
Abstract
Although studies have shown that endothelial nitric oxide synthase (eNOS) homozygous knockout mice (eNOS−/−) develop left ventricular (LV) hypertrophy, well compensated at least to 24 wks, uncertainty still exists as to the cardiac functional and molecular mechanistic consequences of eNOS deficiency at later time-points. To bridge the gap in existent data, we examined whole hearts from eNOS−/− and age-matched wild-type (WT) control mice ranging in age from 18 to 52 wks for macroscopic and microscopic histopathology, LV mRNA and protein expression using RNA Dot blots and Western blots, respectively, and LV function using isolated perfused work-performing heart preparations. Heart weight to body weight (HW/BW in mg/g) ratio increased significantly as eNOS−/− mice aged (82.2%, P < 0.001). Multi-focal replacement fibrosis and myocyte degeneration/death were first apparent in eNOS−/− mouse hearts at 40 wks. Progressive increases in LV atrial natriuretic factor (ANF) and α-skeletal actin mRNA levels both correlated significantly with increasing HW/BW ratio in aged eNOS−/− mice (r = 0.722 and r = 0.648, respectively; P < 0.001). At 52 wks eNOS−/− mouse hearts exhibited basal LV hypercontractility yet blunted beta adrenergic receptor (βAR) responsiveness that coincided with a significant reduction in the LV ratio of phospholamban to sarcoplasmic reticulum Ca2+-ATPase-2a protein levels and was preceded by a significant upregulation in LV steady-state mRNA and protein levels of the 28 kDa membrane-bound form of tumor necrosis factor-alpha. We conclude that absence of eNOS in eNOS−/− mice results in a progressive concentric hypertrophic cardiac phenotype that is functionally compensated with decreased βAR responsiveness, and is associated with a potential cytokine-mediated alteration of calcium handling protein expression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Endothelial nitric oxide synthase (eNOS) homozygous mutant mice (eNOS−/−) develop uniform arterial hypertension secondary to a lack of the eNOS gene [1, 2]. Others have reported that cardiac hypertrophy, consequent to this hypertension, occurs rather early in these mice [3, 4]. Therefore, eNOS−/− mice are particularly useful not only as tools to elucidate the functional consequences surrounding the absence of the eNOS gene, but also as instruments to study the long-term effects of chronic pressure overload-induced myocardial remodeling. However, the vast majority of studies, in this regard, have limited their focus to younger [3, 5–7] or very old [8–10] eNOS−/− age groups, and a great number of these reports point almost exclusively to eNOS deficiency as the mechanistic basis for altered cardiac function in these mice [5–7, 9]. Accordingly, as these mice are known to live well beyond 1 year [8, 10], some of the functional features related to the hypertrophic response in eNOS−/− mice have yet to be defined.
A unique aspect of the myocardial hypertrophic response is an attempt by the heart to return to the fetal gene program. This is characterized by alterations in expression patterns of sarcomeric structural proteins and further adaptive modifications at the subcellular level that eventuate, ultimately, in a concentric hypertrophic cardiac phenotype [11]. Although the biochemical bases for the structural and functional alterations occurring during this adaptive response are well documented [11–13], to our knowledge, no study has fully addressed the developmental aspects of these molecular phenomena in naïve eNOS−/− mouse hearts. Hence, many questions also remain regarding the particular biochemical nature of the hypertrophic response in these mice.
Typical of this hypertrophic response and well described in models of hypertrophic heart disease are specific subcellular alterations in expression patterns of key excitation–contraction coupling regulatory molecules, namely SERCA2a (the primary Ca2+-ATPase isoform in cardiac sarcoplasmic reticulum) and phospholamban (PLB; its specific inhibitor) [13–15]. For instance, whereas reductions in SERCA2a protein levels are known to accompany cardiac decompensation and progression toward heart failure [13, 14], other reports suggest that specific stoichiometric changes in levels of its repressor PLB (relative to SERCA2a) may rescue this functional decline in favor of enhanced contractility [16–18]. The precise pathways regulating these changes are not clearly understood. However, in light of recent reports, one possibility is that tumor necrosis factor-alpha (TNF-α), an inducible pro-inflammatory cytokine with pleiotropic effects, may be partially contributing to this biochemical dysregulation [19–21].
Until recently, the primary role of TNF-α in the development of heart failure has been viewed primarily as a contributor to cardiodepression and LV (left ventricular) dilatation [22]. New data suggests, however, that TNF-α has salutary effects when temporally and differentially expressed in the heart and, in this regard, may confer cytoprotective responses [23, 24]. Indeed, evidence from studies performed in genetically modified mice has linked TNF-α expression to the development of a compensatory concentric hypertrophic phenotype and to improvement in cardiac function and survival [25, 26].
In view of the above findings and the discrepant reports in the literature regarding LV hypertrophy and function in eNOS−/− mice, the present study was designed to provide a more detailed characterization of the progressive myocardial remodeling occurring in naïve eNOS−/− mouse hearts. Moreover, we sought to investigate the relationship between TNF-α upregulation, levels of calcium handling proteins and the structural, biochemical and functional alterations that are characteristic of myocardial remodeling in eNOS−/− mice.
Materials and Methods
Mice
Several independent eNOS−/− mouse models exist (eNOSHuang−/−, eNOSShesely−/− and eNOSGodecke−/−). Though each line was originally generated with a slightly different genetic background and subsequently backcrossed onto different hybrid backgrounds (eNOSHuang−/− on C57BL/6/129Sv, eNOSShesely−/− and eNOSGodecke−/− on C57BL/6), it is well established that mice from all lines exhibit significantly elevated blood pressure [1, 2, 5]. Moreover, detailed hemodynamic analysis of C57BL/6, Sv129 and mixed Sv129/C57BL/6 WT mouse strains has revealed no significant differences [6]. eNOS−/− mice utilized here (a gracious gift from Paul Huang; Harvard Medical School, Boston, MA) were back-crossed onto the C57BL/6129/J background for several generations to generate breeding colonies. Age-matched wild-type controls utilized in this study were 18- to 52-week-old male C57BL/6/129 (C57BL/6/129F2/J; Jackson Laboratory, Bar Harbor, Maine, stock #100903) mice. The eNOS−/− mice utilized here were also male mice ranging from 18 to 52 wks of age. All genotypes were preserved by sib–sib breeding. Mouse groups were established by age; group 1: 18–23 wks (12 eNOS−/− and 12 age-matched WT controls); group 2: 27–30 wks (12 eNOS−/− and 12 age-matched WT controls); group 3: 40 wks (12 eNOS−/− and 12 age-matched WT controls); and group 4: 52 wks (6 eNOS−/− and 8 age-matched WT controls). Additional eNOS−/− and WT mice underwent histological analyses at 27–30 wks (2 eNOS−/− and 2 age-matched WT controls) and at 40 wks (2 eNOS−/− and 2 age-matched WT controls), and Western blotting at 18 wks (2 eNOS−/− and 3 age-matched WT controls), 30 wks (3 eNOS−/− and 3 age-matched WT controls), 40 wks (3 eNOS−/− and 2 age-matched WT controls) and 52 wks (4 eNOS−/− and 4 age-matched WT controls). A total of 121 mice were utilized in this study: group 1, n = 32; group 2, n = 34; group 3, n = 33; and group 4, n = 22.
Mice were maintained at the University of Louisville and at the University of Cincinnati in strict accordance with institutional guidelines and the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). Experimental protocols were approved by the Animal Care and Use Committees of the University of Louisville School of Medicine and the University of Cincinnati College of Medicine.
In vivo Measurement of Blood Pressure and Heart Rate
Groups of eNOS−/− and age-matched WT control mice (n = 4) at 18 and 30 wks underwent conscious systolic blood pressure (SBP) and heart rate (HR) measurement with computerized tail cuff manometers coupled with a photoelectric sensor (Visitech Systems, Apex, NC), allowing measurement of SBP in four mice simultaneously, minimizing observer bias. Mice are trained daily for 7 consecutive days followed by SBP recordings for 5 consecutive days. The validity of this system, including correlation of the data with that obtained using intra-arterial measurement has been previously demonstrated [27, 28].
Cardiac Hypertrophy
Heart and Body Weight Measurements
Mice were weighed and euthanized. The hearts were excised while still beating and immediately placed into an RNAse-free ice bath. The right and left ventricles were separated, opened and then rinsed with ice-cold DEPC-treated PBS. Each piece of tissue was blotted and its dry-weight was recorded before being snap-frozen in liquid nitrogen. Measurements were expressed as heart weight to body weight (HW/BW) ratios in mg/g.
Gross Pathology and Histology
Mice were euthanized, hearts were excised while still beating and flushed with PBS (using a 20-gauge needle), followed by cardioplegic solution (25 mM KCl, 5% dextrose, PBS) and finally, fixed by perfusion (at 80 mmHg pressure) using 4% paraformaldehyde (in PBS). The hearts were dehydrated through a graded series of alcohol concentrations and then paraffin-embedded. Step-wise serial sections (5 μm thick) were taken every 75 μm from the base and stained with hematoxylin and eosin or with trichrome and examined by light microscopy. The entire procedure, including examination by an experienced pathologist, was done in a blinded manner.
Cardiac Functional Analysis
Isolated Perfused Work-performing Heart Preparation
The working heart preparation was as described previously [29]. Left intraventricular pressure (IVP) was measured and custom software calculated HR, mean aortic pressure (MAP), IVP, LV systolic pressure (LVSP), time to peak pressure (TPP), half time of relaxation (RT1/2), the first derivatives of the change in LVSP with respect to time (±dP/dt), aortic and coronary flow, venous return (cardiac output), LV minute work (MAP × CO), stroke volume (SV = CO/HR), stroke work (SV × MAP), and perfusate temperature. Frank–Starling curves were generated by linear regression [29].
Analysis of Steady-state mRNA Levels
Dot Blotting
Total RNA was isolated from frozen LV tissue samples and analyzed by quantitative dot blot hybridization as described previously [30]. The synthetic oligonucleotides used as transcript-specific probes were as previously described [30]. A radiolabeled cDNA probe (ATCC, Manassas, VA) was used for TNF-α. Hybridization signals were quantitated using a Storm 840 PhosphorImager and Imagequant software (Molecular Dynamics, Sunnyvale, CA). Samples were assayed in duplicate and signal intensity of each dot was normalized to that produced by a GAPDH probe [30].
Analysis of Cytokine and Calcium Handling Protein Expression
Western Blotting
Protein lysates were prepared for Western blotting of SERCA/PLB and for the membrane-bound form of tumor necrosis factor-alpha (m-TNF-α) as previously described in detail [31, 32]. Protein levels were quantitated by Western blotting as previously described [31] using primary antibodies against the following proteins: TNF-α (Sigma T0938, specifically, we quantitated the 28 kDa m-TNF-α); the 110 kDa protein SERCA2a (Santa Cruz G210); the 24–27 kDa phosphoprotein PLB (Upstate biotechnology). LV tissue protein lysates from age-matched WT control mice and eNOS−/− mice from each group were prepared and analyzed as previously described [32] and bands corresponding to each protein were quantitated by laser densitometry. Signal intensities were normalized to total protein loading determined by densitometry of Ponceau S stained filters. Importantly, SERCA2a and PLB levels were determined simultaneously on the same gels; the gels were cut between 110 kDa and 30 kDa and the upper half blotted and probed for SERCA2A, generally as described above for TNF-α. The bottom portion was blotted to a special nitrocellulose of 2-micron pore size (Schleicher and Shull), this is necessary to retain PLB upon the filter for Western analysis [31]. Reference samples and dilutions to confirm linearity of loading to signal were run on all blots and the signal intensity was normalized to loading (determined by Ponceau S staining) and to the reference sample signal intensity, as previously described [31].
Statistical Analysis
Results are reported as means ± SEM. Differences between age-matched WT control mice and eNOS−/− mice within the same age groups and pair-wise multiple comparisons of intergroup data were analyzed by two-way (time and group) ANOVA. Overall differences were further analyzed by post hoc contrasts with unpaired Student’s t-tests using the Bonferroni correction. Correlation between HW/BW ratio and steady-state mRNA levels was determined by linear regression. Differences were considered significant at P < 0.05 (and P < 0.001). All statistical analyses were performed using the SPSS statistical software (version 10; SPSS, Inc., Chicago, IL).
Results
In vivo Measurement of Blood Pressure and Heart Rate
Systolic blood pressure was significantly elevated in eNOS−/− mice at both 18 (116.1 ± 8.5 vs. 93.9 ± 5.5, in eNOS−/− vs. WT, respectively, P < 0.05) and 30 (122 ± 4.5 vs. 104.6 ± 6.8, in eNOS−/− vs. WT, respectively, P < 0.05) wks of age consistent with published reports [1, 2, 31, 33]. We found no significant change in HR in eNOS−/− mice relative to age-matched WT control, as has been previously reported [34].
Cardiac Hypertrophy
HW/BW ratio was significantly increased in eNOS−/− mice from groups 2 and 3, relative to age-matched WT controls (41.5% and 82.2%, respectively; P < 0.001, Fig. 1). eNOS−/− mouse hearts from group 3 showed a significant increase in HW/BW ratio relative to eNOS−/− mice from groups 1 and 2 (P < 0.001, Fig. 1).

Heart-to-body weight (HW/BW) ratios. eNOS−/− mice exhibited an increase in HW/BW at 27–30 and 40 wks relative to controls. There was a progressive increase of HW/BW in eNOS−/− mice as mice aged
Macroscopic examination of whole hearts and cross-sections from eNOS−/− mice at 27–30 wks (group 2) and 40 wks of age (group 3) confirmed the presence of cardiomegaly and marked LV hypertrophy, respectively, relative to age-matched WT controls. Figure 2 reveals substantial thickening of the LV wall and septum and a smaller LV cavity in eNOS−/− mice from group 3 (40 wks), relative to age-matched WT controls (Fig. 2A–D). Hypertrophy of the RV wall was not appreciated in any of the groups. Likewise, microscopic histological examination revealed cardiomyocyte hypertrophy throughout the LV of eNOS−/− mouse hearts from groups 2 and 3. Myocyte hypertrophy in group 3 was associated with changes in nuclear morphology (Fig. 2E). Many of these nuclei were enlarged and box-shaped (Fig. 2E), and compared to age-matched WT controls (Fig. 2F), 40-wk-old eNOS−/− mouse hearts had an increased number of multi-nuclear cardiomyocytes with cardiomyocyte degeneration/death (Fig. 2E, H). We also observed multiple regions of focal replacement fibrosis (Fig. 2G–H) in hearts from 40-wk-old eNOS−/− mice but not in group 2 or either group’s age-matched WT counterparts. There was no evidence of significant infiltrate in any of the hearts from eNOS−/− or age-matched WT mice.

Cardiac hypertrophy in eNOS−/− mice. Whole hearts and representative cross-sections of eNOS−/− (A and C, respectively) and age-matched WT control (B and D, respectively) mice at 40 wks of age (group 3). Sections were stained with hematoxylin and eosin (×40 magnification). Note the increased thickness of LV wall and smaller LV cavity in eNOS−/− mice. Representative fields (×400, E and F; ×100, G and H) from 40-wk-old eNOS−/− (E, G, and H) and age-matched WT control (F) hearts stained with hematoxylin/eosin (E, F, and H) or trichrome (G). Note cardiomyocyte hypertrophy, enlarged box-shaped nuclei with cardiomyocyte degeneration/death (E and H) and numerous regions of focal fibrosis (G and H) in eNOS−/− mice
LV Re-expression of Fetal Genes
Relative to controls, steady-state α-skeletal actin transcript levels were significantly (P < 0.001) increased in the hearts of eNOS−/− mice from groups 1, 2, and 3 (by 67%, 90%, and 92%, respectively, Fig. 3). Steady-state ANF mRNA levels were also elevated significantly (P < 0.001) by 68%, 89%, and 93.2% in hearts from eNOS−/− mice (groups 1, 2, and 3, respectively, Fig. 3) relative to age-matched controls. Moreover, α-skeletal actin and ANF mRNA levels in eNOS−/− mouse hearts (groups 2 and 3) progressively increased relative to hearts of eNOS−/− mice from group 1 (P < 0.05, Fig. 3). These data were plotted verses HW/BW ratio in aged eNOS−/− mice and linear regression analysis was performed. A significant positive linear relationship was observed between LV steady-state levels of both ANF (r = 0.722; P < 0.001) and α-skeletal actin (r = 0.648; P < 0.001) mRNA (relative to age-matched WT controls) and HW/BW ratio in eNOS−/− mice, indicating a hypertrophic response to increased afterload [35]. The LV α-MyHC steady-state transcript levels were decreased significantly (P < 0.001) by 23%, 35%, and 40% in eNOS−/− mice (groups 1, 2, and 3, respectively) relative to age-matched controls (Fig. 3). Conversely, LV β-MyHC steady-state mRNA levels in eNOS−/− mouse hearts were significantly (P < 0.001) increased in groups 1, 2, and 3 (by 70.8%, 79.2%, and 84.7%, respectively, Fig. 3), relative to age-matched WT controls.

Modulation of LV transcript levels. Steady-state LV mRNA levels were measured for α-skeletal actin, ANF, α-MyHC and β-MyHC in naïve eNOS−/− and wild-type hearts at the indicated ages. Levels of α-skeletal actin and ANF mRNAs were significantly increased in eNOS−/− mice at 18–23 (group 1), 27–30 (group 2) and 40 wks (group 3) of age, relative to controls (P < 0.001). The levels of β-MyHC and α-MyHC mRNA were significantly increased and decreased, respectively, in groups 1–3 eNOS−/− mice, relative to controls (P < 0.001)
LV Function
Left intraventricular pressure tracings were acquired under identical baseline loading conditions in hearts from age-matched WT control and eNOS−/− mice at 52 wks of age; values and loading conditions are listed in Table 1. Figure 4A–B demonstrate significantly increased parameters of cardiac contractility in eNOS−/− hearts as compared to age-matched WT hearts (+dP/dt = 4,532 ± 118 vs. 4,175 ± 73, P < 0.05; TPP = 0.365 ± 0.01 vs. 0.397 ± 0.01, P < 0.05). Both half time to relaxation (RT1/2 = 0.475 ± 0.01 vs. 0.527 ± 0.02; P = 0.110) and −dP/dt (3,625 ± 64 vs. 3,335 ± 90, P = 0.153) (Fig. 4A–B) were not significantly different. The slopes of Frank–Starling function curves (Fig. 4C–D) were unchanged in eNOS−/− mice, relative to age-matched WT controls. Though not significant, peak LVSP was slightly higher in eNOS−/− mice (103.11 ± 2.18 vs. 100.91 ± 1.75, P = 0.50). However, there were no significant differences in HR (336.12 ± 12.35 vs. 357.06 ± 12.77, P = 0.274), LV diastolic (−6.83 ± 1.16 vs. −6.35 ± 1.26, P = 0.788), or LV end-diastolic (5.50 ± 0.80 vs. 5.32 ± 0.99, P = 0.897) pressures in eNOS−/− mouse hearts, relative to age-matched WT controls. Beta-adrenergic receptor (βAR) response to isoproterenol was significantly (P < 0.05) decreased in aged eNOS−/− mice relative to age-matched WT controls (Fig. 4E–F). Figure 4E shows the contractility curves for eNOS−/− and age-matched WT control hearts. The isoproterenol response curve in eNOS−/− mouse hearts lies to the right of the age-matched WT control curve (Fig. 4E, right shift), indicating significant blunting of contractile reserve function in eNOS−/− mouse hearts, relative to age-matched WT controls.

Cardiac function in eNOS−/− mice. The isolated work-performing heart preparation was used to examine cardiac function at 52 wks of age. Under baseline conditions, LV +dP/dt (A) was significantly higher and TPP (B) significantly shorter in eNOS−/− mice. LV RT1/2 (B) and −dP/dt (A) were not significantly different relative to age-matched WT control mice. Although the slopes of Frank–Starling curves (C and D) were not different between eNOS−/− and age-matched WT control mice, the LV response to isoproterenol (E and F) demonstrated a right-shift, unmasking the reduced cardiac functional response in eNOS−/− mouse hearts
LV SERCA2a and Cytokine mRNA Levels
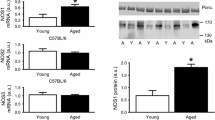
SERCA2a levels were significantly reduced in both groups 2 and 3, relative to age-matched WT controls (Fig. 5A, P < 0.001). Conversely, TNF-α steady-state mRNA levels were significantly increased in both groups 1 and 2, relative to age-matched WT controls (Fig. 5B, P < 0.001). Interestingly, despite a nearly 5-fold increase in TNF-α mRNA levels at 18–23 wks (group 1) and a 29-fold increase at 27–30 wks (group 2), mRNA levels were not significantly elevated in eNOS−/− mice at 40 wks (group 3, Fig. 5B). This was due to a >2-fold increase in TNF-α mRNA levels in age-matched WT control, and a >60% decrease in eNOS−/− mice. The surge in TNF-α mRNA expression at 27–30 wks (group 2) corresponded to a concomitant 18-fold decrease in SERCA2a transcript level that remained suppressed up to 40 wks (Fig. 5A–B).

Quantitative analysis of LV SERCA2a and m-TNF-α mRNA levels. (A) Steady-state LV levels of SERCA2a mRNA demonstrated with a trend toward decreasing levels at 18–23 wks and were significantly decreased at 27–30 wks (P < 0.001) and at 40 wks (P < 0.001). (B) Steady-state LV levels of m-TNF-α mRNA were increased at 18–23 and peaked at 27–30 wks (P < 0.001) in eNOS−/− mouse hearts, but were not significantly different from controls at 40 wks of age
LV Cytokine and Calcium Handling Protein Expression
Western immunoblotting analysis of lysates from eNOS−/− mouse hearts revealed that LV levels of SERCA2a and PLB (Fig. 6A) were approximately 78% (P = 0.36) and 36% (P < 0.05), respectively, of age-matched WT control levels at 52 wks. Overall, the ratio of PLB:SERCA2a protein levels at 52 wks of age was significantly (P < 0.05) decreased by 2-fold in eNOS−/− mice, relative to age-matched WT controls (Fig. 6A).

Western blot analysis of LV SERCA2a, PLB, and m-TNF-α expression. (A) TNF-α, SERCA2a and PLB protein levels were determined in LV tissue samples from eNOS−/− and age-matched WT control mice at different ages by Western blot (representatives shown). Bar graph in panel A shows the relative densitometric levels of SERCA2a and PLB proteins expressed as percent of levels in age-matched WT control hearts. The lower panel represents the absolute densitometric units of the PLB:SERCA2a ratio. (B) m-TNF-α protein levels were determined in LV tissue samples from eNOS−/− and age-matched WT control mice at different ages by Western blot (representatives shown). Bar graph represents the densitometric units ×1,000
Further analysis of protein expression in eNOS−/− mouse hearts revealed that LV TNF-α protein levels were significantly increased at 18 wks, 30 wks (∼10-fold), and 40 wks, relative to age-matched WT controls (Fig. 6B, P < 0.05). Interestingly, though significantly elevated still, the level of TNF-α protein in eNOS−/− hearts dropped markedly from 30 to 40 wks (38% of the 30 wk level at 40 wks; Fig. 6B).
Discussion
This study describes three important observations in eNOS−/− mice. First, we demonstrate that the development of a LV hypertrophic phenotype in naïve eNOS−/− mice is progressive and is characteristic of chronic pressure overload-induced myocardial remodeling as established by others [11–13]. Second, the key finding in this study was that this phenotype has functional effects that are associated with further molecular modifications at the subcellular level, namely modulation in PLB:SERCA2a stoichiometry, suggesting a mechanism that may partially underlie the functional attenuation observed in aged eNOS−/− mouse hearts. Finally, we describe a significant upregulation of TNF-α that corresponds temporally to the development of cardiac hypertrophy, occurs prior to down-regulation of SERCA2A mRNA levels and precedes the significant reduction of SERCA2A protein levels and perturbation of the PLB/SERCA2A ratio.
Concentric Hypertrophic Cardiac Phenotype in eNOS−/− Mice
Re-expression of the fetal gene program, in part, defines pressure-overload myocardial remodeling, as previously described [11]. Accordingly, beginning at 18–23 wk, eNOS−/− mice demonstrated reciprocal coordinate alterations in LV levels of the α-MyHC and β-MyHC transcripts consistent with similar isoform-specific changes observed by others in hypertrophied and failing hearts [11–13] and a significant developing increase in both ANF and α-skeletal actin mRNA (Fig. 3). Though these data appear to be somewhat congruent with a recent study by Bubikat et al. who reported a significant transcriptional increase in ANF and β/α-MyHC ratios at 24–32 wks in eNOS−/− mouse hearts, they observed no significant alteration in α-MyHC or α-skeletal actin at these time-points [36]. Moreover, we observed a progressive upregulation of α-skeletal actin and ANF mRNA and a positive correlation between hypertrophy and these transcript levels, substantiates a genuine LV hypertrophic response to increased afterload in naïve eNOS−/− mouse hearts [35]. Unlike others who have reported LV hypertrophy in eNOS−/− mouse hearts as early as 12 wks [3, 4], here we observed no gross evidence of cardiac hypertrophy until 27–30 wks, relative to age-matched WT controls (Fig. 1). Moreover, though pathohistological evidence of concentric LVH was present in eNOS−/− mice at 27–30 wks, at the cardiac cellular level, multi-focal replacement fibrosis and signs of cardiomyocyte degeneration/death were not apparent until 40 wks in eNOS−/− mice (Fig. 2E, G–H). These findings suggest that eNOS−/− mouse hearts undergo early adaptive molecular remodeling that later becomes structurally maladaptive.
Altered Function and Dysregulation in SERCA2a and PLB Expression in eNOS−/− Hearts
Cardiac function has been studied extensively in eNOS−/− mice, yet most studies have focused attention on relatively young or relatively old mice, with somewhat conflicting results. For instance, whereas previous investigators found no difference in basal contractility in eNOS−/− hearts relative to WT at 8–12 wks [6], others demonstrate enhanced contractility beginning at 14 wks of age [9]. Since eNOS−/− mice are known to live much longer (≥83 wks) and others have reported echocardiographic evidence of significant LV dilation and dysfunction in these mice at 21 months [10], we evaluated cardiac function in hypertrophic eNOS−/− mouse hearts at 52 wks; a considerably aged time-point [8, 10]. Our results clearly demonstrated that LV basal contractility in hypertrophic eNOS−/− mouse hearts is enhanced up to 1 year (Fig. 4A–B). Interestingly, according to studies performed by Barouch et al. in eNOS−/− mice 14 and 20 wks of age, predication of this enhanced contractile response is based upon corresponding NO-dependent changes in SR Ca2+ transient dynamics [9]. To investigate whether regulation of calcium handling proteins was associated, in a similar manner, to these functional changes, we studied SERCA2a and PLB protein expression in aged eNOS−/− mouse hearts.
Several reports clearly document that disruption, particularly down-regulation, in SERCA2a expression occurs during heart failure progression and can negatively alter Ca2+ transient dynamics with commensurate effects upon cardiac function [13, 14, 37, 38]. We demonstrate that progressive transcriptional down-regulation of SERCA2a occurs in eNOS−/− hearts between 27 and 40 weeks (Fig. 5A). We also observe a modest translational down-regulation in SERCA2a at 52 wks (78% of age-matched WT controls; Fig. 6A). It may seem confounding then that we report enhanced contractility in eNOS−/− mice at these same time-points. This could be explained potentially by altered levels of the specific repressor of SERCA2a, PLB. Indeed, investigators have shown that gene-targeted ablation of PLB removes this repression and enhances parameters of LV contractility [16]. Furthermore, Koss et al. demonstrated, using transgenesis, that a low PLB:SERCA2a stoichiometry is actually a critical determinant of myocardial contractility. In fact, these investigators showed that a 33% reduction in the PLB:SERCA2a ratio resulted in a 35% increase in +dP/dt [18]. In accordance with these data, we observed a striking reduction in PLB protein at 52 wks in eNOS−/− mouse hearts (Fig. 6A), 36% of age-matched WT controls (SERCA2a protein was 78% of age-matched WT controls, Fig. 6A). This represents an overall 2-fold (50%) decrease in the PLB:SERCA2a ratio relative to age-matched WT control mice (Fig. 6A). This 50% decrease in PLB:SERCA2a ratio coincided temporally with a significant increase in basal contractility at 1 year, despite reduction in SERCA2a expression. One explanation for this may be that in aged eNOS−/− mouse hearts, the reduction in SERCA2a protein levels is compensated by the concomitant reduction in PLB levels. When considered together with findings by Barouche et al. [9] and others [16–18], it is tempting to postulate that changes in SR Ca2+ release along with a significant reduction in PLB:SERCA2a ratio alleviated the PLB-mediated repression of remaining SERCA2a channels, thereby favoring improved calcium handling and enhanced base-line cardiac performance.
Further functional analysis in the current study revealed depressed βAR responsiveness in eNOS−/− mice, occurring at 1 year (Fig. 4E); a novel finding in this model. Gyurko et al. and Barouche et al. both reported enhanced βAR-induced inotropy in eNOS−/− mouse hearts at early time-points, 8 wks and 20 wks, respectively [6, 9]. However, Vandecasteele et al., Godecke et al., and Han et al. found that βAR-mediated responses in papillary muscle strips, isolated hearts and cardiomyocytes [5, 7, 39], from eNOS−/− mouse hearts were indistinguishable from age-matched WT controls at a later time-point, approximately 6 months. It therefore appears that the reported early enhanced βAR-mediated response [6, 9] declines as eNOS−/− mice age and, as shown here, is significantly blunted by 52 wks of age.
Several mechanisms potentially participate in the decreased βAR responsiveness in aged eNOS−/− mice. The mechanisms by which NOS ablation affects inotropy in eNOS−/− mice have been previously addressed in depth by Barouche et al. and Guyrko et al. and include enhanced calcium cycling, the removal of the inhibitory effect of NO upon myofilament contractility and effects upon G-protein activity. Collectively, these may explain enhanced βAR responsiveness in young eNOS−/− mice [6, 9]. Yet, studies performed in other models of hypertrophic heart disease, including some in eNOS−/− mice, suggest that adaptive alterations in catecholamine release lead to the development of a hyperadrenergic state, resulting in chronic βAR activation. These adaptive changes later become maladaptive and with time attenuate βAR responsiveness, in part, by altering calcium handling but also via remodeling of β-adrenergic receptors themselves [6, 15]. Based on these and other reports [6, 15, 40], it is conceivable that aged eNOS−/− mouse hearts are exposed to chronic βAR stimulation and, as we have demonstrated here, SERCA2a expression is depressed. Consequently, SERCA2a channels may be approaching such low levels that basal contractility is not yet affected but βAR-mediated enhancement of function (via PLB mediated dis-inhibition of remaining SERCA2a channels) is not possible.
Upregulation of m-TNF-α in eNOS−/− Mouse Hearts
The specific functional roles of TNF-α in the myocardium remain controversial. To date, conflicting data exist as to the pathologic significance of myocardial TNF-α signaling and, based on previous evidence, it is likely that a disparate pattern of myocardial remodeling ensues depending upon temporal and spatial features of TNF-α expression within the myocardium [23, 41]. In fact, convincing reports from studies performed with genetically modified rodents clearly support the notion that signaling via specific TNF-α pathways in the heart may confer protection during the pathogenesis of heart failure [24, 25]. However, other reports in models using cardiac-specific transgenic overexpression of TNF-α support a detrimental role for TNF-α in the development of dilated cardiomyopathy, underscoring its negative inotropic effects [22, 42–44].
Explanation of these inconsistencies has proven perplexing. However, recent and quite compelling evidence suggests that posttranslational processing of TNF-α and subsequent production of two distinct proteins (secretory and membrane-bound, s-TNF-α and m-TNF-α, respectively), each with different biological actions, may partially underlie reported discrepancies [23]. Although some have shown that signaling via m-TNF-α pathways results in concentric cardiac hypertrophy [26], a more recent report by Diwan et al. demonstrated that cardiac-specific overexpression of s-TNF-α provokes a dilated cardiomyopathy [45]. Similarly, with regard to TNF-α receptor types 1 and 2 (p55 and p75, respectively; both expressed in the heart), Haas et al. clearly demonstrated that not only is m-TNF-α required by p75 for full activation, it has superior bioactivity on this receptor when compared with s-TNF-α [41]. Consistent with these findings, is a study performed recently by Higuchi et al. using p55 and p75 knockout mice bred with mice overexpressing TNF-α in a cardiac-specific manner. These investigators were able to prolong survival and improve cardiac function in p55 knockout mice, as compared to p75 knockout mice under the same conditions [25]. These data suggest that overexpression of TNF-α, though sufficient to provoke dilatation and heart failure when signaling occurs by way of p55 pathways, may have less injurious and perhaps even cardioprotective effects via p75 signaling.
Accordingly, we measured LV levels of TNF-α mRNA and protein in eNOS−/− mice and questioned whether these expression patterns showed any correlation with the structural or biochemical changes we observed above. Here we demonstrate a definite temporal upregulation of TNF-α in eNOS−/− mouse hearts (Figs. 5B, 6B). We observed a peak in TNF-α expression at the same time-point (27–30 wks) that we began to see gross and histological evidence of a concentric hypertrophic phenotype (Fig. 1). This was also concomitant with decreased SERCA2a mRNA levels (Fig. 5A) and a trend (though not yet significant) toward decreasing PLB:SERCA2a protein ratios (Fig. 6A). These results intimate a temporal association between TNF-α upregulation and the instigation of compensatory regulation of calcium handling proteins and the hypertrophic phenotype in these hearts.
Though published reports have demonstrated that overexpression of TNF-α correlates with repression of both PLB and SERCA2a gene expression [19, 21, 22], the peak in m-TNF-α expression in eNOS-/- mouse hearts observed at 27–30 wks in the present study (Figs. 5A, 6B) preceded the significantly altered, potentially favorable, PLB:SERCA2a protein ratios present at 52 wks (Fig. 6A). However, at the transcriptional level, this peak in m-TNF-α at 27–30wks corresponds directly to an 18-fold decrease in SERCA2a mRNA (Fig. 5A–B). Interestingly, protein levels of m-TNF-α in eNOS−/− mouse hearts begin to normalize with respect to wild-type at 40 wks, yet, remain significantly elevated (Fig. 6B). Whether this relationship is functionally associated with favorable modulation of the PLB:SERCA2a ratio and whether this is a late compensatory mechanism remains to be determined.
Limitations
In the present study, we chose specifically not to address the pleiotropic effects of NO or specific NOS isoforms as these topics have been extensively reported upon. Nevertheless, we concede that an inescapable consequence of this particular model is that any experimental outcomes may, at least partially, be affected by NO-mediated pathways. In addition, we do not specifically address whether enhanced expression of myocardial TNF-α is simply an age-related phenomenon, as suggested by previous reports performed in aging rodents [10). On the contrary, according to our results, cardiac TNF-α protein levels are always higher in eNOS−/− as compared with age-matched WT control levels. Thus, we propose that the upregulation of TNF-α observed in eNOS−/− mice is not a temporal coincidence of aging, but is more likely an adaptive response to or an instigator of cardiac hypertrophy. Nevertheless, whether age-related phenomena significantly alter TNF-α expression in eNOS−/− mice is beyond the scope of this study and remains to be conclusively demonstrated.
Conclusions
Our data demonstrate that naïve eNOS−/− mouse hearts develop a progressive and prolonged compensatory LV hypertrophy, yet, show evidence of transition toward decompensation at 52 wks of age. Interestingly, we found an apparent adaptive stoichiometric alteration in the PLB:SERCA2a ratio that was contemporaneous with both preservation of basal function and loss of cardiac reserve function. One plausible mechanism, though not directly tested herein, is that the resultant stoichiometry at later time-points is favorable and allows for restoration of Ca2+ transient dynamics to maintain basal contractile performance. Yet, as evidenced by blunted βAR responsiveness at 1 year, further attempts to alleviate this repressive effect by PLB (via βAR-mediated recruitment of reserve function) become futile in the face of severely limited SERCA2a substrate and/or modulation of βAR signaling. Additionally, results here are compatible with previous reports demonstrating that TNF-α-mediated signaling contributes to the development of a concentric cardiac hypertrophic phenotype [26, 45], thus implying an early adaptive cytokine-mediated remodeling response in eNOS−/− mouse hearts; whether this response later becomes maladaptive is uncertain. Our results suggest a testable hypothesis that changes in TNF-α levels in eNOS−/− hearts drive altered calcium handling protein expression and the physiological consequences of the same. Future experiments with WT, TNF-α−/−, TNF-α−/−/eNOS−/− double-knockout mice and measurement of calcium transients and determination of pathophysiological effects in these mice will allow us to conclusively determine whether TNF-α-mediated modulation of calcium handling provides the mechanistic basis for prolonged cardiac functional compensation in eNOS−/− mice. Additionally, these studies will allow us to elucidate the role TNF-α plays in age-dependent adaptations in the normal mouse heart.
References
Huang, P. L., Huang, Z., Mashimo, H., Bloch, K. D., Moskowitz, M. A., Bevan, J. A., & Fishman, M. C. (1995). Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature, 377, 239–242.
Shesely, E. G., Maeda, N., Kim, H. S., Desai, K. M., Krege, J. H., Laubach, V. E., Sherman, P. A., Sessa, W. C., & Smithies, O. (1996). Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proceedings of the National Academy of Sciences of the USA, 93, 13176–13181.
Yang, X. P., Liu, Y. H., Shesely, E. G., Bulagannawar, M., Liu, F., & Carretero, O. A. (1999). Endothelial nitric oxide gene knockout mice: cardiac phenotypes and the effect of angiotensin-converting enzyme inhibitor on myocardial ischemia/reperfusion injury. Hypertension, 34, 24–30.
Ruetten, H., Dimmeler, S., Gehring, D., Ihling, C., & Zeiher, A. M. (2005). Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovascular Research, 66, 444–453.
Godecke, A., Heinicke, T., Kamkin, A., Kiseleva, I., Strasser, R. H., Decking, U. K., Stumpe, T., Isenberg, G., & Schrader, J. (2001). Inotropic response to beta-adrenergic receptor stimulation and anti-adrenergic effect of ACh in endothelial NO synthase-deficient mouse hearts. Journal of Physiology, 532, 195–204.
Gyurko, R., Kuhlencordt, P., Fishman, M. C., & Huang, P. L. (2000). Modulation of mouse cardiac function in vivo by eNOS and ANP. American Journal of Physiology–Heart and Circulatory Physiology, 278, H971–H981.
Vandecasteele, G., Eschenhagen, T., Scholz, H., Stein, B., Verde, I., & Fischmeister, R. (1999). Muscarinic and beta-adrenergic regulation of heart rate, force of contraction and calcium current is preserved in mice lacking endothelial nitric oxide synthase. Nature Medicine, 5, 331–334.
Barouch, L. A., Cappola, T. P., Harrison, R. W., Crone, J. K., Rodriguez, E. R., Burnett, A. L., & Hare, J. M. (2003). Combined loss of neuronal and endothelial nitric oxide synthase causes premature mortality and age-related hypertrophic cardiac remodeling in mice. Journal of Molecular and Cellular Cardiology, 35, 637–644.
Barouch, L. A., Harrison, R. W., Skaf, M. W., Rosas, G. O., Cappola, T. P., Kobeissi, Z. A., Hobai, I. A., Lemmon, C. A., Burnett, A. L., O’Rourke, B., Rodriguez, E. R., Huang, P. L., Lima, J. A., Berkowitz, D. E., & Hare, J. M. (2002). Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature, 416, 337–339.
Li, W., Mital, S., Ojaimi, C., Csiszar, A., Kaley, G., & Hintze, T. H. (2004). Premature death and age-related cardiac dysfunction in male eNOS-knockout mice. Journal of Molecular and Cellular Cardiology, 37, 671–680.
Colucci, W. S. (1997). Molecular and cellular mechanisms of myocardial failure. American Journal of Cardiology, 80, 15L–25L.
Siri, F. M., Nordin, C., Factor, S. M., Sonnenblick, E., & Aronson, R. (1989). Compensatory hypertrophy and failure in gradual pressure-overloaded guinea pig heart. American Journal of Physiology, 257, H1016–H1024.
Feldman, A. M., Weinberg, E. O., Ray, P. E., & Lorell, B. H. (1993). Selective changes in cardiac gene expression during compensated hypertrophy and the transition to cardiac decompensation in rats with chronic aortic banding. Circulation Research, 73, 184–192.
Arai, M., Alpert, N. R., MacLennan, D. H., Barton, P., & Periasamy, M. (1993). Alterations in sarcoplasmic reticulum gene expression in human heart failure. A possible mechanism for alterations in systolic and diastolic properties of the failing myocardium. Circulation Research, 72, 463–469.
Marks, A. R. (2003). A guide for the perplexed: Towards an understanding of the molecular basis of heart failure. Circulation, 107, 1456–1459.
Chu, G., Ferguson, D. G., Edes, I., Kiss, E., Sato, Y., & Kranias, E. G. (1998). Phospholamban ablation and compensatory responses in the mammalian heart. Annals of the New York Academy of Sciences, 853, 49–62.
Frank, K. F., Bolck, B., Brixius, K., Kranias, E. G., & Schwinger, R. H. (2002). Modulation of SERCA: Implications for the failing human heart. Basic Research Cardiology, 97(Suppl 1), I72–I78.
Koss, K. L., Grupp, I. L., & Kranias, E. G. (1997). The relative phospholamban and SERCA2 ratio: A critical determinant of myocardial contractility. Basic Research Cardiology, 92(Suppl 1), 17–24.
Janczewski, A. M., Kadokami, T., Lemster, B., Frye, C. S., McTiernan, C. F., & Feldman, A. M. (2003). Morphological and functional changes in cardiac myocytes isolated from mice overexpressing TNF-alpha. American Journal of Physiology–Heart and Circulatory Physiology, 284, H960–H969.
Kilickap, M., Gurlek, A., Dandachi, R., Dincer, I., Tutkak, H., & Oral, D. (2004). Tumour necrosis factor-alpha in diastolic dysfunction. Acta Cardiology, 59, 507–510.
Kubota, T., Bounoutas, G. S., Miyagishima, M., Kadokami, T., Sanders, V. J., Bruton, C., Robbins, P. D., McTiernan, C. F., & Feldman, A. M. (2000). Soluble tumor necrosis factor receptor abrogates myocardial inflammation but not hypertrophy in cytokine-induced cardiomyopathy. Circulation, 101, 2518–2525.
Matsumori, A. (1996). Cytokines in myocarditis and cardiomyopathies. Current Opinion in Cardiology, 11, 302–309.
Grell, M., Douni, E., Wajant, H., Lohden, M., Clauss, M., Maxeiner, B., Georgopoulos, S., Lesslauer, W., Kollias, G., Pfizenmaier, K., & Scheurich, P. (1995). The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell, 83, 793–802.
Mann, D. L. (2003). Stress-activated cytokines and the heart: From adaptation to maladaptation. Annual Review of Physiology, 65, 81–101.
Higuchi, Y., McTiernan, C. F., Frye, C. B., McGowan, B. S., Chan, T. O., & Feldman, A. M. (2004). Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor-alpha-induced cardiomyopathy. Circulation, 109, 1892–1897.
Dibbs, Z. I., Diwan, A., Nemoto, S., DeFreitas, G., Abdellatif, M., Carabello, B. A., Spinale, F. G., Feuerstein, G., Sivasubramanian, N., & Mann, D. L. (2003). Targeted overexpression of transmembrane tumor necrosis factor provokes a concentric cardiac hypertrophic phenotype. Circulation, 108, 1002–1008.
Krege, J. H., Hodgin, J. B., Hagaman, J. R., & Smithies, O. (1995). A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension, 25, 1111–1115.
Athirakul, K., Kim, H. S., Audoly, L. P., Smithies, O., & Coffman, T. M. (2001). Deficiency of COX-1 causes natriuresis and enhanced sensitivity to ACE inhibition. Kidney International, 60, 2324–2329.
Grupp, I. L., Subramaniam, A., Hewett, T. E., Robbins, J., & Grupp, G. (1993). Comparison of normal, hypodynamic, and hyperdynamic mouse hearts using isolated work-performing heart preparations. American Journal of Physiology, 265, H1401–H1410.
Jones, W. K., Grupp, I. L., Doetschman, T., Grupp, G., Osinska, H., Hewett, T. E., Boivin, G., Gulick, J., Ng, W. A., & Robbins, J. (1996). Ablation of the murine alpha myosin heavy chain gene leads to dosage effects and functional deficits in the heart. Journal of Clinical Investigation, 98, 1906–1917.
Chu, G., Li, L., Sato, Y., Harrer, J. M., Kadambi, V. J., Hoit, B. D., Bers, D. M., & Kranias, E. G. (1998). Pentameric assembly of phospholamban facilitates inhibition of cardiac function in vivo. Journal of Biological Chemistry, 273, 33674–33680.
Higuchi, Y., Chan, T. O., Brown, M., McTiernan, C. F., Jones, W. K., Feldman, A. M. (2006). Cardiac-specific blockade of NF-κB activation inhibits hypertrophy, restores Akt activity and improves survival in mice with TNF-α-induced heart failure. American Journal of Physiology–Heart and Circulatory Physiology, 290(2), H590–H598.
Flaherty, M. P., Takano, H., Murphree, S. G., Schultz, J. E., & Jones, W. K. (2000). Hypertensive-cardiac hypertrophy and induction of molecular markers in eNOS knockout mice. Absract. Journal of Molecular Cellular Cardiology, 32, A56–A16.
Sharp, B. R., Jones, S. P., Rimmer, D. M., & Lefer, D. J. (2002). Differential response to myocardial reperfusion injury in eNOS-deficient mice. American Journal of Physiology–Heart and Circulatory Physiology, 282, H2422–H2426.
Boluyt, M. O., O’Neill, L., Meredith, A. L., Bing, O. H., Brooks, W. W., Conrad, C. H., Crow, M. T., & Lakatta, E. G. (1994). Alterations in cardiac gene expression during the transition from stable hypertrophy to heart failure. Marked upregulation of genes encoding extracellular matrix components. Circulation Research, 75, 23–32.
Bubikat, A., De Windt, L. J., Zetsche, B., Fabritz, L., Sickler, H., Eckardt, D., Godecke, A., Baba, H. A., & Kuhn, M. (2005). Local atrial natriuretic peptide signaling prevents hypertensive cardiac hypertrophy in endothelial nitric-oxide synthase-deficient mice. Journal of Biological Chemistry, 280, 21594–21599.
Baker, D. L., Hashimoto, K., Grupp, I. L., Ji, Y., Reed, T., Loukianov, E., Grupp, G., Bhagwhat, A., Hoit, B., Walsh, R., Marban, E., & Periasamy, M. (1998). Targeted overexpression of the sarcoplasmic reticulum Ca2+-ATPase increases cardiac contractility in transgenic mouse hearts. Circulation Research, 83, 1205–1214.
Ji, Y., Lalli, M. J., Babu, G. J., Xu, Y., Kirkpatrick, D. L., Liu, L. H., Chiamvimonvat, N., Walsh, R. A., Shull, G. E., & Periasamy, M. (2000). Disruption of a single copy of the SERCA2 gene results in altered Ca2+ homeostasis and cardiomyocyte function. Journal of Biological Chemistry, 275, 38073–38080.
Han, X., Kubota, I., Feron, O., Opel, D. J., Arstall, M. A., Zhao, Y. Y., Huang, P., Fishman, M. C., Michel, T., & Kelly, R. A. (1998). Muscarinic cholinergic regulation of cardiac myocyte ICa-L is absent in mice with targeted disruption of endothelial nitric oxide synthase. Proceedings of the National Academy of Sciences of the USA, 95, 6510–6515.
Chaudhri, B., Del Monte, F., Hajjar, R. J., & Harding, S. E. (2002). Interaction between increased SERCA2a activity and beta-adrenoceptor stimulation in adult rabbit myocytes. American Journal of Physiology–Heart and Circulatory Physiology, 283, H2450–H2457.
Haas, E., Grell, M., Wajant, H., & Scheurich, P. (1999). Continuous autotropic signaling by membrane-expressed tumor necrosis factor. Journal of Biological Chemistry, 274, 18107–18112.
Finkel, M. S., Oddis, C. V., Jacob, T. D., Watkins, S. C., Hattler, B. G., & Simmons, R. L. (1992). Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science, 257, 387–389.
Bryant, D., Becker, L., Richardson, J., Shelton, J., Franco, F., Peshock, R., Thompson, M., & Giroir, B. (1998). Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation, 97, 1375–1381.
Kubota, T., McTiernan, C. F., Frye, C. S., Slawson, S. E., Lemster, B. H., Koretsky, A. P., Demetris, A. J., & Feldman, A. M. (1997). Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circulation Research, 81, 627–635.
Diwan, A., Dibbs, Z., Nemoto, S., DeFreitas, G., Carabello, B. A., Sivasubramanian, N., Wilson, E. M., Spinale, F. G., & Mann, D. L. (2004). Targeted overexpression of noncleavable and secreted forms of tumor necrosis factor provokes disparate cardiac phenotypes. Circulation, 109, 262–268.
Acknowledgments
This study was supported by NIH R01HL-63034 (WKJ) and AHA 9930195N (WKJ). We gratefully acknowledge Dr. Paul Huang for the gift of the eNOS−/− mice and Drs. E.G. Kranias and G. Chu for their consultation and assistance with the measurement of PLB and SERCA2a protein levels and discussion of these results.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Flaherty, M.P., Brown, M., Grupp, I.L. et al. eNOS Deficient Mice Develop Progressive Cardiac Hypertrophy with Altered Cytokine and Calcium Handling Protein Expression. Cardiovasc Toxicol 7, 165–177 (2007). https://doi.org/10.1007/s12012-007-0028-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-007-0028-y