suppresses long-term depression in the lateral amygdala of mice via L-type calcium channels. Neuroscience Letters, 415(1), 68–72. https://doi.org/10.1016/j.neulet.2006.12.040
von Bohlen und Halbach, O., & Albrecht, D. (1998). Angiotensin II inhibits long-term potentiation within the lateral nucleus of the amygdala through AT1 receptors. Peptides, 19(6), 1031–1036. https://doi.org/10.1016/s0196-9781(98)00044-8
Dai, H. L., Hu, W. Y., Jiang, L. H., Li, L., Gaung, X. F., & Xiao, Z. C. (2016). p38 MAPK inhibition improves synaptic plasticity and memory in Angiotensin II-dependent hypertensive mice. Science and Reports, 6, 27600. https://doi.org/10.1038/srep27600
Koch, M., & Holt, M. (2012). Coupling exo- and endocytosis: An essential role for PIP(2) at the synapse. Biochimica et Biophysica Acta, 1821(8), 1114–1132. https://doi.org/10.1016/j.bbalip.2012.02.008
Trovo, L., Ahmed, T., Callaerts-Vegh, Z., Buzzi, A., Bagni, C., Chuah, M., Vandendriessche, T., D’Hooge, R., Balschun, D., & Dotti, C. G. (2013). Low hippocampal PI(4,5)P(2) contributes to reduced cognition in old mice as a result of loss of MARCKS. Nature Neuroscience, 16(4), 449–455. https://doi.org/10.1038/nn.3342
Stokes, C. E., & Hawthorne, J. N. (1987). Reduced phosphoinositide concentrations in anterior temporal cortex of Alzheimer-diseased brains. Journal of Neurochemistry, 48(4), 1018–1021. https://doi.org/10.1111/j.1471-4159.1987.tb05619.x
Zubenko, G. S., Stiffler, J. S., Hughes, H. B., & Martinez, A. J. (1999). Reductions in brain phosphatidylinositol kinase activities in Alzheimer’s disease. Biological Psychiatry, 45(6), 731–736. https://doi.org/10.1016/S0006-3223(98)00073-0
Zambrzycka, A. (2004). Aging decreases phosphatidylinositol-4,5-bisphosphate level but has no effect on activities of phosphoinositide kinases. Polish Journal of Pharmacology, 56(5), 651–654.
Xu, J. X., Si, M., Zhang, H. R., Chen, X. J., Zhang, X. D., Wang, C., Du, X. N., & Zhang, H. L. (2014). Phosphoinositide kinases play key roles in norepinephrine- and angiotensin II-induced increase in phosphatidylinositol 4,5-bisphosphate and modulation of cardiac function. Journal of Biological Chemistry, 289(10), 6941–6948. https://doi.org/10.1074/jbc.M113.527952
Forrester, S. J., Booz, G. W., Sigmund, C. D., Coffman, T. M., Kawai, T., Rizzo, V., Scalia, R., & Eguchi, S. (2018). Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiological Reviews, 98(3), 1627–1738. https://doi.org/10.1152/physrev.00038.2017
Szekeres, M., Nadasy, G. L., Turu, G., Supeki, K., Szidonya, L., Buday, L., Chaplin, T., Clark, A. J., & Hunyady, L. (2010). Angiotensin II-induced expression of brain-derived neurotrophic factor in human and rat adrenocortical cells. Endocrinology, 151(4), 1695–1703. https://doi.org/10.1210/en.2009-1060
Becker, B. K., Wang, H. J., Tian, C., & Zucker, I. H. (2015). BDNF contributes to angiotensin II-mediated reductions in peak voltage-gated K+ current in cultured CATH.a cells. Physiological Reports. https://doi.org/10.14814/phy2.12598
Choe, K. Y., Han, S. Y., Gaub, P., Shell, B., Voisin, D. L., Knapp, B. A., Barker, P. A., Brown, C. H., Cunningham, J. T., & Bourque, C. W. (2015). High salt intake increases blood pressure via BDNF-mediated downregulation of KCC2 and impaired baroreflex inhibition of vasopressin neurons. Neuron, 85(3), 549–560. https://doi.org/10.1016/j.neuron.2014.12.048
Zhang, Z., Zhang, Y., Wang, Y., Ding, S., Wang, C., Gao, L., Johnson, A., & Xue, B. (2019). Genetic knockdown of brain-derived neurotrophic factor in the nervous system attenuates angiotensin II-induced hypertension in mice. Journal of the Renin-Angiotensin-Aldosterone System (JRAAS), 20(1), 1470320319834406. https://doi.org/10.1177/1470320319834406
Lee, T. H., Yang, J. T., Kato, H., & Wu, J. H. (2006). Hypertension downregulates the expression of brain-derived neurotrophic factor in the ischemia-vulnerable hippocampal CA1 and cortical areas after carotid artery occlusion. Brain Research, 1116(1), 31–38. https://doi.org/10.1016/j.brainres.2006.07.117
Thathiah, A., & De Strooper, B. (2011). The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nature Reviews. Neuroscience, 12(2), 73–87. https://doi.org/10.1038/nrn2977
Lecrux, C., Sandoe, C. H., Neupane, S., Kropf, P., Toussay, X., Tong, X. K., Lacalle-Aurioles, M., Shmuel, A., & Hamel, E. (2017). Impact of altered cholinergic tones on the neurovascular coupling response to whisker stimulation. The Journal of Neuroscience: Official Journal of the Society for Neuroscience, 37(6), 1518–1531. https://doi.org/10.1523/JNEUROSCI.1784-16.2016
Barnes, J. M., Barnes, N. M., Costall, B., Horovitz, Z. P., & Naylor, R. J. (1989). Angiotensin II inhibits the release of [3H]acetylcholine from rat entorhinal cortex in vitro. Brain Research, 491(1), 136–143. https://doi.org/10.1016/0006-8993(89)90095-4
Barnes, J. M., Barnes, N. M., Costall, B., Horovitz, Z. P., Ironside, J. W., Naylor, R. J., & Williams, T. J. (1990). Angiotensin II inhibits acetylcholine release from human temporal cortex: Implications for cognition. Brain Research, 507(2), 341–343. https://doi.org/10.1016/0006-8993(90)90294-l
Slack, B. E., Ma, L. K., & Seah, C. C. (2001). Constitutive shedding of the amyloid precursor protein ectodomain is up-regulated by tumour necrosis factor-alpha converting enzyme. The Biochemical Journal, 357(Pt 3), 787–794. https://doi.org/10.1042/0264-6021:3570787
Davis, A. A., Fritz, J. J., Wess, J., Lah, J. J., & Levey, A. I. (2010). Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. The Journal of Neuroscience Official Journal of the Society for Neuroscience, 30(12), 4190–4196. https://doi.org/10.1523/JNEUROSCI.6393-09.2010
Caccamo, A., Oddo, S., Billings, L. M., Green, K. N., Martinez-Coria, H., Fisher, A., & LaFerla, F. M. (2006). M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron, 49(5), 671–682. https://doi.org/10.1016/j.neuron.2006.01.020
Zhao, H. R., Jiang, T., Tian, Y. Y., Gao, Q., Li, Z., Pan, Y., Wu, L., Lu, J., & Zhang, Y. D. (2015). Angiotensin II triggers apoptosis via enhancement of NADPH oxidase-dependent oxidative stress in a dopaminergic neuronal cell line. Neurochemical Research, 40(4), 854–863. https://doi.org/10.1007/s11064-015-1536-y
Gao, Q., Jiang, T., Zhao, H. R., Wu, L., Tian, Y. Y., Ou, Z., Zhang, L., Pan, Y., Lu, J., & Zhang, Y. D. (2016). Activation of autophagy contributes to the angiotensin II-triggered apoptosis in a dopaminergic neuronal cell line. Molecular Neurobiology, 53(5), 2911–2919. https://doi.org/10.1007/s12035-015-9177-3
Kim, M. S., Lee, G. H., Kim, Y. M., Lee, B. W., Nam, H. Y., Sim, U. C., Choo, S. J., Yu, S. W., Kim, J. J., Kim Kwon, Y., & Who Kim, S. (2017). Angiotensin II causes apoptosis of adult hippocampal neural stem cells and memory impairment through the action on AMPK-PGC1alpha signaling in heart failure. Stem Cells Translational Medicine, 6(6), 1491–1503. https://doi.org/10.1002/sctm.16-0382
Ou, Z., Jiang, T., Gao, Q., Tian, Y. Y., Zhou, J. S., Wu, L., Shi, J. Q., & Zhang, Y. D. (2016). Mitochondrial-dependent mechanisms are involved in angiotensin II-induced apoptosis in dopaminergic neurons. Journal of the Renin-Angiotensin-Aldosterone System (JRAAS). https://doi.org/10.1177/1470320316672349
Shen, K., Mao, Q., Yin, X., Zhang, C., Jin, Y., Deng, A., Gu, Z., & Chen, B. (2018). NLRP3 inflammasome activation leads to epileptic neuronal apoptosis. Current Neurovascular Research, 15(4), 276–281. https://doi.org/10.2174/1567202616666181122165540
Fann, D. Y., Lim, Y. A., Cheng, Y. L., Lok, K. Z., Chunduri, P., Baik, S. H., Drummond, G. R., Dheen, S. T., Sobey, C. G., Jo, D. G., Chen, C. L., & Arumugam, T. V. (2018). Evidence that NF-kappaB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Molecular Neurobiology, 55(2), 1082–1096. https://doi.org/10.1007/s12035-017-0394-9
Sweeney, M. D., Sagare, A. P., & Zlokovic, B. V. (2018). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nature Reviews. Neurology, 14(3), 133–150. https://doi.org/10.1038/nrneurol.2017.188
van de Haar, H. J., Burgmans, S., Jansen, J. F., van Osch, M. J., van Buchem, M. A., Muller, M., Hofman, P. A., Verhey, F. R., & Backes, W. H. (2016). Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology, 281(2), 527–535. https://doi.org/10.1148/radiol.2016152244
Li, Z., Mo, N., Li, L., Cao, Y., Wang, W., Liang, Y., Deng, H., Xing, R., Yang, L., Ni, C., Chui, D., & Guo, X. (2016). Surgery-induced hippocampal angiotensin II elevation causes blood–brain barrier disruption via MMP/TIMP in aged rats. Frontiers in Cellular Neuroscience, 10, 105. https://doi.org/10.3389/fncel.2016.00105
Buttler, L., Jordao, M. T., Fragas, M. G., Ruggeri, A., Ceroni, A., & Michelini, L. C. (2017). Maintenance of blood–brain barrier integrity in hypertension: a novel benefit of exercise training for autonomic control. Frontiers in Physiology, 8, 1048. https://doi.org/10.3389/fphys.2017.01048
Fleegal-DeMotta, M. A., Doghu, S., & Banks, W. A. (2009). Angiotensin II modulates BBB permeability via activation of the AT(1) receptor in brain endothelial cells. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism, 29(3), 640–647. https://doi.org/10.1038/jcbfm.2008.158
Festoff, B. W., Sajja, R. K., van Dreden, P., & Cucullo, L. (2016). HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. Journal of Neuroinflammation, 13(1), 194. https://doi.org/10.1186/s12974-016-0670-z
Tilley, D. G. (2011). G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circulation Research, 109(2), 217–230. https://doi.org/10.1161/CIRCRESAHA.110.231225
Wang, T., Li, G. Q., Zhang, H. P., Zhang, Y., & Li, Q. (2017). Overactivation of cannabinoid receptor type 1 in rostral ventrolateral medulla promotes cardiovascular responses in spontaneously hypertensive rats. Journal of Hypertension, 35(3), 538–545. https://doi.org/10.1097/HJH.0000000000001179
Carvalho-Galvao, A., Ogunlade, B., Xu, J., Silva-Alves, C. R. A., Mendes-Junior, L. G., Guimaraes, D. D., Cruz, J. C., Queiroz, T. M., Balarini, C. M., Braga, V. A., Filipeanu, C. M., Lazartigues, E., & Franca-Silva, M. S. (2018). Central administration of TRV027 improves baroreflex sensitivity and vascular reactivity in spontaneously hypertensive rats. Clinical Science (London, England). https://doi.org/10.1042/CS20180222
Ji, H., de Souza, A. M. A., Bajaj, B., Zheng, W., Wu, X., Speth, R. C., & Sandberg, K. (2020). Sex-specific modulation of blood pressure and the renin-angiotensin system by ACE (angiotensin-converting enzyme) 2. Hypertension, 76(2), 478–487. https://doi.org/10.1161/HYPERTENSIONAHA.120.15276
Jia, H., Yue, X., & Lazartigues, E. (2020). ACE2 mouse models: A toolbox for cardiovascular and pulmonary research. Nature Communications, 11(1), 5165. https://doi.org/10.1038/s41467-020-18880-0
Silva-Antonialli, M. M., Tostes, R. C., Fernandes, L., Fior-Chadi, D. R., Akamine, E. H., Carvalho, M. H., Fortes, Z. B., & Nigro, D. (2004). A lower ratio of AT1/AT2 receptors of angiotensin II is found in female than in male spontaneously hypertensive rats. Cardiovascular Research, 62(3), 587–593. https://doi.org/10.1016/j.cardiores.2004.01.020
Iadecola, C., & Gottesman, R. F. (2019). Neurovascular and cognitive dysfunction in hypertension. Circulation Research, 124(7), 1025–1044. https://doi.org/10.1161/CIRCRESAHA.118.313260
Koide, M., Harraz, O. F., Dabertrand, F., Longden, T. A., Ferris, H. R., Wellman, G. C., Hill-Eubanks, D. C., Greenstein, A. S., & Nelson, M. T. (2021). Differential restoration of functional hyperemia by antihypertensive drug classes in hypertension-related cerebral small vessel disease. The Journal of Clinical Investigation. https://doi.org/10.1172/JCI149029
Choksi, T. T., Zhang, H., Chen, T., & Malhotra, N. (2021). Outcomes of hospitalized COVID-19 patients receiving renin angiotensin system blockers and calcium channel blockers. American Journal of Nephrology, 52(3), 250–260. https://doi.org/10.1159/000515232
Nunez-Gil, I. J., Olier, I., Feltes, G., Viana-Llamas, M. C., Maroun-Eid, C., Romero, R., Fernandez-Rozas, I., Uribarri, A., Becerra-Munoz, V. M., Alfonso-Rodriguez, E., Garcia-Aguado, M., Elola, J., Castro-Mejia, A., Pepe, M., Garcia-Prieto, J. F., Gonzalez, A., Ugo, F., Cerrato, E., Bondia, E., … HOPE COVID-19 Investigators. (2021). Renin-angiotensin system inhibitors effect before and during hospitalization in COVID-19 outcomes: Final analysis of the international HOPE COVID-19 (Health Outcome Predictive Evaluation for COVID-19) registry. American Heart Journal, 237, 104–115. https://doi.org/10.1016/j.ahj.2021.04.001
Gao, C., Cai, Y., Zhang, K., Zhou, L., Zhang, Y., Zhang, X., Li, Q., Li, W., Yang, S., Zhao, X., Zhao, Y., Wang, H., Liu, Y., Yin, Z., Zhang, R., Wang, R., Yang, M., Hui, C., Wijns, W., … Li, F. (2020). Association of hypertension and antihypertensive treatment with COVID-19 mortality: A retrospective observational study. European Heart Journal, 41(22), 2058–2066. https://doi.org/10.1093/eurheartj/ehaa433
Gurwitz, D. (2020). Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Development Research, 81(5), 537–540. https://doi.org/10.1002/ddr.21656
Ogunlade, B. O., Lazartigues, E., & Filipeanu, C. M. (2021). angiotensin type 1 receptor-dependent internalization of SARS-CoV-2 by angiotensin-converting enzyme 2. Hypertension, 77(4), e42–e43. https://doi.org/10.1161/HYPERTENSIONAHA.120.16795
Johansson, A., Mohamed, M. S., Moulin, T. C., & Schioth, H. B. (2021). Neurological manifestations of COVID-19: A comprehensive literature review and discussion of mechanisms. Journal of Neuroimmunology, 358, 577658. https://doi.org/10.1016/j.jneuroim.2021.577658
Wagner, T., Shweta, F., Murugadoss, K., Awasthi, S., Venkatakrishnan, A. J., Bade, S., Puranik, A., Kang, M., Pickering, B. W., O’Horo, J. C., Bauer, P. R., Razonable, R. R., Vergidis, P., Temesgen, Z., Rizza, S., Mahmood, M., Wilson, W. R., Challener, D., Anand, P., … Soundararajan, V. (2020). Augmented curation of clinical notes from a massive EHR system reveals symptoms of impending COVID-19 diagnosis. eLife. https://doi.org/10.7554/eLife.58227
Butowt, R., & von Bartheld, C. S. (2020). Anosmia in COVID-19: Underlying mechanisms and assessment of an olfactory route to brain infection. The Neuroscientist: A Review Journal Bringing Neurobiology, Neurology and Psychiatry. https://doi.org/10.1177/1073858420956905
Xu, J., & Lazartigues, E. (2020). Expression of ACE2 in Human Neurons Supports the Neuro-Invasive Potential of COVID-19 Virus. Cellular and Molecular Neurobiology. https://doi.org/10.1007/s10571-020-00915-1
Daroische, R., Hemminghyth, M. S., Eilertsen, T. H., Breitve, M. H., & Chwiszczuk, L. J. (2021). Cognitive impairment after COVID-19—a review on objective test data. Frontiers in Neurology, 12, 699582. https://doi.org/10.3389/fneur.2021.699582
Song, E., Zhang, C., Israelow, B., Lu-Culligan, A., Prado, A. V., Skriabine, S., Lu, P., Weizman, O. E., Liu, F., Dai, Y., Szigeti-Buck, K., Yasumoto, Y., Wang, G., Castaldi, C., Heltke, J., Ng, E., Wheeler, J., Alfajaro, M. M., Levavasseur, E., … Iwasaki, A. (2021). Neuroinvasion of SARS-CoV-2 in human and mouse brain. The Journal of Experimental Medicine. https://doi.org/10.1084/jem.20202135
Acknowledgements
This project was supported by Xi’an Jiaotong University (TX6J007 to JX).
Author information
Authors and Affiliations
Contributions
XC, RY, and JX conceived and completed the general structure of the review. HW, QS, SY, JW, and JX drafted the manuscript. HW and HG prepared the schematic figures. FL and JX critically revised the work.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that there is no conflict of interest.
Ethical Approval
Not applicable.
Consent to Participate
All authors reviewed and approved the manuscript for publication.
Data Availability
Not applicable.
Code Availability
Not applicable.
Additional information
Handling Editor: Y. James Kang.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, H., Sun, Q., Yuan, S. et al. AT1 Receptors: Their Actions from Hypertension to Cognitive Impairment. Cardiovasc Toxicol 22, 311–325 (2022). https://doi.org/10.1007/s12012-022-09730-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-022-09730-0