
Abstract
Before menopause, a woman has a relatively low risk for developing cardiovascular disease. After menopause, however, the risk increases nearly twofold and cardiovascular disease remains the number one cause of death among women. Observational trials and studies in animal models of cardiovascular disease suggested that females have reduced injury after myocardial ischemia and reperfusion injury. However, two large clinical trials, the women’s health initiative (WHI) and the heart estrogen and progestin replacement study (HERS), found an increase in cardiovascular incidences in women taking hormone replacement therapy. The discrepancy between these data highlights the need for further research on the mechanism of estrogen in the cardiovascular system. Animal studies have demonstrated protective effects by endogenous estrogen (gender differences) and also by the administration of exogenous estrogen. In vivo studies suggest a possible anti-inflammatory mechanism of estrogen. Exogenous estrogen has been shown to have anti-oxidant activities. Pre-treatment with estrogen prior to myocardial ischemia and reperfusion causes a decrease in neutrophil infiltration into the irreversibly injured myocardium, decrease in C-reactive protein expression, and deposition of the membrane attack complex. This review will summarize the protection afforded by estrogen as well as discuss several possible mechanisms of protection for exogenous estrogen administration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease (CVD) is the number one cause of death among women. On average, women develop cardiovascular disease 10–15 years later in life than men and the risk of CVD increases more than twofold after menopause. This observation lead to the speculation that the decrease in estrogen associated with menopause is responsible for the increase in cardiovascular disease, osteoporosis, and the decline in both cognitive and sexual function. The observed increased risk in cardiovascular disease correlates to the decrease in estrogen levels during menopause. This, in addition to the fact that estrogen is both a cardioprotective agent and an immune modulator, strengthens the belief that estrogen has a potential role in the prevention of CVD. Early observational studies, such as the Nurses’ Health Study, demonstrated estrogen’s ability to reduce the risk of heart disease [8] as well as improve blood lipid levels [74] and other surrogate markers [112] in menopausal women. However, the randomized clinical trials with hormone therapy that followed were neutral or even harmful in the ability of estrogen to prevent primary or secondary cardiovascular disease. Several factors may play a role in the discrepancy between the initial observational studies and the later randomized clinical trails. These factors include the age and level of pre-existing disease of patients at the initiation of therapy.
Despite the disappointing outcomes of the clinical trials, there is extensive experimental evidence that estrogen has beneficial effects on the cardiovascular system, particularly in myocardial ischemia-reperfusion injury, atherosclerosis, and arrhythmia [21, 49, 113]. The remainder of this review will focus on the actions of endogenous and exogenous estrogen on myocardial reperfusion injury.
Overview of Myocardial Reperfusion Injury
Acute myocardial infarction with extensive irreversible tissue injury occurs in response to an extended period of reduced regional myocardial blood flow. A prolonged ischemic event results in permanent loss of function and cell death. The return of blood flow within a critical time period is capable of maintaining cell viability, which provides the justification for early interventions designed to restore blood flow and myocardial perfusion. Undeniably, restoration of blood flow is necessary to reverse the progression of cell death and to maintain tissue viability. The reintroduction of oxygenated blood to previously ischemic tissue can paradoxically cause irreversible cellular damage and depressed myocardial function. Reperfusion also elicits a deleterious inflammatory response against host tissue, which is mediated by free radicals, neutrophils, complement activation, and multiple mediators of inflammation. The term “reperfusion injury” is defined as the conversion of reversibly injured cells to a state of irreversible injury resulting from reintroduction of oxygenated blood to an ischemic area [82]. Several agents, including free-radical scavengers and inhibitors of the immune system, have been shown to protect the heart from injury after reperfusion.
Estrogen Biology
Estradiol (E2) is a steroid hormone involved in the regulation of a wide array of processes, including normal cell growth and development, tissue-specific gene regulation, reproduction, sexual development, behavior, stress response, bone integrity, neuroprotection, and cardiovascular health [24, 85, 110]. Initially, it was thought that E2 was a metabolic cofactor until the discovery of the first specific estrogen receptor (ER), now known as ERα [46]. A second receptor subtype, ERβ, also exists and may play a role in the cell specific actions of estrogens [63]. The expression patterns of ERα and ERβ and the receptor distribution differ throughout the body, suggesting that each has a distinct role in ER pharmacology [24, 62].
Genomic and Non-genomic Effects
The ligand-dependent genomic mechanism of ER action is the defining element of the nuclear steroid/thyroid receptor superfamily to which E2 belongs. The binding of estrogen to the ER induces a conformational change in the receptor that causes ER activation. These changes include receptor phosphorylation on serine and tyrosine residues, dissociation from the 90-kDa heat shock protein (hsp90), and subsequent ER dimerization. The ERα and ERβ can either homodimerize or heterodimerize to generate distinct functions. The ER dimer then binds with high affinity to the estrogen response element (ERE), a specific regulatory DNA sequence present in the promoter of target genes [110]. The DNA-bound receptors contact co-activators or co-repressors depending on the cellular environment and can exert either positive or negative effects on the expression of downstream target genes.
Although the ER is classically a ligand-dependent transcription factor, many of the effects of estrogen treatment cannot be explained by the genomic scheme of action. It is apparent that the receptor also modulates the activity of intracellular second messengers, membrane-associated receptors, and signaling complexes, which may enhance the classic activity of the ER. Non-genomic actions of E2 have been described in several model systems and include changes in Ca2+ flux [124], mitogen activated protein kinase (MAPK) activation [57], and activation of the PI3K-AKT pathway [83]. These effects are believed to be mediated by estrogen receptors, possibly the classical ER, located in or close to the plasma membrane [20]. Evidence for the presence of a membrane bound estrogen receptor has been shown with estrogens rendered membrane-impermeant by conjugation to albumin [95], fluorescent labeled membrane-impermeant estrogen conjugates [81], and overexpression of the receptor [89]. Antibodies directed against various regions of the nuclear ERα are reported to react with a protein in the plasma membrane of cells [81]. Transfection of cells with cDNA for both ERα and ERβ demonstrated that both receptor subtypes may be expressed (although significantly less than in the nucleus) in the plasma membrane [89].
Despite abundant data now supporting responses to estrogen through membrane-localized receptors, it is not currently known if the classical estrogen receptor or a closely related protein transduces these rapid estrogen signals. Several reports have detailed the identification of putative membrane ERs, which are distinct from nuclear ERs [70] other investigators have demonstrated localization of nuclear ERs at membrane sites [23, 66]. Recent evidence has demonstrated the involvement of the orphan G protein-coupled receptor GPR30 in estrogen-induced transactivation suggesting that this may be a novel membrane ER [90]. The G protein GαI was reported to facilitate ERα-eNOS coupling and E2-stimulated eNOS activation in both transfected cells and endothelial cells [122]. In cells transfected with ERα and eNOS, both proteins associated at the membrane in a caveolin-1-dependent manner [20]. A variant form of ER, ER46, has been shown to translocate to the caveolar fraction in response to E2 in endothelial cells [68]. Palmitoylation has been shown to be critical for membrane localization and rapid signaling for both the mutant ER46 and ERα [1, 2, 68] and the rapid signaling responses are reduced when palmitoylation is blocked or the critical palmitoylation sites in ERα are mutated [2]. Taken together, these observations suggest that there may exist cellular membranous receptor/binding sites for estrogens that are responsible for their acute non-genomic actions.
Estrogen’s Effects in Ischemia-Reperfusion Injury and Possible Mechanisms
It is known that estrogen has favorable effects on some risk factors related to the development of atherosclerosis and cardiovascular disease. Although there is a tendency to increase plasma triglyceride levels [116], in postmenopausal women, HRT also reduces levels of low-density lipoprotein (LDL) and increases levels of high-density lipoprotein (HDL) [120]. Following HRT, plasma lipid levels are altered so that they are again identical to those of pre-menopausal women who have a lower rate of CVD [76]. Estrogens have also demonstrated beneficial effects in animal models of cardiovascular disease. Estrogen administration decreases the levels of collagen production in rabbits [39], and has a favorable effect on insulin resistance [43]. When monkeys were treated with conjugated equine estrogen (CEE), an increase in coronary artery vasodilation was observed [117] and a reduction was seen in the accumulation of cholesterol in the atherosclerotic vessels [3]. Furthermore, estrogen treatment inhibited balloon-mediated intimal thickening in the rat carotid artery, suggesting protective actions of estrogen on vascular cells [58]. Acute and chronic E2 administration reduces infarct size after an ischemic insult [14, 16, 86].
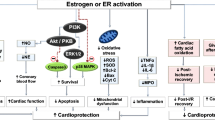
Studies with knockout animals have given insight into the function of the estrogen receptors in various cardiovascular diseases. It has been shown that administration of estradiol blocks cardiac hypertrophy in mice and that ERβ, but not ERα, was required for this protection [98]. ERβ knockout mice also have a higher mortality after MI than wild-type mice [84]. In addition, studies by Forster et al., have reported that ERβ knockout mice exhibit a cardiac phenotype similar to human hypertensive cardiomyopathy [40]. It has been suggested that ERα may play a role in reperfusion injury by improving NO release, attenuating calcium accumulation, and preserving mitochondria [123]. Studies with the ERα agonist PPT also suggest that activation of this receptor decreases the inflammatory response to reperfusion injury leading to protection [16]. Although knockout mice and receptor subtype-specific agonists have aided in understanding the complex nature of estrogen in the cardiovascular system, we still do not have a full understanding of how the receptors interact to cause protection. Several possible mechanisms of estrogens protection are listed below and outlined in Figure 1. The exact mechanism or mechanisms by which estrogen reduces tissue injury is unknown.

Possible mechanism of myocardial protection after E2 treatment
Sex Differences in Reperfusion Injury
Results from animal studies have suggested a gender difference in reperfusion injury without the addition of exogenous estrogen. Female isolated perfused rat hearts had smaller infarcts than male after global ischemia [6]. In contrast to these studies, other studies using both in vivo and ex vivo models of myocardial reperfusion injury did not demonstrate a difference between male and female hearts [69, 94]. Although these studies did not demonstrate a gender difference in wild-type animals, females showed a decrease in reperfusion injury under conditions of increased contractility and/or increased cellular calcium [25, 26, 28]. It has been suggested that the inconsistencies regarding protection observed in females compared to males may be related to the level of contractility between the different models. The protection may relate to the ability of the female heart to reduce calcium loading during ischemia and reperfusion, as it is known that elevated calcium levels increase reperfusion injury [102]. It was found that hearts from intact females have a lower expression of β1-adrenergic receptor compared to oophorectomized females, which may cause a reduced calcium overload and protect the hearts from injury [27]. A reduction in calcium overload can also explain the reduction in injury compared to males when the hearts are in a hypercontractile state. In fact, female transgenic hearts overexpressing the plasma membrane sodium–calcium exchanger and WT hearts treated with isoproterenol have been shown to have less calcium loading [26]. These data suggest that estrogen reduces calcium loading, such that there is less of an increase in intracellular calcium during ischemia and reperfusion, which results in less injury. One possible mechanism for estrogen’s action is that increased calcium in hearts of intact females causes an increase in NOS and results in increased S-nitrysolation of the L-type calcium channel resulting in a decrease in intracellular calcium entry during ischemia and reperfusion [79, 105].
Addition of Exogenous Estrogen
In addition to sex differences in the response to reperfusion injury, studies have also demonstrated the protective effects of exogenous estrogen on the heart after ischemia and reperfusion [14, 69]. In contrast to previous studies, gender difference in response to exogenous estrogen was not observed when male and female rabbits were treated with 17 β-estradiol 15 min prior to in vivo coronary artery ligation. In this study, a reduction in infarct size was observed in both sexes [50]. Pretreatment of male rabbits with estradiol prior to in vivo coronary artery ligation significantly reduced infarct size [29]. The observed protection appeared to be mediated by activation of the mitoKATP channel as pretreatment with a mitoKATP channel inhibitor prevented the infarct size reduction afforded by estradiol. Similarly, our laboratory has demonstrated that treatment of oophorectomized rabbits with 20 μg of β-estradiol 30 min before in vivo coronary artery occlusion reduced infarct size compared to vehicle treated animals [14, 16]. The observed protection was demonstrated to require activation of the estrogen receptor and the results suggest that estrogen protects the myocardium, in part, by reducing the inflammatory response that is activated during reperfusion. Specifically, we have demonstrated the ability of exogenous estrogen to inhibit neutrophil infiltration into the area at risk, C-reactive protein (CRP) deposition, and deposition of the membrane attack complex (MAC). Similar studies have implicated estrogen’s anti-inflammatory action as a mechanism for the protection observed in myocardial reperfusion injury as outlined below.
Estrogens Action on the Inflammatory System
Inflammation is recognized to be an important component of the pathophysiology of myocardial ischemia [71]. In the heart, the inflammatory response is produced not only by invading macrophages, but also in the cardiomyocytes [73, 108] and methods to inhibit the inflammatory response might protect the myocardium after ischemia and reperfusion [107]. Estrogen can act on multiple immune processes including lymphocyte and monocyte development, dendritic cell function, T cell responses, and cytokine regulation [65]. Estrogen can elicit a response by acting directly on the ER in inflammatory cells or via activation of the ER and subsequent activation of second messenger systems in endothelial cells.
Antioxidant
Oxidant stress is one of the stimuli for production of inflammatory cytokines and upregulation of adhesion molecule expression. Differences in antioxidant levels with endogenous estrogen or estrogen replacement following menopause could lead to differences in inflammatory response after acute injury. There is a sudden increase in free radical production during ischemia and reperfusion. In studies using in vivo models of myocardial ischemia and reperfusion, E2 was shown to decrease hydroxyl radical production and decrease lipid peroxidation [21]. In vitro studies demonstrated an estrogen-mediated reduction in superoxide anion production from coronary artery segments after hypoxia and reoxygenation [60]. Possible mechanisms for the antioxidant effect of estrogen include increased levels of superoxide dismutase in cardiac myocytes. Hearts from female animals are reported to have an increase in superoxide dismutase compared with males until ovariectomy when the levels decrease to a value significantly lower than intact animals [7].
Prostaglandins
Prostaglandins (PGs) are short-lived lipid-derived autacoids with diverse biological functions in many systems, including the immune system and cardiovascular systems. Prostaglandins have been implicated in several diseases, one of which is cardiovascular disease. [5]. Cyclooxygenase [58], the enzyme that synthesizes prostaglandins, is expressed in two isoforms, COX-1 and COX-2. COX-1 is constitutively expressed in most tissues and synthesizes PGs to maintain physiological function [34], whereas COX-2 is inducible in response to pro-inflammatory stimuli and cytokines, which results in increased PG levels [114]. Recently, there has been accumulating evidence that COX-2 has anti-inflammatory properties, specifically in the mononuclear cell dominated phase of inflammation [42]. It is also well known that COX-2 activation is required for both the early and late phases of ischemic preconditioning [5, 12]. The importance of COX-2 in the cardiovascular system was highlighted in the Vioxx™ gastrointestinal outcome research trial in which the rate of myocardial infarction was greater for patients taking rofecoxib than the non-selective COX inhibitor naproxin [13, 56]. Recent results call attention to the importance of estrogen-mediated COX-2 activation by demonstrating the requirement of ERα-dependent increases of PGI2 production for atheroprotection in female mice [36].
Nuclear Factor kappa B (NF-κB)
Nuclear factor kappa B (NF-κB) has been shown to be involved in both the early and late stages of inflammatory-proliferate processes [109]. Activation of NF-κB during ischemia and reperfusion induces the transcription of genes encoding proteins, such as cytokines, chemokines, and adhesion molecules, all of which are important mediators of reperfusion injury. The functional interaction, or “cross talk,” between the estrogen receptor and the pro-inflammatory transcription factor NF-κB has been demonstrated both in vitro and in vivo and is suggested to play a role in estradiol’s reduction in cardiovascular disease [38, 52, 99]. The anti-inflammatory activity of estradiol has been attributed, in part, to the interference of NF-κB activity. Inhibition of NF-κB by estradiol requires activation of the estrogen receptor and has been reported to occur through multiple mechanisms, including direct protein-protein interactions [87, 103], inhibition of NF-κB DNA binding [33, 88], induction of IκB expression [106], or via coactivator sharing [52, 99].
Cell Adhesion Molecules
Leukocyte accumulation in the jeopardized myocardium has been suggested to amplify tissue damage following ischemia by producing cell activation of the myocytes and by releasing cytotoxic substances, such as thromboxane A2 [22], oxygen free radicals [72], and platelet activating factor [18]. Adhesion molecules also serve an important role in the localization and development of an inflammatory response to tissue injury. Intracellular adhesion molecule-1 (ICAM-1) expression is enhanced by several inflammatory mediators, including interleukin-1 (IL-1) and tumor necrosis factor alpha (TNF-α), and serves as an adhesive molecule for neutrophils. Estrogen decreased serum and macrophage TNF levels and decreased ICAM-1 expression in the myocardium after coronary artery occlusion and reperfusion [100]. This led to decreased leukocyte accumulation and smaller infarcts in the estradiol-treated group. Other studies have shown that estrogen decreases neutrophil accumulation after myocardial ischemia and reperfusion [32].
C-Reactive Protein and the Membrane Attack Complex
CRP is one of the best-characterized biomarkers of cardiovascular risk. CRP levels are an independent predictor of risk for future CV events [41, 96]. This protein is secreted by hepatocytes in response to interleukin 6 (IL-6) and tumor necrosis factor a (TNF-α). CRP may also act as a pro-coagulant by inducing the expression of the monocyte tissue factor that plays an important role in atherosclerosis [77]. CRP activates the complement cascade, the components of which have been implicated in early stages of atherogenesis and are mediators of myocardial damage after ischemia and reperfusion. CRP colocalizes with complement components in atherosclerotic lesions of human coronary arteries [111] as well as in human infarcted myocardium [78]. It has been suggested that hormone replacement therapy in postmenopausal women is associated with an increased inflammatory response that may trigger acute cardiovascular events. This suggestion is based on the findings of elevated CRP levels after HRT. However, recent findings show that although there is an increase in CRP associated with HRT, other plasma markers of inflammation are reduced, suggesting that increased CRP levels following oral HRT might be related to metabolic hepatic activation and not to an acute-phase response [96]. Moreover, the increase in CRP does not occur in women using transdermal HRT [31]. Recently, E2 has been linked to the inflammatory response in that acute treatment inhibits the deposition of CRP and the MAC in rabbit hearts subject to ischemia and reperfusion [16].
Overview of Observational and Clinical Trials—Does Hormone Replacement Therapy Provide Protection Against Cardiovascular Events?
Observational and Clinical trials were utilized to determine the effectiveness of estrogen replacement on the prevention of cardiovascular disease. The trials presented divergent data and raised more questions than they answered. Several factors play a role in the discrepancy between observational trials and the clinical trials. The results of three trials as well as some reasons for the inconsistencies are discussed below.
The Nurses’ Health Study
The Nurses’ Health Study is one of the longest prospective investigations of hormone therapy and CHD incidence. The study began in 1976 when 121,700 nurses aged between 30 and 55 completed a mailed questionnaire about their postmenopausal hormone use and medical history, including cardiovascular risk factors [47, 48]. The information was updated in biennial follow-up questionnaires. Type and dose of hormone used were recorded as well as first occurrences of nonfatal myocardial infarction, fatal coronary disease, and fatal or non-fatal stroke. A 20-year follow-up study suggested that women who take estrogen are less likely to develop CHD than women who do not take estrogen. Importantly, the follow-up study demonstrated an association between menopause and initiation of hormone therapy. The results suggest that women who initiate hormone therapy near the beginning of menopause receive benefits and beginning therapy at longer intervals since menopause may be associated with less cardiovascular benefits.
Heart and Estrogen/Progestin Replacement Study (HERS)
One of the first large-scale studies to determine the cardioprotective role of estrogen replacement after menopause investigated the ability of hormone replacement therapy (HRT) to prevent secondary coronary events. The Heart and Estrogen/Progestin Replacement Study (HERS) [54] was a randomized trial involving nearly 3,000 postmenopausal women who had a history of coronary artery disease. The mean age of the participants was 66.7 years and the women were treated with either placebo or combination hormone therapy (0.625 mg of conjugated estrogen (CEE) and 2.5 mg of medroxyprogesterone acetate (MPA)). Despite a favorable effect of hormone therapy on lipid levels, no significant differences in primary outcome (cardio vascular deaths or nonfatal myocardial infarction) or secondary outcome (stroke, peripheral artery disease, congestive heart failure, unstable angina, or resuscitated arrest) were noted after a 4-year follow-up. Surprisingly, in the first year of treatment, it was found that more women in the hormone-treated group suffered coronary events despite the decrease in cardiovascular events during years 4 and 5. At the completion of the study, HERS investigators found no overall benefit for HRT after menopause.
The HERS II study was undertaken primarily to determine if the apparent decrease in the risk of CAD observed in the later years of the HERS trial persisted or became more marked resulting in overall benefit. The follow-up of the HERS cohort was extended with women voluntarily continuing treatment for a total (HERS and HERS II) of almost 7 years [45]. The study showed no evidence of overall benefit for any cardiovascular outcome with the use of hormone replacement therapy. These findings gave additional support to the recommendations that postmenopausal hormone therapy should not be used for the purpose of reducing risk for CAD events in women with a history of disease [75].
Women’s Health Initiative (WHI)
The Women’s Health Initiative (WHI) [92] was a primary prevention study that administered combination hormone therapy (CEE and MPA) or unopposed estrogen to healthy postmenopausal women. Four years after the conclusion of HERS, the combination treatment arm of the WHI was terminated prematurely due to an increased incidence of breast cancer and trends toward a worsening of cardiovascular events. At the 5-year follow up, patients on the combination treatment had a higher occurrence of stroke, cardiac events, thromboembolic events, and invasive breast cancer compared to the placebo group. These harmful outcomes far outweighed the benefits of a decrease in hip fractures and colon cancer.
The estrogen alone arm, consisting of 10,739 women who had hysterectomies, did not show the same adverse effects as the combination treatment and was determined safe to continue [55]. As in the combination arm, the hypothesis was that estrogen treatment would reduce the risk of cardiovascular events. Although the increase in cardiovascular disease in the first year of treatment was less pronounced, the estrogen alone arm of the study was stopped almost 1 year before the predicted end date. None of the pre-defined stopping boundaries had been reached. The increase in stroke, however, outweighed the ability to obtain more data about the potential beneficial outcomes in the 50–59-year-old group [55].
Potential Factors Responsible for the Clinical Trial Outcomes
Age and Pre-existing Disease
The major difference between the observational and clinical trials is the age of the patients at initiation of hormone replacement. Patients in the observational trials generally start therapy early in menopause, whereas patients enrolled in the clinical trials began therapy long after menses had ended. The mean age of menopause in the US is 51 years. The ages of patients in the Nurses’ Health Study ranged from 30 to 55 years and most of the members who used hormone replacement within 2 years of menopause. In contrast, the mean age of the patients enrolled in the WHI and HERS was 63 and 67 years, respectively, and most of the women had been post-menopausal for several years before beginning hormone therapy.
The fact that HRT did not confer cardioprotective effects in the recent randomized controlled trials may be explained by the fact that atherosclerosis and vascular remodeling is an age-dependent process and the delay in initiation of therapy may have influenced the outcome [104]. If this is the case, HRT may be beneficial in younger women before plaque complication begins. In fact, the subgroup of women in the youngest decade (aged 50–59) appeared to respond to hormone replacement more favorably than older women. Only one clinical outcome was significantly lower in the HRT group. This is possibly due to lower disease incidence in a younger population, but larger trials with younger participants must be performed to determine if there is a clear effect with replacement therapy. Experimental studies further support an age-dependent effect of estrogen inhibition on cardiovascular protection. Non-human primate studies have demonstrated the importance of timing of treatment and status of atherosclerosis. Estrogen replacement therapy (ERT) and HRT have been shown to be effective in inhibiting the early stage atherosclerosis but much less effective in inhibiting more advanced atherosclerosis (advanced plaques) [4, 118]. Based on these findings, the subjects in HERS may have received no benefit from HRT because of the advanced stage of their atherosclerosis at the time that HRT was initiated. It is possible that the results from these trials may not apply to a HRT regimen initiated early in the menopause or during the peri-menopause.
Type of Estrogen and Route of Administration
The type of estrogen used in the clinical studies may also affect the outcome. Both the HERS and WHI used conjugated equine estrogen, which is a mixture of steroids extracted from pregnant mare urine. The exact composition of CEE is unknown, but the principal active ingredients are sodium estrone sulfate, sodium equilin sulfate, and sodium 17α-dihydroequilinenin. The major endogenous form of estrogen is estradiol. After menopause, women lose the major ovarian hormone estradiol and the level of estrone, which is produced primarily in the peripheral tissues, and remains the same. CEE does not replace estradiol. Both estradiol and CEE are estrogens; however, estradiol and CEE differ chemically and the pharmacological properties of these estrogens vary and may influence the outcome of studies evaluating the effects of hormone replacement [35]. The estrogens in CEE have different binding affinities for estrogen receptors (ERs), selectivity for ER subtypes, and agonist activities for ERs compared with estradiol [35]. CEE and other estrogens may not mimic the cardiovascular protective effects of estradiol because of the different pharmacological actions of estradiol and CEE on the cardiovascular system.
Because CEE is given orally, while estradiol is often administered transdermally to humans and subcutaneously, intravenously, or in release pellets to animals, it is possible that the differences in outcome between CEE and estradiol are related in part to the route of administration. The importance of the route of administration is demonstrated by the fact that CEE administered orally increases the circulating levels of C-reactive protein, a protein upregulated during the acute phase response to injury. However, when CEE is administered via a transdermal patch, CRP levels remain unchanged [31]. Although it is possible that other forms or routes of administration could be more beneficial, this must be demonstrated in disease end point trials before any hormone treatment regimen can be recommended for disease prevention. However, the observational studies demonstrating the benefits of estrogen primarily involved the oral use of conjugated equine estrogen. Therefore, although the type of estrogen and route of administration may play a small role, it is likely not the primary contributor to the adverse effects observed in the clinical trials.
Progestin in the HRT Regimen
The WHI trial suggests that estrogen alone provides potential advantages over the estrogen/progestin combination for treating postmenopausal women. Estrogen alone had only one or two adverse outcomes (increased stroke and probably pulmonary embolism), whereas the combination therapy had four adverse outcomes (increased stroke, pulmonary embolism, CHD, and breast cancer) [55]. However, it is important to note that estrogen alone produced no overall benefit in the global index. A possible reason for the greater number of adverse effects seen in the population of women treated with combination therapy than treated with estrogen alone is the addition of progesterone to the hormone regimen. In the 1980s it was recognized that postmenopausal estrogen treatment was causing endometrial cancer, which could be prevented by antagonizing estrogen with a progestin [44]. It was determined that when conjugated equine estrogen was combined with medroxyprogesterone acetate (MPA), the benefits of estrogen, mainly the retention of favorable lipid effects, were preserved and this particular regime has become the most widely used combination in the United States [120].
There are a variety of experimental studies outlining the effects of the addition of progestin to the estrogen regimen. The findings are controversial in that some demonstrate that progestin inhibits estrogen’s effects and others support the fact that progestin has no adverse effect on estrogen treatment when added to estrogen therapy. MPA has been shown to attenuate estrogen-mediated inhibition of balloon-mediated intimal thickening in the rat carotid artery [67]. In primate studies, the addition of MPA abolished the protective effects of unopposed CEE on atherogenesis [4] and inhibited acetylcholine induced vasodilation by 50% [117].
Contrary to the above studies, it has also been shown that when combined with a progestin, estrogen is still able to positively effect coronary vasodilation [21] or the decrease in LDL levels [115] and infarct size [93]. The significant amount of data on both sides indicates that the addition of the progestin MPA may affect the cardiovascular response to estrogen treatment. However, the addition of progestin to the treatment regimen was not the only reason hormone replacement failed to provide cardiovascular protection as evidenced by the fact that the unopposed estrogen arm of the WHI was terminated.
Alternatives to Traditional Hormone Replacement Therapy
Although experimental models of hormone replacement have demonstrated a definite benefit of estrogen on the cardiovascular system, the adverse effects associated with long-term treatment limit its clinical use. Efforts to understand the mechanisms governing estrogen’s protective effects after ischemia and reperfusion have yielded several possible strategies to reduce tissue injury after estrogen loss. Two possible therapies to specifically target cardiovascular protection are currently under investigation: the selective estrogen receptor modulators (SERMs) and the pathway selective ER ligands. The cardiovascular actions of some SERMs are listed in Table 1.
Selective Estrogen Receptor Modulators (SERMs)
SERMs are steroidal or non-steroidal compounds that activate estrogen receptors in target tissues while inhibiting receptors in others to target the protective effects of estrogen [91]. The ideal SERM would exhibit agonist activity in the heart, bone, and brain without activating receptors in the breast and uterus. The SERMs currently approved for human use are clomophene for management of infertility in women [11], tamoxifen and toremifene for the treatment of breast cancer [80], and raloxifene for the treatment and prevention of postmenopausal osteoporosis [97].
Tamoxifen is perhaps the best known SERM and is used as an adjuvant therapy for breast cancer [80]. Recent clinical trials using tamoxifen in a breast cancer prevention trial have suggested its estrogen-like cardioprotective properties. Tamoxifen was shown to reduce the incidence of acute MI or angina [17], improve endothelial function [101], and reduce carotid artery intima-media thickness [101] in postmenopausal women. Animal models of tamoxifen therapy have demonstrated a reduction in ischemic damage after cerebral occlusion [61] and a decrease in the progression of atherosclerosis [119]. Tamoxifen has its drawbacks for long-term use, namely, the increased risk of venous thromboembolic events in women with conventional risk factors for atherosclerosis [30] as well as an increased risk of endometrial cancer in women on tamoxifen therapy [121].
Toremifene was originally developed because it was thought to have fewer toxic effects than its parent tamoxifien [53]. Few clinical trials have been conducted with toremifene. It has demonstrated that cardiovascular effects similar to those observed with Tamoxifen reduce serum LDL cholesterol levels, but, unlike tamoxifen, toremifene modestly raises serum HDL cholesterol levels and lowers serum triglyceride levels [64]. The risk for stroke and pulmonary embolism may be less for toremifene than tamoxifen [53].
Originally developed for breast cancer, raloxifene is currently prescribed for the prevention and treatment of osteoporosis. As with tamoxifen, raloxifene has been studied in several clinical trials. Raloxifene treatment had a neutral effect on cardiovascular event outcomes in the MORE trial [37], but was found to have a positive effect on a subset of women at high-risk for cardiovascular disease [9]. However, raloxifene was shown to have no overall benefit in women at high risk for cardiovascular disease after a 5-year follow-up of the raloxifene use in the heart (RUTH) trial [10].
Pathway Selective Estrogen Receptor Ligands
Another way of taking advantage of estrogen’s beneficial effects is to selectively target the non-genomic effects of estrogen receptor activation, one of which is the ability to inhibit the activity of nuclear factor kappa B (NF-κB) [5–8]. Estrogen’s anti-inflammatory action is attributed to inhibition of NF-κB activity [15, 16] in animal models of inflammatory disease such as arthritis [59]. Pathway-selective ER ligands are non-steroidal compounds that would retain the anti-inflammatory function of estrogen receptor activation while devoid of conventional estrogenic activity. The pathway selective ER ligand WAY-169916 is a non-steroidal ER-dependent inhibitor of NF-κB transcriptional activity. Although it inhibits the expression of a range of inflammatory proteins that are expressed after NF-κB activation, WAY-169916 lacks estrogenic activity such as the stimulation of uterine proliferation [19]. Additionally, this compound has proved to be effective in reducing myocardial infarct size after ischemia and reperfusion [15, 51]. Compounds such as WAY-169916 lack classical estrogenic effects; it and others in its class may be therapeutically useful in the preventive management of cardiovascular disease in post-menopausal patients while avoiding the unwanted side effects.
Conclusions
Although the clinical trials provided important conclusions regarding the treatment of post-menopausal women with hormone replacement therapy, it also raised many questions regarding the effects of hormone therapy on younger women without existing cardiovascular disease. These trials have provided the framework for future studies in the field of hormone replacement to better determine the mechanism by which estrogen protects the cardiovascular system as well as the most appropriate time to treat after menopause.
There is compelling evidence to suggest a role for estrogen in modulating the immune system and protecting the myocyte after ischemia and reperfusion injury. Estrogen responses are diverse and depend on the type of estrogen administered, the binding affinity of the estrogen to the two known estrogen receptors as well as possible membrane targets, the age at which hormone replacement is initiated, and the inhibitory effects of other concurrently administered therapeutic interventions (such as selective and non-selective COX-2 inhibitors). A number of questions must still be addressed in this constantly evolving field. The Kronos Early Estrogen Prevention Study (KEEPS) was designed to answer two of the unresolved questions: (a) early initiation of hormone replacement and (b) the route of administration. This is a multi center, 5-year, randomized, double-blind, placebo-controlled clinical trial that will evaluate the effectiveness of oral CEE and transdermal estradiol in preventing progression of atherosclerosis in recent post-menopausal women.
The understanding of the molecular mechanisms of estrogen will be the basis of new pharmacological developments to prevent the deleterious effects of exogenous estrogen while preserving the desired responses. The design of selective estrogen receptor modulators that prevent breast cancer and cardiovascular disease while maintaining bone mass is a challenge for the future of menopause treatment.
References
Acconcia, F., Ascenzi, P., Bocedi, A., Spisni, E., Tomasi, V., Trentalance, A., et al. (2005). Palmitoylation-dependent estrogen receptor alpha membrane localization: Regulation by 17beta-estradiol. Molecular Biology of the Cell, 16, 231–237. doi:10.1091/mbc.E04-07-0547.
Acconcia, F., Ascenzi, P., Fabozzi, G., Visca, P., & Marino, M. (2004). S-palmitoylation modulates human estrogen receptor-alpha functions. Biochemical and Biophysical Research Communications, 316, 878–883. doi:10.1016/j.bbrc.2004.02.129.
Adams, M. R., Kaplan, J. R., Manuck, S. B., Koritnik, D. R., Parks, J. S., Wolfe, M. S., et al. (1990). Inhibition of coronary artery atherosclerosis by 17-beta estradiol in ovariectomized monkeys. Lack of an effect of added progesterone. Arteriosclerosis (Dallas, Tex.), 10, 1051–1057.
Adams, M. R., Register, T. C., Golden, D. L., Wagner, J. D., & Williams, J. K. (1997). Medroxyprogesterone acetate antagonizes inhibitory effects of conjugated equine estrogens on coronary artery atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 17, 217–221.
Alcindor, D., Krolikowski, J. G., Pagel, P. S., Warltier, D. C., & Kersten, J. R. (2004). Cyclooxygenase-2 mediates ischemic, anesthetic, and pharmacologic preconditioning in vivo. Anesthesiology, 100, 547–554. doi:10.1097/00000542-200403000-00013.
Bae, S., & Zhang, L. (2005). Gender differences in cardioprotection against ischemia/reperfusion injury in adult rat hearts: Focus on Akt and protein kinase C signaling. The Journal of Pharmacology and Experimental Therapeutics, 315, 1125–1135. doi:10.1124/jpet.105.090803.
Barp, J., Araujo, A. S., Fernandes, T. R., Rigatto, K. V., Llesuy, S., Bello-Klein, A., et al. (2002). Myocardial antioxidant and oxidative stress changes due to sex hormones. Brazilian Journal of Medical and Biological Research, 35, 1075–1081. doi:10.1590/S0100-879X2002000900008.
Barrett-Connor, E., & Grady, D. (1998). Hormone replacement therapy, heart disease, and other considerations. Annual Review of Public Health, 19, 55–72. doi:10.1146/annurev.publhealth.19.1.55.
Barrett-Connor, E., Grady, D., Sashegyi, A., Anderson, P. W., Cox, D. A., Hoszowski, K., et al. (2002). Raloxifene and cardiovascular events in osteoporotic postmenopausal women: Four-year results from the MORE (Multiple Outcomes of Raloxifene Evaluation) randomized trial. Journal of the American Medical Association, 287, 847–857. doi:10.1001/jama.287.7.847.
Barrett-Connor, E., Mosca, L., Collins, P., Geiger, M. J., Grady, D., Kornitzer, M., et al. (2006). Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. The New England Journal of Medicine, 355, 125–137. doi:10.1056/NEJMoa062462.
Beck, J. I., Boothroyd, C., Proctor, M., Farquhar, C., & Hughes, E. (2005). Oral anti-oestrogens and medical adjuncts for subfertility associated with anovulation. Cochrane Database of Systematic Reviews (Online: Update Software), CD002249.
Bolli, R., Shinmura, K., Tang, X. L., Kodani, E., Xuan, Y. T., Guo, Y., et al. (2002). Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning. Cardiovascular Research, 55, 506–519. doi:10.1016/S0008-6363(02)00414-5.
Bombardier, C., Laine, L., Reicin, A., Shapiro, D., Burgos-Vargas, R., Davis, B., et al. (2000). Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. New England Journal of Medicine, 343, 1520–1528, 1522 p following 1528.
Booth, E. A., Marchesi, M., Kilbourne, E. J., & Lucchesi, B. R. (2003). 17Beta-estradiol as a receptor-mediated cardioprotective agent. The Journal of Pharmacology and Experimental Therapeutics, 307, 395–401. doi:10.1124/jpet.103.054205.
Booth, E. A., Marchesi, M., Knittel, A. K., Kilbourne, E. J., & Lucchesi, B. R. (2007). The pathway-selective estrogen receptor ligand WAY-169916 reduces infarct size after myocardial ischemia and reperfusion by an estrogen receptor dependent mechanism. Journal of Cardiovascular Pharmacology, 49, 401–407. doi:10.1097/FJC.0b013e3180544527.
Booth, E. A., Obeid, N. R., & Lucchesi, B. R. (2005). Activation of estrogen receptor-alpha protects the in vivo rabbit heart from ischemia-reperfusion injury. American Journal of Physiology. Heart and Circulatory Physiology, 289(5), H2039–H2047. doi:10.1152/ajpheart.00479.2005.
Bradbury, B. D., Lash, T. L., Kaye, J. A., & Jick, S. S. (2005). Tamoxifen-treated breast carcinoma patients and the risk of acute myocardial infarction and newly-diagnosed angina. Cancer, 103, 1114–1121. doi:10.1002/cncr.20900.
Braquet, P., Touqui, L., Shen, T. Y., & Vargaftig, B. B. (1987). Perspectives in platelet-activating factor research. Pharmacological Reviews, 39, 97–145.
Chadwick, C. C., Chippari, S., Matelan, E., Borges-Marcucci, L., Eckert, A. M., Keith, J. C., Jr., et al. (2005). Identification of pathway-selective estrogen receptor ligands that inhibit NF-kappaB transcriptional activity. Proceedings of the National Academy of Sciences of the United States of America, 102, 2543–2548. doi:10.1073/pnas.0405841102.
Chambliss, K. L., Yuhanna, I. S., Mineo, C., Liu, P., German, Z., Sherman, T. S., et al. (2000). Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circulation Research, 87, E44–E52.
Clarkson, T. B., Anthony, M. S., & Wagner, J. D. (2001). A comparison of tibolone and conjugated equine estrogens effects on coronary artery atherosclerosis and bone density of postmenopausal monkeys. The Journal of Clinical Endocrinology and Metabolism, 86, 5396–5404. doi:10.1210/jc.86.11.5396.
Coker, S. J., & Parratt, J. R. (1985). AH23848, a thromboxane antagonist, suppresses ischaemia and reperfusion-induced arrhythmias in anaesthetized greyhounds. British Journal of Pharmacology, 86, 259–264.
Collins, P., & Webb, C. (1999). Estrogen hits the surface. Nature Medicine, 5, 1130–1131. doi:10.1038/13453.
Couse, J. F., & Korach, K. S. (1999). Estrogen receptor null mice: What have we learned and where will they lead us? Endocrine Reviews, 20, 358–417. doi:10.1210/er.20.3.358.
Cross, H. R., Kranias, E. G., Murphy, E., & Steenbergen, C. (2003). Ablation of PLB exacerbates ischemic injury to a lesser extent in female than male mice: Protective role of NO. American Journal of Physiology. Heart and Circulatory Physiology, 284, H683–H690.
Cross, H. R., Lu, L., Steenbergen, C., Philipson, K. D., & Murphy, E. (1998). Overexpression of the cardiac Na+/Ca2+ exchanger increases susceptibility to ischemia/reperfusion injury in male, but not female, transgenic mice. Circulation Research, 83, 1215–1223.
Cross, H. R., Murphy, E., Koch, W. J., & Steenbergen, C. (2002). Male and female mice overexpressing the beta(2)-adrenergic receptor exhibit differences in ischemia/reperfusion injury: Role of nitric oxide. Cardiovascular Research, 53, 662–671. doi:10.1016/S0008-6363(01)00528-4.
Cross, H. R., Steenbergen, C., Lefkowitz, R. J., Koch, W. J., & Murphy, E. (1999). Overexpression of the cardiac beta(2)-adrenergic receptor and expression of a beta-adrenergic receptor kinase-1 (betaARK1) inhibitor both increase myocardial contractility but have differential effects on susceptibility to ischemic injury. Circulation Research, 85, 1077–1084.
Das, B., & Sarkar, C. (2006). Similarities between ischemic preconditioning and 17beta-estradiol mediated cardiomyocyte KATP channel activation leading to cardioprotective and antiarrhythmic effects during ischemia/reperfusion in the intact rabbit heart. Journal of Cardiovascular Pharmacology, 47, 277–286. doi:10.1097/01.fjc.0000202563.54043.d6.
Decensi, A., Maisonneuve, P., Rotmensz, N., Bettega, D., Costa, A., Sacchini, V., et al. (2005). Effect of tamoxifen on venous thromboembolic events in a breast cancer prevention trial. Circulation, 111, 650–656. doi:10.1161/01.CIR.0000154545.84124.AC.
Decensi, A., Omodei, U., Robertson, C., Bonanni, B., Guerrieri-Gonzaga, A., Ramazzotto, F., et al. (2002). Effect of transdermal estradiol and oral conjugated estrogen on C-reactive protein in retinoid-placebo trial in healthy women. Circulation, 106, 1224–1228. doi:10.1161/01.CIR.0000028463.74880.EA.
Delyani, J. A., Murohara, T., Nossuli, T. O., & Lefer, A. M. (1996). Protection from myocardial reperfusion injury by acute administration of 17 beta-estradiol. Journal of Molecular and Cellular Cardiology, 28, 1001–1008. doi:10.1006/jmcc.1996.0093.
Deshpande, R., Khalili, H., Pergolizzi, R. G., Michael, S. D., & Chang, M. D. (1997). Estradiol down-regulates LPS-induced cytokine production and NFκB activation in murine macrophages. American Journal of Reproductive Immunology (New York, N.Y.), 38, 46–54.
DeWitt, D. L. (1991). Prostaglandin endoperoxide synthase: Regulation of enzyme expression. Biochimica et Biophysica Acta, 1083, 121–134.
Dubey, R. K., Jackson, E. K., Gillespie, D. G., Zacharia, L. C., Imthurn, B., & Keller, P. J. (2000). Clinically used estrogens differentially inhibit human aortic smooth muscle cell growth and mitogen-activated protein kinase activity. Arteriosclerosis, Thrombosis, and Vascular Biology, 20, 964–972.
Egan, K. M., Lawson, J. A., Fries, S., Koller, B., Rader, D. J., Smyth, E. M., et al. (2005). Cyclooxygenase-2-derived prostacyclin confers atheroprotection on female mice. Obstetrical & Gynecological Survey, 60, 309–310. doi:10.1097/01.ogx.0000160574.33752.4f.
Ensrud, K., Genazzani, A. R., Geiger, M. J., McNabb, M., Dowsett, S. A., Cox, D. A., et al. (2006). Effect of raloxifene on cardiovascular adverse events in postmenopausal women with osteoporosis. The American Journal of Cardiology, 97, 520–527. doi:10.1016/j.amjcard.2005.09.083.
Evans, M. J., Eckert, A., Lai, K., Adelman, S. J., & Harnish, D. C. (2001). Reciprocal antagonism between estrogen receptor and NF-kappaB activity in vivo. Circulation Research, 89, 823–830. doi:10.1161/hh2101.098543.
Fischer, G. M., & Swain, M. L. (1985). Effects of estradiol and progesterone on the increased synthesis of collagen in atherosclerotic rabbit aortas. Atherosclerosis, 54, 177–185. doi:10.1016/0021-9150(85)90177-7.
Forster, C., Kietz, S., Hultenby, K., Warner, M., & Gustafsson, J. A. (2004). Characterization of the ERbeta−/−mouse heart. Proceedings of the National Academy of Sciences of the United States of America, 101, 14234–14239. doi:10.1073/pnas.0405571101.
Garcia-Moll, X., Zouridakis, E., Cole, D., & Kaski, J. C. (2000). C-reactive protein in patients with chronic stable angina: Differences in baseline serum concentration between women and men. European Heart Journal, 21, 1598–1606. doi:10.1053/euhj.2000.2128.
Gilroy, D. W., Colville-Nash, P. R., Willis, D., Chivers, J., Paul-Clark, M. J., & Willoughby, D. A. (1999). Inducible cyclooxygenase may have anti-inflammatory properties. Nature Medicine, 5, 698–701. doi:10.1038/9550.
Godsland, I. F., Crook, D., Worthington, M., Proudler, A. J., Felton, C., Sidhu, M., et al. (1993). Effects of a low-estrogen, desogestrel-containing oral contraceptive on lipid and carbohydrate metabolism. Contraception, 48, 217–227.
Grady, D., Gebretsadik, T., Kerlikowske, K., Ernster, V., & Petitti, D. (1995). Hormone replacement therapy and endometrial cancer risk: A meta-analysis. Obstetrics and Gynecology, 85, 304–313. doi:10.1016/0029-7844(94)00383-O.
Grady, D., Herrington, D., Bittner, V., Blumenthal, R., Davidson, M., Hlatky, M., et al. (2002). Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II). Journal of the American Medical Association, 288, 49–57. doi:10.1001/jama.288.1.49.
Greene, G. L., Gilna, P., Waterfield, M., Baker, A., Hort, Y., & Shine, J. (1986). Sequence and expression of human estrogen receptor complementary DNA. Science, 231, 1150–1154. doi:10.1126/science.3753802.
Grodstein, F., Manson, J. E., Colditz, G. A., Willett, W. C., Speizer, F. E., & Stampfer, M. J. (2000). A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Annals of Internal Medicine, 133, 933–941.
Grodstein, F., Manson, J. E., & Stampfer, M. J. (2006). Hormone therapy and coronary heart disease: The role of time since menopause and age at hormone initiation. Journal of Women’s Health, 15, 35–44. doi:10.1089/jwh.2006.15.35.
Hale, S. L., Birnbaum, Y., & Kloner, R. A. (1996). beta-Estradiol, but not alpha-estradiol, reduced myocardial necrosis in rabbits after ischemia and reperfusion. American Heart Journal, 132, 258–262. doi:10.1016/S0002-8703(96)90419-6.
Hale, S. L., Birnbaum, Y., & Kloner, R. A. (1997). Estradiol, administered acutely, protects ischemic myocardium in both female and male rabbits. Journal of Cardiovascular Pharmacology and Therapeutics, 2, 47–52. doi:10.1177/107424849700200106.
Harnish, D. C., Liu, X., Kenney, T., Winneker, R. C., Chadwick, C., Friedrichs, G. S., et al. (2006). The pathway-selective estrogen receptor ligand WAY-169916 displays differential activity in ischemia-reperfusion injury models. Journal of Cardiovascular Pharmacology, 47, 788–795. doi:10.1097/01.fjc.0000211793.60528.f7.
Harnish, D. C., Scicchitano, M. S., Adelman, S. J., Lyttle, C. R., & Karathanasis, S. K. (2000). The role of CBP in estrogen receptor cross-talk with nuclear factor-kappaB in HepG2 cells. Endocrinology, 141, 3403–3411. doi:10.1210/en.141.9.3403.
Harvey, H. A., Kimura, M., & Hajba, A. (2006). Toremifene: An evaluation of its safety profile. The Breast, 15, 142–157. doi:10.1016/j.breast.2005.09.007.
Hulley, S., Grady, D., Bush, T., Furberg, C., Herrington, D., Riggs, B., et al. (1998). Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) research group. Journal of the American Medical Association, 280, 605–613. doi:10.1001/jama.280.7.605.
Hulley, S. B., & Grady, D. (2004). The WHI estrogen-alone trial—do things look any better? Journal of the American Medical Association, 291, 1769–1771. doi:10.1001/jama.291.14.1769.
Juni, P., Nartey, L., Reichenbach, S., Sterchi, R., Dieppe, P. A., & Egger, M. (2004). Risk of cardiovascular events and rofecoxib: Cumulative meta-analysis. Lancet, 364, 2021–2029. doi:10.1016/S0140-6736(04)17514-4.
Karas, R. H., Patterson, B. L., & Mendelsohn, M. E. (1994). Human vascular smooth muscle cells contain functional estrogen receptor. Circulation, 89, 1943–1950.
Kauffman, R. F., Bean, J. S., Fahey, K. J., Cullinan, G. J., Cox, D. A., & Bensch, W. R. (2000). Raloxifene and estrogen inhibit neointimal thickening after balloon injury in the carotid artery of male and ovariectomized female rats. Journal of Cardiovascular Pharmacology, 36, 459–465. doi:10.1097/00005344-200010000-00007.
Keith, J. C., Jr., Albert, L. M., Leathurby, Y., Follettie, M., Wang, L., Borges-Marcucci, L., et al. (2005). The utility of pathway selective estrogen receptor ligands that inhibit nuclear factor-kappaB transcriptional activity in models of rheumatoid arthritis. Arthritis Research & Therapy, 7, R427–R438. doi:10.1186/ar1692.
Kim, Y. D., Chen, B., Beauregard, J., Kouretas, P., Thomas, G., Farhat, M. Y., et al. (1996). 17 beta-Estradiol prevents dysfunction of canine coronary endothelium and myocardium and reperfusion arrhythmias after brief ischemia/reperfusion. Circulation, 94, 2901–2908.
Kimelberg, H. K., Feustel, P. J., Jin, Y., Paquette, J., Boulos, A., Keller, R. W., Jr., et al. (2000). Acute treatment with tamoxifen reduces ischemic damage following middle cerebral artery occlusion. Neuroreport, 11, 2675–2679. doi:10.1097/00001756-200008210-00014.
Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Haggblad, J., Nilsson, S., et al. (1997). Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology, 138, 863–870. doi:10.1210/en.138.3.863.
Kuiper, G. G., & Gustafsson, J. A. (1997). The novel estrogen receptor-beta subtype: Potential role in the cell- and promoter-specific actions of estrogens and anti-estrogens. FEBS Letters, 410, 87–90. doi:10.1016/S0014-5793(97)00413-4.
Kusama, M., Miyauchi, K., Aoyama, H., Sano, M., Kimura, M., Mitsuyama, S., et al. (2004). Effects of toremifene (TOR) and tamoxifen (TAM) on serum lipids in postmenopausal patients with breast cancer. Breast Cancer Research and Treatment, 88, 1–8. doi:10.1007/s10549-004-4384-z.
Lang, T. J. (2004). Estrogen as an immunomodulator. Clinical Immunology (Orlando, Fla.), 113, 224–230. doi:10.1016/j.clim.2004.05.011.
Levin, E. R. (1999). Cellular functions of the plasma membrane estrogen receptor. Trends in Endocrinology and Metabolism, 10, 374–377. doi:10.1016/S1043-2760(99)00192-7.
Levine, R. L., Chen, S. J., Durand, J., Chen, Y. F., & Oparil, S. (1996). Medroxyprogesterone attenuates estrogen-mediated inhibition of neointima formation after balloon injury of the rat carotid artery. Circulation, 94, 2221–2227.
Li, L., Haynes, M. P., & Bender, J. R. (2003). Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proceedings of the National Academy of Sciences of the United States of America, 100, 4807–4812. doi:10.1073/pnas.0831079100.
Li, Y., & Kloner, R. A. (1995). Is there a gender difference in infarct size and arrhythmias following experimental coronary occlusion and reperfusion? Journal of Thrombosis and Thrombolysis, 2, 221–225.
Losordo, D. W., Kearney, M., Kim, E. A., Jekanowski, J., & Isner, J. M. (1994). Variable expression of the estrogen receptor in normal and atherosclerotic coronary arteries of premenopausal women. Circulation, 89, 1501–1510.
Lucchesi, B. R. (1990). Modulation of leukocyte-mediated myocardial reperfusion injury. Annual Review of Physiology, 52, 561–576. doi:10.1146/annurev.ph.52.030190.003021.
McCord, J. M. (1985). Oxygen-derived free radicals in postischemic tissue injury. The New England Journal of Medicine, 312, 159–163.
Meldrum, D. R. (1998). Tumor necrosis factor in the heart. The American Journal of Physiology, 274, R577–R595.
Mendelsohn, M. E., & Karas, R. H. (1999). The protective effects of estrogen on the cardiovascular system. The New England Journal of Medicine, 340, 1801–1811. doi:10.1056/NEJM199906103402306.
Mosca, L., Collins, P., Herrington, D. M., Mendelsohn, M. E., Pasternak, R. C., Robertson, R. M., et al. (2001). Hormone replacement therapy and cardiovascular disease: A statement for healthcare professionals from the American Heart Association. Circulation, 104, 499–503. doi:10.1161/hc2901.092200.
Nabulsi, A. A., Folsom, A. R., White, A., Patsch, W., Heiss, G., Wu, K. K., et al. (1993). Association of hormone-replacement therapy with various cardiovascular risk factors in postmenopausal women. The atherosclerosis risk in communities study investigators. The New England Journal of Medicine, 328, 1069–1075. doi:10.1056/NEJM199304153281501.
Nakagomi, A., Freedman, S. B., & Geczy, C. L. (2000). Interferon-gamma and lipopolysaccharide potentiate monocyte tissue factor induction by C-reactive protein: Relationship with age, sex, and hormone replacement treatment. Circulation, 101, 1785–1791.
Nijmeijer, R., Lagrand, W. K., Lubbers, Y. T., Visser, C. A., Meijer, C. J., Niessen, H. W., et al. (2003). C-reactive protein activates complement in infarcted human myocardium. American Journal of Pathology, 163, 269–275.
Nuedling, S., Karas, R. H., Mendelsohn, M. E., Katzenellenbogen, J. A., Katzenellenbogen, B. S., Meyer, R., et al. (2001). Activation of estrogen receptor beta is a prerequisite for estrogen-dependent upregulation of nitric oxide synthases in neonatal rat cardiac myocytes. FEBS Letters, 502, 103–108. doi:10.1016/S0014-5793(01)02675-8.
Osborne, C. K. (1998). Tamoxifen in the treatment of breast cancer. The New England Journal of Medicine, 339, 1609–1618. doi:10.1056/NEJM199811263392207.
Pappas, T. C., Gametchu, B., & Watson, C. S. (1995). Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. The FASEB Journal, 9, 404–410.
Park, J. L., & Lucchesi, B. R. (1999). Mechanisms of myocardial reperfusion injury. The Annals of Thoracic Surgery, 68, 1905–1912. doi:10.1016/S0003-4975(99)01073-5.
Patten, R. D., Pourati, I., Aronovitz, M. J., Baur, J., Celestin, F., Chen, X., et al. (2004). 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circulation Research, 95, 692–699. doi:10.1161/01.RES.0000144126.57786.89.
Pelzer, T., Loza, P. A., Hu, K., Bayer, B., Dienesch, C., Calvillo, L., et al. (2005). Increased mortality and aggravation of heart failure in estrogen receptor-beta knockout mice after myocardial infarction. Circulation, 111, 1492–1498. doi:10.1161/01.CIR.0000159262.18512.46.
Pettersson, K., & Gustafsson, J. A. (2001). Role of estrogen receptor beta in estrogen action. Annual Review of Physiology, 63, 165–192. doi:10.1146/annurev.physiol.63.1.165.
Rau, S. W., Dubal, D. B., Bottner, M., Gerhold, L. M., & Wise, P. M. (2003). Estradiol attenuates programmed cell death after stroke-like injury. The Journal of Neuroscience, 23, 11420–11426.
Ray, A., Prefontaine, K. E., & Ray, P. (1994). Down-modulation of interleukin-6 gene expression by 17 beta-estradiol in the absence of high affinity DNA binding by the estrogen receptor. The Journal of Biological Chemistry, 269, 12940–12946.
Ray, P., Ghosh, S. K., Zhang, D. H., & Ray, A. (1997). Repression of interleukin-6 gene expression by 17 beta-estradiol: Inhibition of the DNA-binding activity of the transcription factors NF-IL6 and NF-kappa B by the estrogen receptor. FEBS Letters, 409, 79–85. doi:10.1016/S0014-5793(97)00487-0.
Razandi, M., Pedram, A., Greene, G. L., & Levin, E. R. (1999). Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: Studies of ERalpha and ERbeta expressed in Chinese hamster ovary cells. Molecular Endocrinology (Baltimore, Md.), 13, 307–319. doi:10.1210/me.13.2.307.
Revankar, C. M., Cimino, D. F., Sklar, L. A., Arterburn, J. B., & Prossnitz, E. R. (2005). A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science, 307, 1625–1630. doi:10.1126/science.1106943.
Riggs, B. L., & Hartmann, L. C. (2003). Selective estrogen-receptor modulators—Mechanisms of action and application to clinical practice. The New England Journal of Medicine, 348, 618–629. doi:10.1056/NEJMra022219.
Rossouw, J. E., Anderson, G. L., Prentice, R. L., LaCroix, A. Z., Kooperberg, C., Stefanick, M. L., et al. (2002). Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. Journal of the American Medical Association, 288, 321–333. doi:10.1001/jama.288.3.321.
Sbarouni, E., Iliodromitis, E. K., Bofilis, E., Kyriakides, Z. S., & Kremastinos, D. T. (2003). Estrogen alone or combined with medroxyprogesterone but not raloxifene reduce myocardial infarct size. European Journal of Pharmacology, 467, 163–168. doi:10.1016/S0014-2999(03)01627-3.
Schuit, S. C., de Jong, F. H., Stolk, L., Koek, W. N., van Meurs, J. B., Schoofs, M. W., et al. (2005). Estrogen receptor alpha gene polymorphisms are associated with estradiol levels in postmenopausal women. European Journal of Endocrinology, 153, 327–334. doi:10.1530/eje.1.01973.
Shaw, L., Taggart, M. J., & Austin, C. (2000). Mechanisms of 17 beta-oestradiol induced vasodilatation in isolated pressurized rat small arteries. British Journal of Pharmacology, 129, 555–565. doi:10.1038/sj.bjp.0703084.
Silvestri, A., Gebara, O., Vitale, C., Wajngarten, M., Leonardo, F., Ramires, J. A., et al. (2003). Increased levels of C-reactive protein after oral hormone replacement therapy may not be related to an increased inflammatory response. Circulation, 107, 3165–3169. doi:10.1161/01.CIR.0000074208.02226.5E.
Siris, E. S., Harris, S. T., Eastell, R., Zanchetta, J. R., Goemaere, S., Diez-Perez, A., et al. (2005). Skeletal effects of raloxifene after 8 years: Results from the continuing outcomes relevant to Evista (CORE) study. Journal of Bone and Mineral Research, 20, 1514–1524. doi:10.1359/JBMR.050509.
Skavdahl, M., Steenbergen, C., Clark, J., Myers, P., Demianenko, T., Mao, L., et al. (2005). Estrogen receptor-beta mediates male–female differences in the development of pressure overload hypertrophy. American Journal of Physiology. Heart and Circulatory Physiology, 288, H469–H476. doi:10.1152/ajpheart.00723.2004.
Speir, E., Yu, Z. X., Takeda, K., Ferrans, V. J., & Cannon, R. O., III. (2000). Competition for p300 regulates transcription by estrogen receptors and nuclear factor-kappaB in human coronary smooth muscle cells. Circulation Research, 87, 1006–1011.
Squadrito, F., Altavilla, D., Squadrito, G., Campo, G. M., Arlotta, M., Arcoraci, V., et al. (1997). 17Beta-oestradiol reduces cardiac leukocyte accumulation in myocardial ischaemia reperfusion injury in rat. European Journal of Pharmacology, 335, 185–192. doi:10.1016/S0014-2999(97)01201-6.
Stamatelopoulos, K. S., Lekakis, J. P., Poulakaki, N. A., Papamichael, C. M., Venetsanou, K., Aznaouridis, K., et al. (2004). Tamoxifen improves endothelial function and reduces carotid intima-media thickness in postmenopausal women. American Heart Journal, 147, 1093–1099. doi:10.1016/j.ahj.2003.12.029.
Steenbergen, C., Murphy, E., Levy, L., & London, R. E. (1987). Elevation in cytosolic free calcium concentration early in myocardial ischemia in perfused rat heart. Circulation Research, 60, 700–707.
Stein, B., & Yang, M. X. (1995). Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-kappa B and C/EBP beta. Molecular and Cellular Biology, 15, 4971–4979.
Strong, J. P. (1999). Atherosclerosis in the young: Risk and prevention. Hospital Practice (Off Ed), 34, 15–16, 19.
Sun, J., Picht, E., Ginsburg, K. S., Bers, D. M., Steenbergen, C., & Murphy, E. (2006). Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circulation Research, 98, 403–411. doi:10.1161/01.RES.0000202707.79018.0a.
Sun, W. H., Keller, E. T., Stebler, B. S., & Ershler, W. B. (1998). Estrogen inhibits phorbol ester-induced I kappa B alpha transcription and protein degradation. Biochemical and Biophysical Research Communications, 244, 691–695. doi:10.1006/bbrc.1998.8324.
Tanhehco, E. J., Lee, H., & Lucchesi, B. R. (2000). Sublytic complement attack reduces infarct size in rabbit isolated hearts: Evidence for C5a-mediated cardioprotection. Immunopharmacology, 49, 391–399. doi:10.1016/S0162-3109(00)00258-7.
Tanhehco, E. J., Yasojima, K., McGeer, P. L., Washington, R. A., Kilgore, K. S., Homeister, J. W., et al. (1999). Preconditioning reduces tissue complement gene expression in the rabbit isolated heart. The American Journal of Physiology, 277, H2373–H2380.
Thurberg, B. L., & Collins, T. (1998). The nuclear factor-kappa B/inhibitor of kappa B autoregulatory system and atherosclerosis. Current Opinion in Lipidology, 9, 387–396. doi:10.1097/00041433-199810000-00002.
Toran-Allerand, C. D. (2004). Minireview: A plethora of estrogen receptors in the brain: Where will it end? Endocrinology, 145, 1069–1074. doi:10.1210/en.2003-1462.
Torzewski, M., Torzewski, J., Bowyer, D. E., Waltenberger, J., Fitzsimmons, C., Hombach, V., et al. (1997). Immunohistochemical colocalization of the terminal complex of human complement and smooth muscle cell alpha-actin in early atherosclerotic lesions. Arteriosclerosis, Thrombosis, and Vascular Biology, 17, 2448–2452.
trial WGftP. (1995). Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI). Trial the writing group for the PEPI Trial. Journal of the American Medical Association, 273, 199–208. doi:10.1001/jama.273.3.199.
Tsai, C. H., Su, S. F., Chou, T. F., & Lee, T. M. (2002). Differential effects of sarcolemmal and mitochondrial K(ATP) channels activated by 17 beta-estradiol on reperfusion arrhythmias and infarct sizes in canine hearts. The Journal of Pharmacology and Experimental Therapeutics, 301, 234–240. doi:10.1124/jpet.301.1.234.
Vane, J. (1994). Towards a better aspirin. Nature, 367, 215–216. doi:10.1038/367215a0.
Wagner, J. D., Clarkson, T. B., St Clair, R. W., Schwenke, D. C., Shively, C. A., & Adams, M. R. (1991). Estrogen and progesterone replacement therapy reduces low density lipoprotein accumulation in the coronary arteries of surgically postmenopausal cynomolgus monkeys. The Journal of Clinical Investigation, 88, 1995–2002. doi:10.1172/JCI115526.
Walsh, B. W., Schiff, I., Rosner, B., Greenberg, L., Ravnikar, V., & Sacks, F. M. (1991). Effects of postmenopausal estrogen replacement on the concentrations and metabolism of plasma lipoproteins. The New England Journal of Medicine, 325, 1196–1204.
Williams, J. K., Adams, M. R., Herrington, D. M., & Clarkson, T. B. (1992). Short-term administration of estrogen and vascular responses of atherosclerotic coronary arteries. Journal of the American College of Cardiology, 20, 452–457.
Williams, J. K., Anthony, M. S., Honore, E. K., Herrington, D. M., Morgan, T. M., Register, T. C., et al. (1995). Regression of atherosclerosis in female monkeys. Arteriosclerosis, Thrombosis, and Vascular Biology, 15, 827–836.
Williams, J. K., Wagner, J. D., Li, Z., Golden, D. L., & Adams, M. R. (1997). Tamoxifen inhibits arterial accumulation of LDL degradation products and progression of coronary artery atherosclerosis in monkeys. Arteriosclerosis, Thrombosis, and Vascular Biology, 17, 403–408.
Writing, G. (1995). Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. The writing group for the PEPI Trial. Journal of the American Medical Association, 273, 199–208. doi:10.1001/jama.273.3.199.
Writing, G. (1998). Tamoxifen for early breast cancer: An overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet, 351, 1451–1467. doi:10.1016/S0140-6736(97)11423-4.
Wyckoff, M. H., Chambliss, K. L., Mineo, C., Yuhanna, I. S., Mendelsohn, M. E., Mumby, S. M., et al. (2001). Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Galpha(i). The Journal of Biological Chemistry, 276, 27071–27076. doi:10.1074/jbc.M100312200.
Zhai, P., Eurell, T. E., Cooke, P. S., Lubahn, D. B., & Gross, D. R. (2000). Myocardial ischemia-reperfusion injury in estrogen receptor-alpha knockout and wild-type mice. American Journal of Physiology. Heart and Circulatory Physiology, 278, H1640–H1647.
Zhao, X., Macbride, M. M., Peterson, B. R., Pfaff, D. W., & Vasudevan, N. (2005). Calcium flux in neuroblastoma cells is a coupling mechanism between non-genomic and genomic modes of estrogens. Neuroendocrinology, 81, 174–182. doi:10.1159/000087000.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Booth, E.A., Lucchesi, B.R. Estrogen-Mediated Protection in Myocardial Ischemia-Reperfusion Injury. Cardiovasc Toxicol 8, 101–113 (2008). https://doi.org/10.1007/s12012-008-9022-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-008-9022-2