
Abstract
Alcoholic cardiomyopathy (ACM) is a specific heart muscle disease found in individuals with a history of long-term heavy alcohol consumption. ACM is associated with a number of adverse histological, cellular, and structural changes within the myocardium. Several mechanisms are implicated in mediating the adverse effects of ethanol, including the generation of oxidative stress, apoptotic cell death, impaired mitochondrial bioenergetics/stress, derangements in fatty acid metabolism and transport, and accelerated protein catabolism. In this review, we discuss the evidence for such mechanisms and present the potential importance of drinking patterns, genetic susceptibility, nutritional factors, race, and sex. The purpose of this review is to provide a mechanistic paradigm for future research in the area of ACM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The term alcoholic cardiomyopathy (ACM) has been widely used to describe a specific heart muscle disease found in individuals with a history of long-term heavy alcohol (ethanol) consumption. Data from human and animal studies have revealed that within the myocardium, a number of adverse histological, cellular, and structural changes occur in response to and over the course of long-term heavy alcohol consumption. The most important unresolved question, however, relates to the primary injury/mechanism by which ethanol stimulates or initiates this array of adverse changes within the myocardium. Several interrelated mechanisms may include oxidative stress, apoptotic cell death, impaired mitochondrial bioenergetics/stress, derangements in fatty acid metabolism and transport, and accelerated protein catabolism. In this review, we discuss these mechanisms, as well as the potential importance of drinking patterns, genetic susceptibility, nutritional factors, ethnicity, and sex in the development of ACM.
Clinical Characteristics and Prevalence
Alcoholic cardiomyopathy is characterized by dilated left ventricle (LV), normal or reduced LV wall thickness, increased LV mass, and (in advanced stages) a reduced LV ejection fraction (<40 %). There are no specific immunohistochemical, immunological biomarkers or other criteria for the diagnosis of ACM. Therefore, the diagnosis of ACM is often considered presumptive and is usually one of exclusion. A key factor in ruling in ACM is a long-term history of heavy alcohol abuse in the absence of coronary artery disease or other cardiac conditions such as myocarditis. Contemporary cardiomyopathy classification schemas take into account molecular genetics and strictly emphasize cardiomyopathies as conditions involving the myocardium and not arising secondary to other cardiovascular conditions (i.e., atherosclerotic or coronary heart disease or alcohol abuse) [1]. Consequently, the use of other terminologies such as “alcoholic heart muscle disease” has been recommended, while others have suggested the term “alcohol muscle disease” because skeletal muscle changes are also present [2]. However, more than likely, the term ACM will continue to be used because ACM is characterized by a dilated LV phenotype, which is similar to genetically linked dilated cardiomyopathies.
The exact amount and duration of alcohol consumption associated with the development of ACM remain unknown [3]. Also, at least in humans, the point at which alcohol-induced abnormalities appear during the course of an individual’s lifetime of drinking is not well established and is highly individualized, suggesting either the protective or adverse interaction effects of genetic or lifestyle factors [3]. Though there is a lack of a specific alcohol dose–response myocardial injury relationship, some general conclusions can be made based on data derived from prospective studies enrolling subjects with a history of alcohol consumption. Consuming >90–120 g/day of alcohol (approximately 7–15 standard drinks per day) over a 5- to 15-year period is associated with changes in cardiac structure and function [4–9]. Those with a history of heavy alcohol consumption can present with diastolic or systolic dysfunction and may have no symptoms (preclinical and asymptomatic) or symptomatic ACM (signs and symptoms of heart failure). However, the duration of drinking appears to be an important variable, with longer durations more often associated with symptomatic ACM. For example, both Matthews et al. [10] and Urbano-Marquez et al. [11] found that among alcoholics consuming the same amount of alcohol, those with a longer duration of drinking had more heart failure symptoms [10, 11]. Another study examining the duration of alcohol use reported that LV dilation occurs within 5–9 years in those consuming >90 g alcohol/day for ≥4 days/week and precedes the development of diastolic dysfunction and LV enlargement, which occurred with drinking durations between 10 and 15 years [9]. Finally, in a hallmark, prospective, cross-sectional study, Urbano-Marquez et al. [11] reported that among alcoholics (n = 52), there was a significant negative correlation (r = −0.46, p < 0.001) between ejection fraction and lifetime alcohol intake and a positive correlation (r = 0.42, p < 0.001) between LV mass and lifetime alcohol consumption.
The exact prevalence of ACM remains elusive. This in part relates to how the diagnostic code for ACM [International Classification of Diseases (ICD)-9, 425.5 or ICD-10, I42.5] is often not listed individually, but rather is subsumed in other broader diagnostic categories such as “other forms of heart disease” [12] or other categories of alcohol-related diagnoses such as “alcohol dependence syndrome.” Among cases of individuals with the diagnosis of idiopathic dilated cardiomyopathy, the number of cardiomyopathy cases attributable to alcohol abuse varies between 23 and 40 % [4, 13, 14]. Recently, Hookana et al. [15] reported that among non-ischemic causes of sudden death in northern Finland (N = 2,661), ACM accounted for 19 %. It is possible that other factors, most notably drinking patterns and other lifestyle patterns, give rise to the variance in the incidence of ACM.
Oxidative Stress Contributes to ACM
There is evidence that within the myocardium, repeated and long-term alcohol consumption/exposure is associated with the development of oxidative stress either directly via stimulating the generation of free radicals or indirectly via activating other systems or hormones, such as angiotensin II. Oxidative stress is involved in the etiology of several other types of cardiovascular insults, such as ischemia–reperfusion injury and cardiomyopathies [16]. In the setting of alcohol-induced pathologies, others have suggested that excess free radical generation and oxidative stress may arise from several processes or mechanisms and these are noted in Table 1 [17].
Interestingly, many of the adverse cardiac intracellular effects found after chronic ethanol consumption in both contemporary and older investigations are typical of oxidative stress situations. These effects include myocyte loss and disarray [18], sarcoplasmic reticulum dysfunction [19–21] and changes in intracellular Ca2+ handling [22], depressed/disturbed mitochondrial function [20, 23–26], decreased myofibrillar ATPase activity [25, 27], decreased myofibrillar calcium sensitivity [28], contractile protein fragmentation and disarray [11, 29, 30], and fatty acid accumulation within intracellular organelles [31–33]. Accumulation of reactive oxygen species (ROS) can induce changes in intracellular organelles or processes through lipid peroxidation and/or other chemical modifications of structural proteins, cytoskeletal proteins, transport proteins, and enzymes [34].
In various biologic systems, oxidative stress can be measured or inferred by several biologic indexes that can include measurement of antioxidant enzymes (e.g., catalase, glutathione peroxidase) or scavenging proteins (e.g., glutathione), oxidative damage (e.g., increase/presence of protein carbonyl and conjugated diene levels), or the measurement of circulating oxidative products (e.g., isoprostanes) [34]. There are several reports of ethanol-induced changes in oxidative enzyme activity and levels. For example, others have reported evidence of increased catalase activity, an antioxidant enzyme important for stimulating the breakdown of ROS such as hydrogen peroxide into water and oxygen. Vikhert et al. [35] found an increase in catalase activity in autopsy heart samples from individuals with the diagnosis of ACM. Fahimi et al. [36] found both 5 and 18 weeks of an ethanol diet (Lieber DeCarli diet, 5 % w/v ethanol) was associated with significant increase in myocardial catalase levels and catalase activity as well as the number of peroxisomes in ethanol-fed rats compared to control rats. In another study, Kino [37] found an increase in myocardial catalase levels and catalase activity in ethanol-fed rats compared to control-fed rats (5–6 weeks of ethanol feeding, Lieber DeCarli diet, 5 % w/v). When compared to ethanol alone, rats that received ethanol concurrent with amino-1,2,4-triazole (AT) to inhibit catalase activity showed marked ultrastructural changes in the hearts, such as dilation of the sarcoplasmic reticulum, increased numbers of lysosomes, dehiscence of intercalated disks, and various mitochondrial alterations [37]. These findings suggest that increased catalase activity may be both an adaptive and protective response to ethanol in the heart.
Using a rodent model, Ribiere et al. [38] examined the effects of 4 weeks of ethanol consumption (10 % v/v in drinking water) on catalase, as well as different antioxidant enzymes such as copper/zinc (Cu, Zn) superoxide dismutase and selenium-glutathione peroxidase. These antioxidant enzymes are also important cellular defenses against free radical injury. Cu, Zn superoxide dismutase catalyzes the breakdown of superoxide anions. Ribiere et al. [38] found a significant increase in heart cytosolic Cu, Zn superoxide dismutase activity and no change in mitochondrial superoxide dismutase activity in ethanol-fed rats compared to control rats. Neither cytosolic catalase nor selenium-glutathione peroxidase activities were altered in the ethanol-fed rats compared to control rats. There was no evidence of lipid peroxidation as evidenced by no change in malondialdehyde levels. However, cytosolic and membrane fraction protein thiol levels were lower in the ethanol group compared to the control group, suggesting some degree of free radical attack on proteins [38]. Vendemiale et al. [39] found that 8 weeks of ethanol consumption (3 % v/v in drinking water) in rodents was associated with a significant decrease in heart cytosolic and mitochondria total glutathione levels and no change in cytosolic glutathione levels (mitochondrial levels of the latter were not measured) [39]. However, there were significant increases in cytosolic and mitochondrial levels of malondialdehyde and protein carbonyls and decrease in protein sulfhydryl levels in the ethanol-fed rats, indicating some degree of lipid peroxidation and protein damage, respectively [39]. Heart weight-to-body weight ratios were similar between groups; however, in the ethanol-fed group, there was increased accumulation of fibronectin in subepicardial and subendocardial layers of the heart.
Edes et al. [40] found increased levels of LV conjugated dienes and decreased glutathione levels in rats that received ethanol (27 % v/v, in drinking water) for 6 weeks. In this same study, the daily coadministration of vitamin E (10 mg/kg) or another antioxidant, cyanidanol-3 (300 mg/kg), prevented these changes. No ultrastructural changes were detected in the myocardium of alcohol-fed rats compared to controls [40]. In another study, this same group of investigators examined the effects of 15 weeks of ethanol consumption (15 % v/v in drinking water) in adult male turkeys [41]. This duration of alcohol feeding was associated with the development of a dilated cardiomyopathy, exemplified by increases in echocardiograph-derived measures of cardiac structure. They also found significant increases in cytosolic superoxide dismutase, catalase, and glutathione peroxidase enzyme activity.
More recently, using a variety of pharmacologic approaches, transgenic animal models, proteomic approaches, and plasma biomarkers of oxidative stress, investigators have established a more direct role for ethanol-induced oxidative stress in mediating the adverse effects of chronic alcohol consumption. Khanna et al. [42] demonstrated that inducible nitric oxide synthase (iNOS) is increased in cardiomyocytes isolated from rats exposed to 1 month of ethanol (13 g/day, Lieber DeCarli diet). Increased cardiac tissue iNOS levels can lead to the formation of superoxide and peroxynitrite [16]. Ethanol-fed animals had reduced systolic contractility and responses to adrenergic stimuli (isoproterenol) compared to control animals [42]. Pharmacological inhibition of iNOS with N G-monomethyl-l-arginine reversed this depression in systolic function and adrenergic signaling. Zhang et al. [22] found significant increases in myocardial protein carbonyl and superoxide levels in mice fed an ethanol (4 % v/v) diet for 6 weeks. These oxidative stress biomarkers corresponded to myocardial fibrosis development and decreases in fractional shortening and cardiac output. As detailed below, there was also evidence of apoptosis. Interestingly, these changes were prevented by the coadministration of the cytochrome P450 2E1 (CYP2E1) inhibitor, diallyl sulfate (100 mg/kg/day). Data from Jing et al. [30] also support a role for CYP2E1 activation and changes in oxidative stress markers, such as superoxide dismutase, glutathione peroxidase, and malondialdehyde protein levels. These investigators found that cardiac microsomal CYP2E1 activity was increased and corresponded to decreased superoxide dismutase and glutathione peroxidase activities and increased malondialdehyde levels in dogs that received alcohol (22 %) in their water once per day for 6 months [30]. No changes in heart weight-to-body weight ratios were found; however, the myocardium from ethanol-treated animals showed fibrosis and an irregular, disorganized myocyte pattern. All of these latter changes were prevented by the administration of either valsartan (angiotensin II receptor blocker, 5 mg/kg/day) or carnitine (antioxidant, 2 g/day), suggesting a role for angiotensin II and oxidative stress [30]. Using both pharmacologic and transgenic approaches, Tan et al. [43] have shown that the administration of a reactive oxygen species scavenger (a superoxide dismutase mimetic) to wild-type mice receiving ethanol (5.4 % w/v, Lieber DeCarli diet) for 2 months significantly reduced nitrative damage (i.e., 3-nitrotyrosine accumulation). In addition, there was also no evidence of nitrative damage in transgenic mice with knockout of the angiotensin I receptor (AT1-KO) fed ethanol for a similar amount of time [43]. In these studies, there was also evidence of ethanol-induced collagen and fibronectin accumulation, apoptotic cell death, and ventricular remodeling, all of which were blocked with the administration of the superoxide dismutase mimetic or not present in the AT1-KO ethanol-fed group [43].
Zhang et al. [44], using a wild-type FVB and transgenic overexpressing insulin-like growth factor (IGF-1) mouse model, found that 16 weeks of alcohol consumption in the control FVB mice resulted in decreased myocardial tissue superoxide dismutase 1 expression and increased superoxide production, which was accompanied by depressed intracellular systolic calcium transients and contractile function. However, these changes were not found in the transgenic animals overexpressing IGF-1, suggesting a protective and perhaps antioxidant effect of IGF-1 [44]. Using a proteomic approach, Fogle et al. [45] found that 16 weeks of ethanol consumption (40 % ethanol-agar blocks) in a rodent model was associated with marked reductions in the expression of myocardial levels of the antioxidant proteins, peroxiredoxin 5, antioxidant protein 2, and glutathione transferase 5.
There are several plasma biomarkers of oxidative stress, such as 8-isoprostane [34]. In a non-human primate model (cynomolgus and rhesus monkeys), following 12 months of alcohol consumption (3.3 ± 0.2 g/kg alcohol/day), Cheng et al. [46] found marked increases in plasma 8-isoprostane levels in animals consuming alcohol (124.8 ± 11 pg/ml) compared to controls (28 ± 5.1 pg/ml).
There are several summary points from the above studies. Every study reviewed provided some evidence of oxidative stress, as exemplified by a change in antioxidant substrate (e.g., glutathione) or enzyme activity [38–41] evidence of oxidative damage, such as the presence of conjugated dienes or protein carbonyls or 3-nitrotyrosine accumulation [22, 38–41, 43]. Except for Cheng et al. [46], nearly every study exposed animals for a short period of time (2–18 weeks). These data suggest that the onset of antioxidant defense mechanisms that attempt to protect the heart against oxidative damage appears to be initiated soon after ethanol drinking. Several studies concomitantly evaluated changes in cardiac structure or function, providing evidence of a potential cause-and-effect relationship between oxidative stress and the development of ACM. Also, data from several studies demonstrated a protective role of administered antioxidants, such as superoxide dismutase, vitamin E or IGF-1, or agents that inhibit CYP2E1 activity or inhibit angiotensin II, strongly supporting either a direct or an indirect role for ethanol-mediated oxidative stress in the heart. Table 2 summarizes evidence from studies reviewed herein as well as other studies using either pharmacologic or transgenic approaches to demonstrate cause-and-affect relationships between alcohol consumption and cardiac changes.
Apoptosis and ACM
Cell death, in particular apoptosis or programmed cell death, has been implicated in the development of ACM. Apoptosis is a consequence of oxidative stress and lipid peroxidation and in many organ systems, including the heart; myocyte loss or cell death may be an important component of organ dysfunction and pathology [50]. Others have shown that apoptosis is an important mechanism underlying ethanol-induced disorders such as fetal alcohol syndrome [51] and alcoholic liver disease [52]. There are several early reports from animal models of ACM and humans with ACM that support a role for myocyte loss as a mechanism underlying alcohol-induced cardiac dysfunction. Using quantitative morphometric analysis, Capasso et al. [18] found a significant loss (14 %) of myocytes in the left ventricle from rats fed ethanol in their drinking water for 8 months. Using contemporary apoptotic assay methods that include evaluation of DNA fragmentation and activation of apoptotic proteases, Tan et al. [43] investigated the effects of ethanol consumption in wild-type C57BL/6 mice and AT1-KO mice. After 2 months of ethanol consumption (Lieber DeCarli diet, 5.4 % w/v alcohol), there was evidence of apoptotic dell death [measured by transferase-mediated dUTP nick-end labeling (TUNNEL)] and caspase-3 activation in the wild-type mice; however, these changes were not found in the ethanol-fed AT1-KO mice [43]. In addition, also examined were cardiac protein kinase C-β1 expression and activation of nicotinamide adenine dinucleotide phosphate oxidase (NOX or NADPH oxidase) isoforms that are predominately expressed in the myocardium (NOX1, NOX2, and NOX4) [43]. NOX1 expression was not altered; however, protein kinase C-β1, NOX2, and NOX4 expression were increased in ethanol-fed wild-type mice, while no changes in these proteins were found in ethanol-fed AT1-KO mice. Corresponding echocardiographic data demonstrated that ethanol consumption in the wild-type group was associated with LV enlargement and dilation (e.g., increased heart weight/tibia length ratio and increased LV area of long axis in diastole and systole, respectively), whereas no changes in any echocardiographic parameters were found in the AT1-KO group [43]. Plasma and cardiac tissue angiotensin II levels were also significantly increased in both ethanol-fed groups. The finding that LV remodeling changes were not present in the AT1-KO ethanol-fed group, despite elevated plasma angiotensin II levels, strongly suggests a role for angiotensin II post-receptor signaling in mediating both remodeling changes and adverse cellular effects, such as apoptosis [43]. Activation of the AT1 receptor also involves PKC-β1-dependent activation of specific NOX isoforms. These mechanisms may be important in the early development of ACM, a time period when LV systolic function remains within normal limits, since even though there was a decrease in the ejection fraction in ethanol-fed wild-type mice, the ejection fraction (67 %) remained within normal limits [43].
There is also evidence of apoptosis occurring in individuals with long-term alcohol consumption and the diagnosis of ACM. In 1965, in a histopathological examination of hearts of patients with the diagnosis of ACM, Hibbs and colleagues reported that myocytes lost their cross-striated appearance and had pyknotic nuclei [31]. The latter, a reduction in the size of the nucleus, can be a characteristic of apoptosis. Fernández-Solà et al. [53] evaluated apoptosis in the hearts of individuals with long-term alcoholism (n = 19), those with long-standing hypertension (n = 20), and those with no known disease as control subjects (n = 7), all of which underwent a heart transplant. Apoptosis was evaluated with TUNNEL and immunohistochemistry to evaluate BAX (pro-apoptotic) and BCL-2 (antiapoptotic) protein expression. All the aforementioned indicators of apoptosis were significantly greater in alcoholic subjects (duration of ethanol intake 26 years) and hypertensive subjects compared to controls; however, no differences were found between alcoholic and hypertensive subjects [53]. These findings demonstrate that evidence of apoptosis was present and was of a similar magnitude in alcoholic subjects compared to those with hypertension. More recent findings from this group corroborate that apoptosis occurs in humans with a long-term history of heavy alcohol consumption [54].
Mitochondrial Bioenergetics/Stress
Among the many ethanol and heart studies, mitochondrial dysfunction or evidence of impaired bioenergetics has been a common finding. This is exemplified by either a change in mitochondrial ultrastructure and/or depressed indices of bioenergetics and oxidative phosphorylation. This is not surprising because mitochondria are a major target for free radical injury; however, dysfunctional mitochondria are not only less bioenergetically efficient, they can also generate increased amounts of ROS and are more likely to initiate apoptosis [55]. As reviewed below, it is possible that mitochondria serve as a site for ethanol-induced ROS generation, but also may be a target of ethanol-induced ROS injury. In particular, mitochondrial DNA is highly susceptible to oxidative stress because of the close proximity to ROS generation and lack of protective histones and DNA repair mechanisms compared to nuclear DNA [55].
In postmortem myocardial biopsies from human alcoholics, others have found microscopic evidence of myocardial mitochondrial enlargement and disorganization and degeneration of the cristae, characteristics of mitochondrial dysfunction [29]. In an early electron microcopy study of autopsy samples obtained from patients with the clinical diagnosis of ACM, Hibbs et al. [31] reported that mitochondria were swollen two to three times their normal size and had few to no cristae, while other mitochondria had deformed cristae. They also reported that myofibrils were disarrayed and fragmented (dissolution of myofilaments). Similarly, using autopsy samples obtained from patients with the clinical diagnosis of ACM, Vikhert et al. [35] found atrophy of myofibrils, dilation of the sarcoplasmic reticulum, increased number of mitochondria as well as mitochondriosis (small mitochondria closely packed together), and an increase in the number of lysosome-like structures. In contrast to the aforementioned studies, Mirό et al. [56] did not find evidence of mitochondrial injury in biopsy samples obtained from long-term ethanol drinkers (n = 10). Specifically, no changes in indices of mitochondrial respiratory chain complexes (i.e., complexes I, II, III, and IV activity) or evidence of lipid peroxidation were found. Differences among human studies may relate to a number of reasons, such as duration of drinking, degree of myocardial dysfunction, and experimental techniques. Interestingly, in the more contemporary study by Mirό et al. [56], ethanol drinkers had been receiving pharmacologic treatments such as beta-blockers, which (depending on the type) have antioxidant effects.
Changes in mitochondrial function have been reported from a number of animal studies and appear to occur in different animal species (dog and rat), as well as under different alcohol consumption paradigms (ethanol in water or liquid diet) and after variable durations of chronic ethanol consumption (6 weeks to 6 months). Oxidative phosphorylation is a key element of mitochondrial bioenergetics and reflects the mechanisms of energy transduction and respiratory control in the electron transport system. In terms of evaluating mitochondrial function, many investigators examined changes in the mitochondrial respiratory control index/ratio (RCI) in the presence of different substrates, mitochondrial oxygen consumption (Qo2) [representing n atoms of oxygen consumed per milligram of mitochondrial protein during rapid adenosine diphosphate phosphorylation (essentially state III respiration)], and the adenosine diphosphate-to-oxygen ratio. The RCI is the ratio of state III/IV respiration, and a decrease indicates an uncoupling of oxidation and phosphorylation. Detailed study design and findings related to investigations reporting changes in oxidative phosphorylation are summarized in Table 3.
Most investigators reported no change in the adenosine diphosphate-to-oxygen ratio; however, every study found a significant decrease in the RCI and Qo2 when using a nicotinamide dinucleotide (NAD)-linked substrate such as glutamate, α-ketoglutarate, or pyruvate [20, 22, 24–26, 57]. The specific finding of a decrease in state III respiration suggests that adenosine triphosphate (ATP) synthesis via oxidative phosphorylation is reduced by ethanol consumption. Recently, Hu et al. [33] found decreased myocardial ATP content levels along with decreased myocardial contractility (e.g., decreased ejection fraction and fractional shortening) in mice receiving ethanol (18 % v/v ethanol in drinking water) for 4 weeks. Although speculative, this reduction in ATP synthesis may be just enough to depress intracellular functions such as sarcoplasmic reticulum uptake of calcium, myofibrillar ATPase activity, and changes in cross-bridge cycling (Table 2).
Others have also found a significant decrease in intramitochondrial isocitrate dehydrogenase activity [20, 24]. Considering that high levels of nicotinamide adenine dinucleotide (NADH) inhibit isocitrate dehydrogenase activity and that RCI and state III respiration were depressed when using NAD-linked substrates, these observations are consistent with the idea of an imbalance between the reducing equivalents NAD and NADH, which in turn could be due to ethanol metabolism. Others have found an increased level of fatty acid ethyl esters in the alcoholic heart, which can attach to the mitochondria and disrupt mitochondria respiratory function [32].
In a proteomic analysis of mitochondrial proteins, Fogle et al. [45] found that 16 weeks of ethanol consumption (40 % ethanol-agar blocks) in rats was associated with changes in the expression of several different types of mitochondrial proteins (those encoded by both mitochondrial and nuclear DNAs). Several mitochondrial proteins, such as NADH dehydrogenase, isocitrate dehydrogenase, and long-chain-specific acyl-CoA dehydrogenase, were reduced, along with mitochondrial proteins associated with the citric acid cycle, citrate synthase, succinyl CoA synthase, and electron transport proteins, such as ATP synthase [45]. These provocative results suggest that ethanol may mediate changes in mitochondrial function at the genomic level.
In summary, there appears to be a number of ways in which mitochondrial perturbations could contribute to both the development and progression of ACM. However, it remains to be determined whether changes in mitochondrial function are cause or consequence. Because the cardiac myocyte relative to other cell types, including the hepatocyte, contains the highest volume of mitochondria, the critical mass of mitochondria negatively impacted by ethanol before significant mitochondrial dysfunction occurs may be higher than other tissues. Furthermore, it is now evident that mitochondria function in networks and that when mitochondria become damaged, their function can possibly be restored by fusion with neighboring mitochondria [55]. Also, others have suggested that in data from animal models of alcoholism, there is an interaction between chronic ethanol consumption and caloric deprivation in eliciting alterations in myocardial energy metabolism [58]. Many of the studies reviewed in this section were published more than 15 years ago and used measurements of respiratory states (1–IV) and respiratory control index ratios. These studies were performed in experimental conditions in which there may be multiple mitochondrial deficits and therefore need to be interpreted with caution. More research is required using more contemporary measures of mitochondrial function as well as determining changes in mitochondrial DNA.
Derangements in Fatty Acid Metabolism and Transport
Aberrations in fatty acid metabolism and uptake have been implicated in several alcohol-induced pathologies, such as hepatic steatosis and cirrhosis. The formation of fatty ethyl esters (FAEE), which are esterification products of fatty acids and ethanol, has also been implicated in ethanol-induced cell injury. More than 50 years ago, Lange and Sobel [59] reported an increase in FAEE content in postmortem myocardium samples obtained from both alcohol abusers (n = 2) and those with history of recent alcohol intoxication (n = 4). The concentration of FAEE was variable and ranged 17–115 μm; the lowest levels were found in those with history of alcohol abuse (19 and 28 μm). FAEE can attach to mitochondria and disrupt mitochondrial function, which might explain in part the adverse effects of ethanol on mitochondrial function. These results were intriguing at the time, but were limited due to the small sample size.
Subsequently, Beckemeier and Bora [32] postulated that increased FAEE production and potential changes in the enzyme FAEE synthase could be a mechanism contributing to ACM. It has also been recently demonstrated that patients (n = 39) admitted for substance abuse (i.e., acute alcohol abusers and alcoholics) had high plasma concentrations of FAEE [60]. Interestingly, plasma FAEE concentrations were correlated with blood alcohol concentrations (BAC) and were twofold higher in patients with a BAC greater than 300 mg %. Ethyl palmitate and ethyl oleate were the main FAEE detected in most subjects [60]. Although the study enrolled a small number of chronic alcohol abusers (n = 5), the chronic alcohol abusers had greater FAEE concentrations (mean 15,086 ng/ml) compared to acute alcohol abusers (4,250 ng/ml) [60]. Importantly, FAEE were either detected in trace amounts or not detectable in the plasma of control subjects. The idea that FAEE are cytotoxic is supported by the fact that increased tissue levels of FAEE are considered the mechanism underlying cell death induced by myocardial ablation: a procedure used to control and prevent the recurrences of cardiac arrhythmias [61].
Using a C56BL/6 J mouse model, Hu et al. [33] investigated the effects of chronic ethanol consumption on long-chain fatty acid (LCFA) uptake, LCFA gene expression (e.g., genes/proteins involved in esterification of triglycerides), LCFA transporters, and cardiac structure and function. Animals received different concentrations of ethanol in their drinking water (10, 14, 18 % v/v) for variable weeks (12, 8, and 4, respectively). In all three ethanol groups, compared to control groups, there was a significant increase in heart weight-to-body weight ratios. In terms of cardiac function and structure, significant decreases in fractional shortening and ejection fraction were found in all ethanol groups, but no other changes were found in other echocardiography-derived parameters between the alcohol and control groups. Intramyocardial lipid accumulation, which was in direct contact with the mitochondria, was found in all ethanol-fed groups and was significantly correlated with increased myocardial triglyceride content. LCFA uptake was evaluated in isolated cardiomyocytes obtained from ethanol-fed rats and was increased in a dose-dependent manner (i.e., greatest in 18 % ethanol group) [33]. Among the LCFA transport genes examined in all ethanol groups, increases were found in Cd36 and Scd-1 expressions. The Cd36 gene encodes for proteins involved with transport of long-chain fatty acids. The Scd-1 gene encodes for stearoyl-CoA desaturase 1, an enzyme that catalyzes the rate-limiting step in monounsaturated fatty acid synthesis. Genes encoding for enzymes important in de novo fatty acid synthesis (e.g., fatty acid synthase) and lipoprotein lipase were unchanged by ethanol consumption [33]. Finally, genes related to oxidative phosphorylation (e.g., those encoding for or part of different mitochondrial complexes, such as complex IV and cytochrome c reductase) and mitochondrial biogenesis and metabolism (e.g., peroxisome proliferator-activated receptor-γ coactivator-1α) were decreased in the ethanol groups [33]. Although only examined in the 18 % ethanol group, ATP production was significantly decreased (5.18 ± 0.54 pg/ml) compared to the control group (7.40 ± 0.64 pg/ml) [33].
Collectively, these findings indicate that ethanol consumption is associated with a dose-dependent increase in LCFA uptake and de novo synthesis and triglyceride accumulation. The latter corresponded to decreased myocardial ATP content levels and decreased myocardial contractility (e.g., decreased ejection fraction and fractional shortening). Collectively, these more recent findings of Hu et al. [33] in a mouse model corroborate those of others [32, 60] and support a toxic role for altered fat metabolism and transport.
Accelerated Protein Catabolism and Autophagy
Others have demonstrated that long-term ethanol administration decreases myocardial protein expression and synthesis and accelerates protein degradation, suggesting that these alterations may represent a key pathophysiologic mechanism underlying the adverse effects of ethanol [62]. Histopathological examination of hearts from individuals with the diagnosis of ACM has revealed contractile protein loss, fragmentation, and disarray, supporting the concept of altered protein physiology/composition [11, 29, 31]. Using a mass spectrometric-based proteomic analysis, Fogle et al. [45] examined the effects of 16 weeks of ethanol consumption on rat cardiac muscle protein expression. Interestingly, protein categories adversely affected included myofibrillar (~39 % decrease in α-myosin and actin), mitochondrial [~30–40 % reduction in mitochondrial dehydrogenases and electron transport proteins (ubiquinone, adenine nucleotide translocator)], glycolytic enzymes (~45–65 % reduction in glycogen phosphorylase, alpha enolase), and fatty acid metabolism proteins (~31–35 % decrease in fatty acid transport protein and long-chain fatty acid acyl-CoA ligase). These investigators also found decreases in peroxiredoxin 5, antioxidant protein 2, and glutathione transferase 5, important antioxidant enzymes. Proteins that were increased were signal transduction proteins such as tyrosine kinase (~2.1-fold increase) and mitogen-activated protein kinase phosphatase (~17.5-fold increase) [45].
Ethanol-induced decreases in myocardial protein synthesis may be mediated in part by decreases in mammalian (or mechanistic) target of rapamycin (mTOR) activity [63–65]. mTOR is a kinase that regulates cell growth, cell proliferation, cell motility, cell survival, protein synthesis, and transcription [66]. mTOR also senses cellular nutrient, oxygen, and energy [66]. Similar to other diseases associated with muscle atrophy, ethanol-induced reductions and dysregulation of mTOR activity may mechanistically be involved in reduced myocardial protein synthesis, ventricular wall thinning, and dilation.
Alterations in protein physiology/content can also be due to accelerated protein degradation. In skeletal muscle, ubiquitin E3 ligases, such as atrogin-1 and muscle RING finger 1 (MuRF1), accelerate protein breakdown and lead to muscle atrophy [67, 68]. Recently, Lang and Korzick [65] reported that 20 weeks of alcohol consumption in female Fischer 344 rats increased myocardial atrogin-1 and MuRF1 expression (e.g., messenger ribonucleic acid levels). In this same study, investigators found increased markers of autophagy, such as LC3B and autophagy-related gene 7 proteins and tumor necrosis factor α, along with a reduction in mTOR activity. Autophagy is a catabolic mechanism carried out by lysosomes and is important for the degradation of unnecessary or damaged intracellular proteins, therefore keeping the cell healthy. This mechanism is also important for cell and organism survival during stress and nutrient deprivation. Under the latter conditions, autophagy via degradation of macromolecular intracellular constituents becomes important in generating and recycling carbons and amino acids. However, there is evidence that there is enhanced autophagy in certain cardiac pathological conditions such as heart failure, cardiomyopathy, and cardiac hypertrophy, conditions in which there are increased levels of angiotensin II [69]. Interestingly, angiotensin II administration induces skeletal muscle atrophy in rodents, and mechanisms include increased expression of the E3 ligases atrogin-1/MuRF-1 [70].
Guo et al. [71] using both FVB wild-type and mice overexpressing the enzyme alcohol dehydrogenase (ADH transgenic mice) showed that 8 weeks of ethanol consumption was associated with increased myocardial markers of autophagy, such as autophagy-related 7 protein. However, increases in autophagy proteins were more robust and significant in the ethanol + ADH transgenic mice [71]. The idea that increased autophagy is a possible mechanism underlying the adverse myocardial effects of ethanol is very interesting, especially in light of the relationship between mTOR, a sensor of stress and nutrient deprivation, and the important role of autophagy and cell survival. Interestingly, activation of mTOR leads to inhibition of autophagy. As noted above, chronic alcohol exposure leads to a decrease in mTOR activity, which corresponds to increased markers of autophagy [65]. Furthermore, the autophagy pathway is rapidly upregulated during ATP depletion, mitochondrial dysfunction, and oxidative stress. Considering the latter, ethanol-mediated increases in autophagy may be an important mechanism underlying the adverse myocardial effects of ethanol.
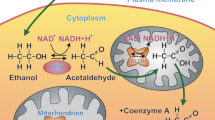
A summary of some of the potential cellular changes related to ethanol consumption is shown in Fig. 1. There may be more than one cellular event happening, and similar to other chronic health conditions, mechanisms may be synergistic and interrelated.

Summary of potential cellular changes related to ethanol. Ethanol-induced changes may be related to oxidative or non-oxidative pathways of ethanol metabolism. More than one mechanism may be activated that leads to the multitude of ethanol-induced changes in cellular proteins and cell function. As reviewed in text, data from pharmacologic and transgenic approaches have revealed an important role for oxidative stress and the hormone, angiotensin II. ANG II angiotensin II, ATP adenosine triphosphate, ATG atrogin, mTOR mammalian (or mechanistic) target of rapamycin (mTOR) activity, CYP2E1 cytochrome P450 2E1, FAEE fatty ethyl esters, NADPH oxidase/NOX—nicotinamide adenine dinucleotide phosphate oxidase, ROS reactive oxygen species
The Pattern of Drinking and Other Mediating Factors
In this section, we briefly discuss the patterns of drinking, specifically binge, as well as genetic variants in certain proteins/enzymes and variability in nutrition or dietary nutrients that may influence the occurrence of ACM. In addition, also reviewed are potential ethnic or sex difference influences.
Patterns of Drinking: Binge Drinking
Nearly, all the studies reviewed herein examined the effects of daily short-term to long-term ethanol consumption on the myocardium. The effects of a binge pattern of drinking or episodic heavy drinking on myocardial structure or function have not been widely investigated. At least in terms of coronary artery disease and ischemic heart disease, binge drinking and irregular heavy drinking are associated with risk for both [72]. Recently, Piano and colleagues examined the effects repeated episodes of binge/bender drinking on cardiac structure and hemodynamics [73]. Animals in the binge group received intragastric administration of 5 g ethanol//kg (30 % w/v solution) × 4 days (Monday, Tuesday, Wednesday, and Thursday), followed by no ethanol on Friday, Saturday, and Sunday. Animals were maintained on the protocol for 5 weeks. This model was developed because it simulates binge/bender drinking behavior, which is characterized by the consumption of large amounts of ethanol within a limited time frame, bringing the blood ethanol levels to >80 mg %, followed by a period of abstinence, which mimics the pattern of drinking in human beings [74]. Five weeks of binge drinking was not associated with changes in cardiac structure as assessed by echocardiography; however, binge drinking was associated with increased phosphorylation of myocardial p38 mitogen-activated protein kinase, which was blocked by administration of the beta-antagonist carvedilol. Interestingly, the binge drinking episodes were associated with transient increases in blood pressure that became progressively greater with repeated episodes of binge drinking. The greater pressor response at 4 and 5 weeks was attenuated but not completely blocked by carvedilol treatment. Binge drinking had no long-term effect on BP because baseline BP (before binge) was unchanged after 5 weeks. To our knowledge, this is the only study to examine the effects of binge drinking on the myocardium. Future studies are required that would examine longer periods of binge drinking and use other techniques to measure in vivo cardiac function.
Genetic Variants
Similar to other diseases, genetic variation in certain myocardial proteins (e.g., angiotensin II) or enzymes related to ethanol metabolism [e.g., alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH)] may influence susceptibility to ethanol-induced myocardial toxicity. To our knowledge, only one study to date has examined the relationship between certain genetic polymorphisms and ACM. Other studies have examined the potential interaction effect of gene variants and the occurrence of other CV risk factors and diseases, such as myocardial infarction. With respect to ACM, Kajander et al. [75] examined polymorphisms in certain genes that code for components of the renin–angiotensin–aldosterone system (RAAS). At autopsy, myocardium from male victims of sudden cardiac death (mean age 56 years) was evaluated for size (weight) and ventricular dimensions. In this study, RAAS alleles (e.g., insertion/deletion polymorphism of angiotensin-converting enzyme and the cytosine allele of −344 cytosine/thymidine polymorphism of aldosterone synthase) and polymorphisms in ADH, ALDH, and microsomal ethanol oxidation system were examined. Other data, such as history of CV risk factors and alcohol consumption, were determined by interviews with a spouse. Even though the daily lifetime alcohol dose was associated with increased myocardial weight and right ventricular size, there were no significant associations with any of the studied genetic polymorphisms [75].
Husemoen et al. [76], in a Danish Caucasian population (N = 1,216 men and women), examined the association between different levels of alcohol drinking and ADH and ALDH genetic variants and CV risk factors. In that study, among all levels of alcohol consumption (no drinking, heavy drinking, and binge drinking) and among the ADH and ALDH polymorphisms studied, no association was found with CV risk factors such as hypertension, increased low-density lipoprotein, or decreased high-density lipoprotein. In contrast, other studies examining ADH variants and CV risk factors among moderate drinkers enrolled in large epidemiologic studies (Physicians’ Health Study and Framingham Offspring Study) have found a reduced risk of myocardial infarction among moderate drinkers with a variant in the slow-oxidizing ADH genotype [77, 78].
Dietary Factors
Interestingly, many decades ago, ACM was thought to arise due to nutritional deficiency, specifically thiamine (vitamin B12). However, when alcoholic patients with ACM received thiamine therapy or other nutritional supplements, myocardial structural and functional changes were often not reversed. Although beyond the scope of this review, it is possible that certain dietary components and/or deficiencies may increase either the susceptibility or progression of ethanol-induced myocardial changes. Reinke et al. [79], using electron paramagnetic resonance (EPR) spectroscopy, found an increased EPR signal, representing elevated free radical generation, in the hearts of female Sprague–Dawley rats (140–150 g) fed a high-fat ethanol-containing diet compared to those fed a control or low-fat ethanol diet. Animals received either the 1982 formulation of the Lieber DeCarli diet (fat 35 % of total calories) or low-fat Lieber DeCarli diet (fat 12 %). Findings from this study suggested that the presence of a moderate-to-high amount of dietary fat increased the production of free radicals over low-fat ethanol-containing diets. Interestingly, the amount of fat deemed high (35 % of calories) is similar to the amount consumed by most Americans.
Others have examined the potential effects of micronutrient deficiencies (such as zinc) on ethanol-induced changes in the heart. Wang et al. [80] found evidence of ethanol-induced changes in mitochondrial structure that were more pronounced in a metallothionein knockout mouse model compared to wild-type mouse. Metallothionein binds zinc within the cell and is important for overall zinc homeostasis. In that study, zinc supplementation suppressed some of the ethanol-induced changes in both the metallothionein knockout mouse model and wild-type; however, ethanol-induced mitochondrial swelling and disorganization remained in both mouse groups.
Edes et al. [40] examined the effects of vitamin E supplementation on ethanol-induced changes in the ultrastructure of the myocardium and indices of oxidative stress and found increased levels of LV conjugated dienes and decreased glutathione levels in male rats that received ethanol (27 % v/v) in their drinking water for 6 weeks. In that study, the daily coadministration of vitamin E (10 mg/kg) or another antioxidant, cyanidanol-3 (300 mg/kg), prevented these changes [40].
Racial and Sex Differences
To date, no studies have examined potential ethnic differences in the occurrence or pathophysiology of ACM. However, two large epidemiologic studies analyzing data from two large national databases (the National Health and Nutrition Examination Survey I Epidemiologic Follow-Up Study and the Atherosclerosis Risk in Communities Study) have examined the cardioprotective effects of different levels of ethanol consumption and incidence of coronary heart disease between black and white individuals. Others have extensively described the J-shaped relationship between alcohol consumption and CV morbidity and mortality and all-cause mortality, with low-to-moderate alcohol consumption associated with lower CV morbidity and mortality and all-cause mortality. However, neither Fuchs et al. [81] nor Sempos et al. [82] found a positive association between ethanol consumption and incident coronary heart disease or all-cause mortality, respectively, for black men or women. Fuchs et al. [81] reported a positive association between ethanol consumption and incident coronary heart disease for black men [for a 13-g/day increment in ethanol consumption, adjusted hazard ratio (HR) 1.13, 95 % confidence interval (CI) 1.01, 1.28] and an inverse association for white men (HR 0.88, 95 % CI 0.79, 0.99). Sempos et al. [82] also did not find a beneficial effect for any level of alcohol consumption in African American men or women on all-cause mortality. However, Sempos et al. [82], after adjusting for systolic blood pressure and use of antihypertensive medications, found that the detrimental effect of alcohol consumption on all-cause mortality risk was reduced for men but not for women. Although the differences in the findings between black and white populations remain unknown, authors have postulated that differences could be attributable or confounded by lifestyle characteristics or drinking patterns among drinkers.
Sex Differences
Long-term heavy ethanol consumption is associated with many adverse medical consequences in both men and women. Ethanol affects women differently and imposes different medical risks compared to men. There are also important interactions with either the presence or absence of hormones such as estrogen. For example, research suggests that as little as one drink per day can increase the risk of breast cancer in some women, especially those who are postmenopausal or have a family history of breast cancer [83]. Some reports in human beings indicate that alcohol-dependent women develop an ACM after consuming less ethanol over a shorter period of time than do age-matched alcohol-dependent men [11, 84]. However, other data, in particular data from epidemiological studies of people with the diagnosis of dilated cardiomyopathy, suggest that women are not more vulnerable to ACM [13, 14].
Results from investigations in animal models have been equivocal. Using an isolated atrial preparation, Kennedy et al. [85] found no negative CV effects of long-term (6 months) ethanol consumption in female rats, whereas tension was significantly decreased in male rats. In a study using exclusively female rats and a Langendorff preparation to evaluate cardiac performance, Lochner et al. [86] found no negative effects of long-term ethanol consumption (18 months). Piano and colleagues examined the effects of long-term (8 months) alcohol consumption in adult male and female sham-operated (sham) and ovariectomized (OVX) Sprague–Dawley rats [87]. All ethanol groups showed echocardiographic evidence of ACM; however, more significant ethanol-elicited differences were found in male group compared to either the female sham or female OVX groups. In addition, the male ethanol group had significant reductions in in vivo measures of contractility, such as the maximum derivative of change in systolic pressure and preload recruitable stroke work [87]. Sex differences were also apparent in the pattern and degree of posterior and septal wall thickness changes, in that the male ethanol group had more posterior and septal wall thinning. Vary et al. [88], using echocardiography, examined the effects of long-term ethanol consumption (26 weeks) on cardiac structure and function in male and female rats and found that chronic alcohol consumption in males, but not in females, was associated with a thinning of the ventricular wall and intraventricular septum. The alterations in cardiac size in male rats occurred, in part, through a lowering of the protein content secondary to a diminished rate of protein synthesis. Fogle et al. [89] examined myocardial structural changes and myocardial protein expression in male and female rats, following 18 weeks of alcohol consumption. Except for a decrease in end-diastolic dimension, no changes in echocardiographic parameters were found between control and alcohol-fed female rats. In contrast, in the male ethanol group, decreases were found in left ventricular mass, stroke volume, cardiac output, and end-diastolic dimension compared to the control group. Using a proteomic approach, these investigators reported that changes in mitochondrial oxidative phosphorylation proteins, such as complexes I (NADH dehydrogenase), III (cytochrome b, c 1), IV (cytochrome c oxidase), and V (ATP synthase), were upregulated in the male ethanol group compared to male controls, whereas no changes (or tendency for downregulation) were found in the female ethanol group [89]. Troponin proteins (troponin C and I) were upregulated in the female ethanol group and downregulated in the male ethanol group, compared to their respective controls [89]. Interestingly, glutathione peroxidase expression and fatty acid oxidation enzymes, such as carnitine palmitoyltransferase 2 and 2,4-dienoyl CoA reductase, were downregulated in the male ethanol group, but no changes were found in the female group. These findings by Fogle et al. [89] support the idea that mitochondrial proteins (those encoded by mitochondrial and nuclear DNA), as well as enzymes involved in protecting against oxidative stress, and modulating fatty acid metabolism are altered by ethanol, supporting the idea that genetic alterations may also be mediating ethanol-induced changes in cardiac structure and function.
Finally, in an interesting study, Mackie et al. [90] examined the interaction effects of estrogen replacement therapy and alcohol consumption on endothelial progenitor cell mobilization in a female mouse model of acute myocardial infarction. Ethanol consumption (6 weeks) decreased the protective effects of estrogen on post-infarct repair and decreased endothelial progenitor cell mobilization and survival. Decreased endothelial progenitor cell mobilization and survival was associated with corresponding decreases in antiapoptotic and cell survival proteins (protein kinase B (Akt)/extracellular signal-regulated kinase signaling) and increases in pro-apoptotic proteins (c-Jun N-terminal kinase) [90]. In the area of ACM, understanding sex differences and potential interaction effects of estrogen remains an important area of inquiry.
Current Therapies for ACM
Treatment of ACM occurs when individuals present with signs and symptoms of heart failure (HF). Individuals with ACM will be treated based on whether they present with signs and symptoms of reduced ejection fraction HF (ejection fraction ≤40 %) or HF with preserved ejection fraction (ejection fraction ≥50 %). Those presenting with signs and symptoms of HF and an ejection fraction ≤40 % should receive an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker and a β-adrenergic blocker [91]. Depending on other signs and symptoms, treatment with diuretics or cardiac glycosides may be warranted. As reviewed above, there is considerable evidence from animal studies indicating that the renin–angiotensin system is a mechanistic component in the development of ACM. Others have reported that current guideline-directed therapy and inclusion of angiotensin-converting enzyme inhibitors are associated with improvement in LV function in individuals with the diagnosis of ACM [4, 13]. Although there is limited information, alcohol abstinence is recommended, since continued drinking despite optimal HF therapies is associated with poorer prognosis and increased cardiovascular mortality [4, 92]. In all patients with HF, regardless of HF etiology, alcohol consumption should be limited to low-to-moderate levels (e.g., no more than 1–2 standard drinks per day) and no one should drink every day [93]. In addition, comprehensive lifestyle management recommendations (incorporated as components of cardiac rehabilitation programs) including diets for weight management and exercise guidelines typically recommended for HF patients are indicated [94]. In terms of HF with preserved ejection fraction, there is no research to support specific therapies such as angiotensin-converting enzyme inhibitors. Treatment should be directed at controlling potential risk factors such as hypertension and the presence of other comorbidities, such as atrial fibrillation and diabetes.
Summary and Future Directions
In the USA, alcohol is the most used and abused substance. Results from the 2010 National Survey on Drug Use and Health indicate that more than half of all Americans greater than 12 years of age (131.3 million people) report being a current drinker of alcohol, and 6.7 % of Americans (16.9 million people) report drinking more than 5 drinks on each of 5 or more days in the past 30 days [95]. ACM is just one of many adverse consequences of long-term heavy alcohol consumption. More than likely, ACM prevalence is underestimated because often in clinical settings, a history of alcohol consumption is either not assessed or derived from self-report. Similar to other chronic health conditions, ACM may develop and evolve due to several interrelated mechanisms, such as oxidative stress, apoptotic cell death, impaired mitochondrial bioenergetics/stress, derangements in fatty acid metabolism and transport, and accelerated protein catabolism (Fig. 2). There are data from pharmacologic and transgenic animal studies that strongly suggest role for oxidative stress early in the development of ACM and which may come about either from ethanol metabolism or from activation of neurohormones such as angiotensin II. Genetic susceptibility, nutritional factors, ethnicity, and sex may moderate development of ACM. Moreover, in the setting of ACM, the potential effects of epigenetic mechanisms may be involved, such as methylation or acetylation of histone complexes associated with deoxyribonucleic acid or increases in the expression of small non-protein-coding ribonucleic acids, but these mechanisms remain to be investigated. Both of these mechanisms influence gene expression and are implicated in ethanol abuse toxicity and cardiovascular disease [96, 97]. Future considerations related to ACM include the following:

Pathophysiologic schema for the development of ACM. As noted in text, the exact amount and duration of alcohol consumption that results in ACM in human beings are variable. The exact sequence for the development of ACM remains incompletely understood, and data from animal models and human beings with a history of long terms suggest that oxidative stress may be an early and initiating mechanism. Many cellular events, such as intrinsic myocyte dysfunction, which is characterized by changes in calcium homeostasis and regulation and decreased myofilament sensitivity, can come about due to oxidative stress. Variables in light blue circles represent potential mediating factors (Color figure online)
-
1.
Alcohol is a major contributor to mortality and years of potential life lost. Therefore, more investigations should be conducted enrolling individuals affected by alcohol dependence to better understand the development and progression of ACM, as well as outcomes related to ACM. Often, studies enrolling alcohol-dependent individuals are subjected to a high level of confounding variables (i.e., hypertension, diabetes, medications, and nutritional deficiencies). Therefore, animal models remain an important method for examining the effects of alcohol. When using animal models, it is important to use an ethanol consumption paradigm that allows for high levels of ethanol consumption and blood ethanol levels and provides adequate nutrition. It is also important for studies to have measures of myocardial structure and function to understand the consequence of tissue and cellular changes.
-
2.
Studies need to be designed to examine the interaction of environmental, nutritional, genetic, and epigenetic factors. Recently, the National Institute on Alcohol Abuse and Alcoholism identified the interaction of alcohol consumption and nutrition as an understudied area of alcohol research (National Institute of Health, PA-13-359, Nutrition and Alcohol-Related Health Outcomes, 2013). Importantly, research questions are warranted that target examination of the interactions among drinking patterns, dietary patterns, and the influence on certain chronic conditions on heart outcomes. These studies should include evaluation of the role of dietary factors, including essential micronutrients and non-nutrient bioactive component of foods, in the development, prevention, and treatment of alcohol-associated organ damage.
-
3.
Understanding the myocardial micro-RNA expression patterns and changes in epigenetic factors/mechanisms.
-
4.
A large amount of evidence indicates that ethanol-related changes occur in the mitochondria. Changes include defects in mitochondrial structure and a decline of mitochondrial oxidative phosphorylation and respiration function and efficacy. Ethanol may interfere with the synthesis of either mitochondrial DNA or nuclear DNA-encoded proteins, thus impairing oxidative phosphorylation and overall function of the mitochondria. Future studies could examine whether these changes are related to corresponding mutations in mitochondrial DNA (mtDNA), such as the 4.9977-bp mtDNA deletion, which often reflects oxidative stress. Mitochondrial DNA mutations caused by ethanol may contribute to the development of ACM. Interventions that target the reversal of mitochondrial dysfunction in the context of ACM appear to be an important future approach to treatment.
-
5.
The effects of a binge-pattern drinking or irregular heavy drinking on myocardial changes have not been widely investigated.
-
6.
Continuing to understand mechanisms that include oxidative stress and activation of intracellular signaling cascades that are involved with mediating cell proliferation, hypertrophy, cell death, and accelerated protein breakdown.
Individuals with alcohol use disorders experience high levels of mortality. Investigating the mechanisms, consequences, and potential treatment options for ACM remains a very important area of research.
References
Maron, B. J., Towbin, J. A., Thiene, G., Antzelevitch, C., Corrado, D., Arnett, D., et al. (2006). Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation, 113, 1807–1816.
Fernandez-Sola, J., Preedy, V. R., Lang, C. H., Gonzalez-Reimers, E., Arno, M., Lin, J. C., et al. (2007). Molecular and cellular events in alcohol-induced muscle disease. Alcoholism, Clinical and Experimental Research, 31, 1953–1962.
Piano, M. R. (2002). Alcoholic cardiomyopathy: Incidence, clinical characteristics, and pathophysiology. Chest, 121, 1638–1650.
Fauchier, L., Babuty, D., Poret, P., Casset-Senon, D., Autret, M. L., Cosnay, P., et al. (2000). Comparison of long-term outcome of alcoholic and idiopathic dilated cardiomyopathy. European Heart Journal, 21, 306–314.
McKenna, C. J., Codd, M. B., McCann, H. A., & Sugrue, D. D. (1998). Alcohol consumption and idiopathic dilated cardiomyopathy: A case control study. American Heart Journal, 135, 833–837.
Kupari, M., Koskinen, P., Suokas, A., & Ventilä, M. (1990). Left ventricular filling impairment in asymptomatic chronic alcoholics. American Journal of Cardiology, 66(20), 1473–1477.
Askanas, A., Udoshi, M., & Sadjadi, S. A. (1980). The heart in chronic alcoholism: A noninvasive study. American Heart Journal, 99(1), 9–16.
Silberbauer, K., Juhasz, M., Ohrenberger, G., & Hess, C. (1988). Noninvasive assessment of left ventricular diastolic function by pulsed Doppler echocardiography in young alcoholics. Cardiology, 75(6), 431–439.
Lazarevic, A. M., Nakatani, S., Neskovic, A. N., Marinkovic, J., Yasumura, Y., Stojicic, D., et al. (2000). Early changes in left ventricular function in chronic asymptomatic alcoholics: Relation to the duration of heavy drinking. Journal of the American College of Cardiology, 35, 1599–1606.
Mathews, E. C, Jr, Gardin, J. M., Henry, W. L., Del Negro, A. A., Fletcher, R. D., Snow, J. A., et al. (1981). Echocardiographic abnormalities in chronic alcoholics with and without overt congestive heart failure. The American Journal of Cardiology, 47, 570–578.
Urbano-Marquez, A., Estruch, R., Navarro-Lopez, F., Grau, J. M., Mont, L., & Rubin, E. (1989). The effects of alcoholism on skeletal and cardiac muscle. The New England Journal of Medicine, 320, 409–415.
Graves, E. J. (1995). Detailed diagnoses and procedures, National Hospital Discharge Survey, 1993. Vital and Health Statistics. Series 13, Data from the National Health Survey (pp. 1–288).
Gavazzi, A., De Maria, R., Parolini, M., & Porcu, M. (2000). Alcohol abuse and dilated cardiomyopathy in men. The American Journal of Cardiology, 85, 1114–1118.
McKenna, C. J., Codd, M. B., McCann, H. A., & Sugrue, D. D. (1998). Alcohol consumption and idiopathic dilated cardiomyopathy: A case control study. American Heart Journal, 135, 833–837.
Hookana, E., Junttila, M. J., Kaikkonen, K. S., Ukkola, O., Kesaniemi, Y. A., Kortelainen, M. L., et al. (2012). Comparison of family history of sudden cardiac death in nonischemic and ischemic heart disease. Circulation. Arrhythmia and Electrophysiology, 5, 757–761.
Ho, E., Karimi Galougahi, K., Liu, C. C., Bhindi, R., & Figtree, G. A. (2013). Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biology, 1(1), 483–491.
Fernandez-Checa, J. C., Kaplowitz, N., Garcia-Ruiz, C., & Colell, A. (1998). Mitochondrial glutathione: Importance and transport. Seminars in Liver Disease, 18, 389–401.
Capasso, J. M., Li, P., Guideri, G., Malhotra, A., Cortese, R., & Anversa, P. (1992). Myocardial mechanical, biochemical, and structural alterations induced by chronic ethanol ingestion in rats. Circulation Research, 71, 346–356.
Segel, L. D., Rendig, S. V., & Mason, D. T. (1981). Alcohol-induced cardiac hemodynamic and Ca2+ flux dysfunctions are reversible. Journal of Molecular and Cellular Cardiology, 13, 443–455.
Sarma, J. S., Ikeda, S., Fischer, R., Maruyama, Y., Weishaar, R., & Bing, R. J. (1976). Biochemical and contractile properties of heart muscle after prolonged alcohol administration. Journal of Molecular and Cellular Cardiology, 8, 951–972.
Bing, R. J., Tillmanns, H., Fauvel, J. M., Seeler, K., & Mao, J. C. (1974). Effect of prolonged alcohol administration on calcium transport in heart muscle of the dog. Circulation Research, 35, 33–38.
Zhang, R. H., Gao, J. Y., Guo, H. T., Scott, G. I., Eason, A. R., Wang, X. M., et al. (2013). Inhibition of CYP2E1 attenuates chronic alcohol intake-induced myocardial contractile dysfunction and apoptosis. Biochimica et Biophysica Acta, 1832(1), 128–141.
Weishaar, R., Sarma, J. S., Maruyama, Y., Fischer, R., Bertuglia, S., & Bing, R. J. (1977). Reversibility of mitochondrial and contractile changes in the myocardium after cessation of prolonged ethanol intake. The American Journal of Cardiology, 40, 556–562.
Pachinger, O. M., Tillmanns, H., Mao, J. C., Fauvel, J. M., & Bing, R. J. (1973). The effect of prolonged administration of ethanol on cardiac metabolism and performance in the dog. Journal of Clinical Investigation, 52, 2690–2696.
Segel, L. D., Rendig, S. V., Choquet, Y., Chacko, K., Amsterdam, E. A., & Mason, D. T. (1975). Effects of chronic graded ethanol consumption on the metabolism, ultrastructure, and mechanical function of the rat heart. Cardiovascular Research, 9, 649–663.
Cederbaum, A. I., & Rubin, E. (1975). Molecular injury to mitochondria produced by ethanol and acetaldehyde. Federation Proceedings, 34, 2045–2051.
Hastillo, A. H., Poland, J., & Hess, M. L. (1980). Mechanical and subcellular function of rat myocardium during chronic ethanol consumption. Proceedings of the Society for Experimental Biology and Medicine, 164, 415–420.
Piano, M. R., Rosenblum, C., Solaro, R. J., & Schwertz, D. (1999). Calcium sensitivity and the effect of the calcium sensitizing drug pimobendan in the alcoholic isolated rat atrium. Journal of Cardiovascular Pharmacology, 33, 237–242.
Tsiplenkova, V. G., Vikhert, A. M., & Cherpachenko, N. M. (1986). Ultrastructural and histochemical observations in human and experimental alcoholic cardiomyopathy. Journal of the American College of Cardiology, 8, 22A–32A.
Jing, L., Jin, C. M., Li, S. S., Zhang, F. M., Yuan, L., Li, W. M., et al. (2012). Chronic alcohol intake-induced oxidative stress and apoptosis: Role of CYP2E1 and calpain-1 in alcoholic cardiomyopathy. Molecular and Cellular Biochemistry, 359(1–2), 283–292.
Hibbs, R. G., Ferrans, V. J., Black, W. C., Weilbaecher, D. G., & Burch, G. E. (1965). Alcoholic cardiomyopathy: An electron microscopic study. American Heart Journal, 69, 766–779.
Beckemeier, M. E., & Bora, P. S. (1998). Fatty acid ethyl esters: Potentially toxic products of myocardial ethanol metabolism. Journal of Molecular and Cellular Cardiology, 30, 2487–2494.
Hu, C., Ge, F., Hyodo, E., Arai, K., Iwata, S., Lobdell, H. T., et al. (2013). Chronic ethanol consumption increases cardiomyocyte fatty acid uptake and decreases ventricular contractile function in C57BL/6 J mice. Journal of Molecular and Cellular Cardiology, 59, 30–40.
Dalle-Donne, I., Rossi, R., Colombo, R., Giustarni, D., & Milzani, A. (2006). Biomarkers of oxidative damage in human disease. Clinical Chemistry, 52(4), 601–623.
Vikhert, A. M., Tsiplenkova, V. G., & Cherpachenko, N. M. (1986). Alcoholic cardiomyopathy and sudden cardiac death. Journal of the American College of Cardiology, 8, 3A–11A.
Fahimi, H. D., Kino, M., Hicks, L., Thorp, K. A., & Abelman, W. H. (1979). Increased myocardial catalase in rats fed ethanol. American Journal of Pathology, 96, 373–390.
Kino, M. (1981). Chronic effects of ethanol under partial inhibition of catalase activity in the rat heart: Light and electron microscopic observations. Journal of Molecular and Cellular Cardiology, 13, 5–21.
Ribiere, C., Hininger, I., Rouach, H., & Nordmann, R. (1992). Effects of chronic ethanol administration on free radical defense in rat myocardium. Biochemical Pharmacology, 44, 1495–1500.
Vendemiale, G., Grattagliano, I., Altomare, E., Serviddio, G., Portincasa, P., Prigigallo, F., et al. (2001). Mitochondrial oxidative damage and myocardial fibrosis in rats chronically intoxicated with moderate doses of ethanol. Toxicology Letters, 123, 209–216.
Edes, I., Toszegi, A., Csanady, M., & Bozoky, B. (1986). Myocardial lipid peroxidation in rats after chronic alcohol ingestion and the effects of different antioxidants. Cardiovascular Research, 20, 542–548.
Edes, I., Piros, G., Forster, T., & Csanady, M. (1987). Alcohol-induced congestive cardiomyopathy in adult turkeys: Effects on myocardial antioxidant defence systems. Basic Research in Cardiology, 82, 551–556.
Khanna, D., Kan, H., Fang, Q., Xie, Z., Underwood, B. L., Jain, A. C., et al. (2007). Inducible nitric oxide synthase attenuates adrenergic signaling in alcohol fed rats. Journal of Cardiovascular Pharmacology, 50, 692–696.
Tan, Y., Li, X., Prabhu, S. D., Brittian, K. R., Chen, Q., Yin, X., et al. (2012). Angiotensin II plays a critical role in alcohol-induced cardiac nitrative damage, cell death, remodeling, and cardiomyopathy in a protein kinase C/nicotinamide adenine dinucleotide phosphate oxidase-dependent manner. Journal of the American College of Cardiology, 59(16), 1477–1486.
Zhang, B., Turdi, S., Li, Q., Lopez, F. L., Eason, A. R., Anversa, P., et al. (2010). Cardiac overexpression of insulin-like growth factor 1 attenuates chronic alcohol intake-induced myocardial contractile dysfunction but not hypertrophy: Roles of Akt, mTOR, GSK3beta, and PTEN. Free Radical Biology & Medicine, 49, 1238–1253.
Fogle, R. L., Lynch, C. J., Palopoli, M., Deiter, G., Stanley, B. A., & Vary, T. C. (2010). Impact of chronic alcohol ingestion on cardiac muscle protein expression. Alcoholism, Clinical and Experimental Research, 34, 1226–1234.
Cheng, H. J., Grant, K. A., Han, Q. H., Daunais, J. B., Friedman, D. P., Masutani, S., et al. (2010). Up-regulation and functional effect of cardiac beta3-adrenoreceptors in alcoholic monkeys. Alcoholism, Clinical and Experimental Research, 34, 1171–1181.
Cheng, C. P., Cheng, H. J., Cunningham, C., Shihabi, Z. K., Sane, D. C., Wannenburg, T., et al. (2006). Angiotensin II type 1 receptor blockade prevents alcoholic cardiomyopathy. Circulation, 114, 226–236.
Li, S. Y., Gilbert, S. A., Li, Q., & Ren, J. (2009). Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. Journal of Molecular and Cellular Cardiology, 47(2), 247–255.
Li, S. Y., & Ren, J. (2008). Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: Role of insulin signaling and ER stress. Journal of Molecular and Cellular Cardiology, 44, 992–1001.
Nadal-Ginard, B., Kajstura, J., Anversa, P., & Leri, A. (2003). A matter of life and death: Cardiac myocyte apoptosis and regeneration. J Clin Investig, 111, 1457–1459.
Farber, N. B., & Olney, J. W. (2003). Drugs of abuse that cause developing neurons to commit suicide. Brain Research. Developmental Brain Research, 147, 37–45.
Zambo, V., Simon-Szabo, L., Szelenyi, P., Kereszturi, E., Banhegyi, G., & Csala, M. (2013). Lipotoxicity in the liver. World Journal of Hepatology, 5, 550–557.
Fernandez-Sola, J., Fatjo, F., Sacanella, E., Estruch, R., Bosch, X., Urbano-Marquez, A., et al. (2006). Evidence of apoptosis in alcoholic cardiomyopathy. Human Pathology, 37, 1100–1110.
Fernandez-Sola, J., Lluis, M., Sacanella, E., Estruch, R., Antunez, E., & Urbano-Marquez, A. (2011). Increased myostatin activity and decreased myocyte proliferation in chronic alcoholic cardiomyopathy. Alcoholism, Clinical and Experimental Research, 35, 1220–1229.
Marzetti, E., Csiszar, A., Dutta, D., Balagopal, G., Calvani, R., & Leeuwenburgh, C. (2013). Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: From mechanisms to therapeutics. American Journal Physiology Heart and Circulatory Physiology, 305(4), H459–H476.
Miro, O., Robert, J., Casademont, J., Alonso, J. R., Nicolas, J. M., Fernandez-Sola, J., et al. (2000). Heart mitochondrial respiratory chain complexes are functionally unaffected in heavy ethanol drinkers without cardiomyopathy. Alcoholism, Clinical and Experimental Research, 24, 859–864.
Williams, E. S., & Li, T. K. (1977). The effect of chronic alcohol administration on fatty acid metabolism and pyruvate oxidation of heart mitochondria. Journal of Molecular and Cellular Cardiology, 9, 1003–1011.
Cunningham, C. C., & Spach, P. I. (1994). Alcoholism and myocardial energy metabolism. Alcoholism, Clinical and Experimental Research, 18, 132–137.
Lange, L. G., & Sobel, B. E. (1983). Myocardial metabolites of ethanol. Circulation Research, 72, 724–731.
Kaphalia, B. S., Cai, P., Khan, M. F., Okorodudu, A. O., & Ansari, G. A. (2004). Fatty acid ethyl esters: Markers of alcohol abuse and alcoholism. Alcohol, 34, 151–158.
Yoerger, D. M., Best, C. A., McQuillan, B. M., Supple, G. E., Guererro, J. L., Cluette-Brown, J. E., et al. (2006). Rapid fatty acid ethyl ester synthesis by porcine myocardium upon ethanol infusion into the left anterior descending coronary artery. American Journal of Pathology, 168, 1435–1442.
Lang, C. H., Frost, R. A., Summer, A. D., & Vary, T. C. (2005). Molecular mechanisms responsible for alcohol-induced myopathy in skeletal muscle and heart. The International Journal of Biochemistry & Cell Biology, 37, 2180–2195.
Vary, T. C., & Deiter, G. (2005). Long-term alcohol administration inhibits synthesis of both myofibrillar and sarcoplasmic proteins in heart. Metabolism, Clinical and Experimental, 54, 212–219.
Vary, T. C., Deiter, G., & Lantry, R. (2008). Chronic alcohol feeding impairs mTOR(Ser 2448) phosphorylation in rat hearts. Alcoholism, Clinical and Experimental Research, 32, 43–51.
Lang, C. H., & Korzick, D. H. (2013). Chronic alcohol consumption disrupts myocardial protein balance and function in aged, but not adult, female F344 rats. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology, 306(1), R23–R33.
Donohue, T. M, Jr. (2009). Autophagy and ethanol-induced liver injury. World Journal of Gastroenterology: WJG, 15, 1178–1185.
Lang, C. H., Huber, D., & Frost, R. A. (2007). Burn-induced increase in atrogin-1 and MuRF-1 in skeletal muscle is glucocorticoid independent but downregulated by IGF-I. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 292, R328–R336.
Lang, S. M., Kazi, A. A., Hong-Brown, L., & Lang, C. H. (2012). Delayed recovery of skeletal muscle mass following hindlimb immobilization in mTOR heterozygous mice. PLoS ONE, 7(6), e38910.
Gustafsson, A. B., & Gottlieb, R. A. (2009). Autophagy in ischemic heart disease. Circulation Research, 104, 150–158.
Sukhanov, S., Semprun-Prieto, L., Yoshida, T., Michael Tabony, A., Higashi, Y., Galvez, S., et al. (2011). Angiotensin II, oxidative stress and skeletal muscle wasting. The American Journal of the Medical Sciences, 342, 143–147.
Guo, R., Hu, N., Kandadi, M. R., & Ren, J. (2012). Facilitated ethanol metabolism promotes cardiomyocyte contractile dysfunction through autophagy in murine hearts. Autophagy, 8, 593–608.
Roerecke, M., & Rehm, J. (2010). Irregular heavy drinking occasions and risk of ischemic heart disease: A systematic review and meta-analysis. American Journal of Epidemiology, 171, 633–644.
Gu, L., Fink, A. M., Chowdhury, S. A., Geenen, D. L., & Piano, M. R. (2013). Cardiovascular responses and differential changes in mitogen-activated protein kinases following repeated episodes of binge drinking. Alcohol and Alcoholism, 48, 131–137.
Thombs, D. L., Olds, R. S., & Snyder, B. M. (2003). Field assessment of BAC data to study late-night college drinking. Journal of Studies on Alcohol, 64, 322–330.
Kajander, O. A., Kupari, M., Perola, M., Pajarinen, J., Savolainen, V., Penttila, A., et al. (2001). Testing genetic susceptibility loci for alcoholic heart muscle disease. Alcoholism, Clinical and Experimental Research, 25, 1409–1413.
Husemoen, L. L., Fenger, M., Friedrich, N., Tolstrup, J. S., Beenfeldt Fredriksen, S., & Linneberg, A. (2008). The association of ADH and ALDH gene variants with alcohol drinking habits and cardiovascular disease risk factors. Alcoholism, Clinical and Experimental Research, 32, 1984–1991.
Djousse, L., Levy, D., Herbert, A. G., Wilson, P. W., D’Agostino, R. B., Cupples, L. A., et al. (2005). Influence of alcohol dehydrogenase 1C polymorphism on the alcohol-cardiovascular disease association (from the Framingham Offspring Study). The American Journal of Cardiology, 96, 227–232.
Hines, L. M., Stampfer, M. J., Ma, J., Gaziano, J. M., Ridker, P. M., Hankinson, S. E., et al. (2001). Genetic variation in alcohol dehydrogenase and the beneficial effect of moderate alcohol consumption on myocardial infarction. The New England Journal of Medicine, 344, 549–555.
Reinke, L. A., Lai, E. K., DuBose, C. M., & McCay, P. B. (1987). Reactive free radical generation in vivo in heart and liver of ethanol-fed rats: Correlation with radical formation in vitro. Proceedings of the National Academy of Sciences of the United States of America, 84, 9223–9227.
Wang, L., Zhou, Z., Saari, J. T., & Kang, Y. J. (2005). Alcohol-induced myocardial fibrosis in metallothionein-null mice: Prevention by zinc supplementation. American Journal of Pathology, 167, 337–344.
Fuchs, F. D., Chambless, L. E., Folsom, A. R., Eigenbrodt, M. L., Duncan, B. B., Gilbert, A., et al. (2004). Association between alcoholic beverage consumption and incidence of coronary heart disease in whites and blacks: The Atherosclerosis Risk in Communities Study. American Journal of Epidemiology, 160, 466–474.
Sempos, C. T., Rehm, J., Wu, T., Crespo, C. J., & Trevisan, M. (2003). Average volume of alcohol consumption and all-cause mortality in African Americans: The NHEFS cohort. Alcoholism, Clinical and Experimental Research, 27, 88–92.
National Institute on Alcohol Abuse and Alcoholism. (2008). Alcohol: A women’s health issue. NIH Publication No. 03 4956. Washington, DC: U.S. Department of Health and Human Services.
Fernandez-Sola, J., Estruch, R., Nicolas, J. M., Pare, J. C., Sacanella, E., Antunez, E., et al. (1997). Comparison of alcoholic cardiomyopathy in women versus men. The American Journal of Cardiology, 80, 481–485.
Kennedy, R. H., Stewart, C., Light, K. E., & Wyeth, R. P. (2002). Effects of gender on the cardiac toxicity elicited by chronic ethanol intake in rats. Toxicology Applied Pharmacology, 179, 111–118.
Lochner, A., Cowley, R., & Brink, A. J. (1969). Effect of ethanol on metabolism and function of perfused rat heart. American Heart Journal, 78, 770–780.
Piano, M. R., Geenen, D. L., Schwertz, D. W., Chowdhury, S. A., & Yuzhakova, M. (2007). Long-term effects of alcohol consumption in male and female rats. Cardiovascular Toxicology, 7, 247–254.
Vary, T. C., Kimball, S. R., & Sumner, A. (2007). Sex-dependent differences in the regulation of myocardial protein synthesis following long-term ethanol consumption. American Journal Physiology-Regulatory Integrative and Comparative Physiology, 292, R778–R787.
Fogle, R. L., Hollenbeak, C. S., Stanley, B. A., Vary, T. C., Kimball, S. R., & Lynch, C. J. (2011). Functional proteomic analysis reveals sex-dependent differences in structural and energy-producing myocardial proteins in rat model of alcoholic cardiomyopathy. Physiological Genomics, 43, 346–356.
Mackie, A. R., Krishnamurthy, P., Verma, S. K., Thorne, T., Ramirez, V., Qin, G., et al. (2013). Alcohol consumption negates estrogen-mediated myocardial repair in ovariectomized mice by inhibiting endothelial progenitor cell mobilization and function. Journal of Biological Chemistry, 288, 18022–18034.
Yancy, C. W., Jessup, M., Bozkurt, B., et al. (2013). ACCF/AHA guideline for the management of heart failure. Journal of the American Association of Cardiology, 62(16), e148–e239.
Iacovoni, A., De Maria, R., & Gavazzi, A. (2010). Alcoholic cardiomyopathy. Journal of Cardiovascular Medicine, 11, 884–892.
Piano, M. R. (2002). Alcohol and heart failure. Journal of Cardiac Failure, 8(4), 239–246.
Balady, G. J., Williams, M. A., Ades, P. A., et al. (2007). Core components of cardiac rehabilitation/secondary prevention programs: 2007 update. Circulation, 115, 2675–2682.
Substance Abuse and Mental Health Services Administration. (2011). Results from the 2010 National Survey on Drug Use and Health: Summary of national findings. NSDUH series H-41. HHS Publication No. (SMA) 11-4658. Rockville, MD: Substance Abuse and Mental Health Services Administration.
Small, E. M., Frost, R. J., & Olson, E. N. (2010). MicroRNAs add a new dimension to cardiovascular disease. Circulation, 121, 1022–1032.
Miranda, R. C., Pietrzykowski, A. Z., Tang, Y., Sathyan, P., Mayfield, D., Keshavarzian, A., et al. (2010). MicroRNAs: Master regulators of ethanol abuse and toxicity? Alcoholism, Clinical and Experimental Research, 34, 575–587.
Acknowledgments
Due to page limitations, we recognize that we have not included all the excellent scientific work completed in the area of alcohol and the cardiovascular system. This study was supported by National Institutes of Health grants AA015578 (MRP), HL85614 (SAP), HL095701 (SAP), and HL095701-02S1 (SAP). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Piano, M.R., Phillips, S.A. Alcoholic Cardiomyopathy: Pathophysiologic Insights. Cardiovasc Toxicol 14, 291–308 (2014). https://doi.org/10.1007/s12012-014-9252-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-014-9252-4