
Abstract
Selenium exerts many, if not most, of its physiological functions as a selenocysteine moiety in proteins. Selenoproteins are involved in many biochemical processes including regulation of cellular redox state, calcium homeostasis, protein biosynthesis, and degradation. A neurodevelopmental syndrome called progressive cerebello-cortical atrophy (PCCA) is caused by mutations in the selenocysteine synthase gene, SEPSECS, demonstrating that selenoproteins are essential for human brain development. While we have shown that selenoproteins are required for correct hippocampal and cortical interneuron development, little is known about the functions of selenoproteins in the cerebellum. Therefore, we have abrogated neuronal selenoprotein biosynthesis by conditional deletion of the gene encoding selenocysteyl tRNA[Ser]Sec (gene symbol Trsp). Enzymatic activity of cellular glutathione peroxidase and cytosolic thioredoxin reductase is reduced in cerebellar extracts from Trsp-mutant mice. These mice grow slowly and fail to gain postural control or to coordinate their movements. Histological analysis reveals marked cerebellar hypoplasia, associated with Purkinje cell death and decreased granule cell proliferation. Purkinje cell death occurs along parasagittal stripes as observed in other models of Purkinje cell loss. Neuron-specific inactivation of glutathione peroxidase 4 (Gpx4) used the same Cre driver phenocopies tRNA[Ser]Sec mutants in several aspects: cerebellar hypoplasia, stripe-like Purkinje cell loss, and reduced granule cell proliferation. Parvalbumin-expressing GABAergic interneurons (stellate and/or basket cells) are virtually absent in tRNA[Ser]Sec-mutant mice, while some remained in Gpx4-mutant mice. Our data show that selenoproteins are specifically required in postmitotic neurons of the developing cerebellum, thus providing a rational explanation for cerebellar hypoplasia as occurring in PCCA patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Selenoproteins are proteins, which specifically have the rare amino acid, selenocysteine (Sec), incorporated in response to UGA codons in their mRNAs. The mouse and human genomes contain 24 and 25 genes encoding selenoproteins [1]. The mouse serves as a good model organism for mammalian brain development, and manipulation of its genome allows for easy studies of the physiological roles of selenoproteins. Most selenoproteins are expressed in the brain, particularly in neurons of the olfactory bulb, cortex, hippocampus, and cerebellum [2]. While dietary selenium (Se) deficiency is not known to induce direct neurological disease or neurodevelopmental delay in mammals [3], inactivation of the plasma Se transport protein, selenoprotein P (SePP), does lead to a neurological disorder which is caused by reduced brain Se content and selenoenzyme activity [4, 5]. Similarly, inactivation of apolipoprotein E receptor 2 (ApoER2, Lrp8), which serves as a SePP receptor in the brain, mimics the phenotype of SePP-KO mice [6, 7].
Selenoprotein biosynthesis relies entirely on the presence of tRNA[Ser]Sec, because Sec is synthetized attached to its tRNA, and this tRNA is also required for Sec incorporation by the ribosome [8]. Previously, we took an advantage of a cell type-specific genetic deficiency model of tRNA[Ser]Sec [9–11] to abolish the expression of all selenoproteins simultaneously. When tRNA[Ser]Sec was specifically inactivated in neurons, cytosolic glutathione peroxidase (Gpx) activity was reduced by 50 % in the forebrain on postnatal day 9 (P9), and Western blot on P12 demonstrated significant reductions of Gpx1, Gpx4, SePH, and SePR proteins approaching the detection limits [12]. This corresponded with predominantly neuronal expression of many selenoproteins. An interesting observation was the specific developmental defect of a certain subgroup of GABAergic interneurons [12], while Se and vitamin E deficiency in vitro also affected principal, glutamatergic neurons [13]. Neuron-specific deletion of Gpx4 reduced forebrain Gpx4 protein by at least 50 % and was sufficient to disrupt interneuron development and mimic most phenotypes observed through the loss of tRNA[Ser]Sec [12, 14]. Neuron-specific inactivation of thioredoxin reductase 1 (Txnrd1) or Txnrd2 alone does not change the cerebellar development, but inactivation of Txnrd1 in neural precursors leads to cerebellar hypoplasia [15]. Recently, a syndrome called progressive cerebello-cerebral atrophy (PCCA) was described which is caused by mutations in the selenocysteine synthase (SEPSECS) gene [16, 17]. Among the phenotypes described in those affected children is a marked cerebellar hypoplasia. Here, we report on the cerebellar development of mice lacking tRNA[Ser]Sec specifically in neurons and demonstrate that the lack of selenoprotein expression in cerebellar neurons specifically entails cerebellar hypoplasia.
Methods
Animals
Mice were maintained according to local regulations as described for the SePP-deficient mice generated in our laboratory [5]. All animal experiments were approved by the local authorities in Berlin and Munich. Conditional Trsp knockout mice (Trsp fl/fl) have been described [9]. These mice were crossed with transgenic mice expressing Cre recombinase under control of the tubulinα1 promotor [18], yielding mice deficient in neuronal selenoprotein biosynthesis, Tα1-Cre/Trsp fl/fl. Mutant mice and littermate controls were analyzed between P3 (not shown, no defects observed) and P15 (histology).
Enzyme Assays
Cerebella were freshly dissected from postnatal mice and immediately frozen on dry ice. Brain tissue was powdered under liquid nitrogen using a dismembrator (Braun Melsungen), and aliquots were homogenized in buffer as described [5]. Activities of selenoenzymes Gpx and Txnrd were measured in cytosolic cerebellar homogenates as described with tBuOOH and bisdithionitrobenzene (DTNB) as substrates, respectively [5].
Immunohistochemistry
Brains from mouse pups were immediately fixed after dissection in 4 % paraformaldehyde in 0.1 M phosphate buffer (PB), pH 7.4, or the mice were perfused transcardially with the same fixative after a washing step with 0.1 M PB. After postfixation at 4 °C over night, brains were cryoprotected in 30 % sucrose in 0.1 M PB for 2 days and frozen at −80 °C. Sections were cut on a cryostat at 20–50 μm. Free floating sections were stained with the indicated antibodies at dilutions of 1:1,000–1:5,000 at 4 °C over night. Polyclonal rabbit α-calbindin, monoclonal mouse α-calbindin, and rabbit α-parvalbumin antibodies were from Swant, Bellinzona, Switzerland, guinea pig monoclonal α-GLAST was from Chemicon, and mouse monoclonal α-glial fibrillary acidic protein (GFAP) antibody was from Sigma. Pre-absorbed secondary Cy2- and Cy3-conjugated antibodies were from Jackson ImmunoResearch. Horseradish peroxidase and diaminobezidine or NovaRed substrate were used in conjunction with the Vectastain ABC kit (Vector, Burlingame, VT). Whole-mount immunohistochemistry was performed as described [19]. Photomicrographs were taken using a Zeiss Axioskop II equipped with AxioCam MRc and operated with AxioVision software. Other photomicrographs were taken using a Leica confocal microscope.
Terminal UTP-Nick End Labeling and Phosphorylated Histone H3 Assay
Terminal UTP-nick end labeling (TUNEL) immunohistochemistry was performed on serial 5-μm paraffin sections as described [20]. Using AxioVision software, TUNEL + cells were counted on at least 10 fields (0.35 mm2) for each genotype. Phosphorylated histone H3 (pH3) + cells were counted in serial sections and the numbers expressed per 1-mm external germinal layer (egl). For each measurement, at least 10 sections from at least three animals per genotype were evaluated. Antibodies against phosphorylated (Ser10) histone H3 were from Cell Signalling.
Nissl Staining
Cresyl violet staining was performed on frozen 20-μm sections as described recently [21] and combined with α-calbindin immunostaining.
Results
Neuron-Specific Disruption of Selenoprotein Biosynthesis Reduces Selenoenzyme Activity
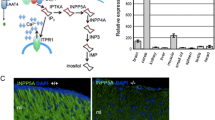
Selenoprotein expression was specifically abrogated in neurons by conditional inactivation of tRNA[Ser]Sec (Trsp) based on neuron-specific expression of Cre recombinase (Fig. 1a). Neuron-specific Trsp-knockout mice (Tα1-Cre/Trsp fl/fl) failed to gain weight after the first week of life and rarely survived beyond P12 (Fig. 1b). Since the knockout animals did not gain postural control, we suspected a cerebellar defect and determined selenoprotein activities in cytosolic extracts of cerebella on P9. Glutathione peroxidase activity was reduced by 30 % (p < 0.01, Student’s t test; Fig. 1c), and cytosolic thioredoxin reductase was reduced by 60 % (p > 0.001; Student’s t test; Fig. 1d). Remaining selenoenzyme activities are likely derived from nonneuronal cells like glia and endothelial cells, which are not targeted by our approach as previously observed in the cerebral cortex [12].

Neuron-specific inactivation of selenoprotein expression. a Schematic drawing shows dependence of selenoprotein synthesis on selenocysteine tRNA. b Body weight in postnatal Tα1-Cre/Trsp fl/fl mice and controls. c Cytosolic glutathione peroxidase activity was determined with tert-butyl hydroperoxide as a substrate in P9 cerebella. d Cytosolic thioredoxin reductase activity was determined with the DTNB assay in P9 cerebella. n = 5–7, **p < 0.01; ***p < 0.001, Student’s t test
Cerebellar Hypoplasia and Stripe-Like Loss of Purkinje Cells
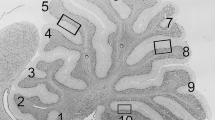
The most striking anatomical phenotype of Tα1-Cre/Trsp fl/fl mice was marked as cerebellar hypoplasia associated with immature foliation (Fig. 2(a)). Staining for Purkinje cells with antibodies against either calbindin or parvalbumin showed a significant loss of Purkinje cells in most folia on P7 and later stages. No Purkinje cell loss or size difference was apparent on P3 (not shown). Purkinje cell dendrites were clearly underdeveloped in Tα1-Cre/Trsp fl/fl cerebella on P12 (Fig. 2(a), insets), and occasionally, Purkinje cells were ectopically distributed within deep layers of the white matter. We noted that the degree of Purkinje cell loss varied among parasagittal sections from the same animal. Since Purkinje cell death is known to occur along parasagittal stripes in several models of Purkinje cell degeneration [22], we performed whole-mount immunohistochemistry using calbindin as a marker for Purkinje cells. Calbindin staining revealed a stripe-like pattern of Purkinje cell loss (Fig. 2(b)) that was readily apparent in a camera lucida representation (Fig. 2(c)). In order to assess whether Purkinje cell loss coincided with the pattern of expression of zebrin II/aldolase C, we stained cerebellar sections with calbindin and zebrin II antibodies (Fig. 2(d)). In the primary fissure, surviving Purkinje cells were zebrin II + on P11.

Cerebellar hypoplasia and Purkinje cell loss in mice lacking neuronal selenoprotein expression. a Nissl staining reveals marked cerebellar hypoplasia and immature foliation pattern in Tα1-Cre/Trsp fl/fl mice on P12. The Purkinje cell layer is partially disrupted. Insets indicate stunted Purkinje cell dendrites and reduced external germinal layer (egl) thickness in Tα1-Cre/Trsp fl/fl mice. α-Calbindin brown. b Whole-mount immunohistochemistry α-calbindin reveals stripe-like Purkinje cell loss on P12. c Camera lucida representation of Purkinje cells. d Co-expression of zebrin II (red) and calbindin (green) on P11. e Left panel TUNEL staining shows apoptosis on P8. Cell death (arrows) occurs in the egl, during migration in the molecular layer, and in the internal granule cell layer (igl). Occasionally, TUNEL + Purkinje cells (PC) are identified based on the large size of their nuclei. Right panel quantification of TUNEL + cells per 0.35 mm2 on P6 and P8. Diamonds indicate wild type and open circles mutant. f Left panel immunostaining for phosphorylated histone H3 (phospho H3+) as an indicator of proliferation. Mitosis in egl is clearly reduced in Tα1-Cre/Trsp fl/fl cerebellum on P8. Right panel quantification of pH3+ cells per 1-mm egl. Diamonds indicate wild type and open circles mutant. ***p < 0.001, Student’s t test. Scale bar = 50 μm
Cell Proliferation and Cell Death
TUNEL staining demonstrated that cell death was significantly increased in Tα1-Cre/Trsp fl/fl cerebella on P6 and P8. Increased TUNEL labeling was not limited to the Purkinje cell layer but was also found in the external germinal layer and internal granule cell layer (Fig. 2(e)). The massive increase in size of the cerebellum during postnatal development is largely caused by the proliferation of granule cell precursors in the secondary neuroepithelium of the external germinal layer. In Tα1-Cre/Trsp fl/fl mice, the external germinal layer was clearly reduced in thickness (Fig. 2(a), insets). In order to test whether granule cell proliferation was reduced in Tα1-Cre/Trsp fl/fl cerebella, we stained serial cerebellar sections on P6 and P8 with antibodies against pH3. The number of proliferating granule cells in the external germinal layer was significantly reduced in Tα1-Cre/Trsp fl/fl mice as compared to controls (Fig. 2(f)).
Inactivation of Gpx4 Recapitulates the Cerebellar Phenotype
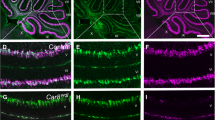
The strong cerebellar phenotype of Tα1-Cre/Trsp fl/fl mice directly prompted the question whether a single selenoprotein could be identified which is responsible for the observed cell death and impaired cell proliferation. We therefore crossed Tα1-Cre with conditional glutathione peroxidase 4 (Gpx4 fl/fl) mice. These mice showed the same growth phenotype as Tα1-Cre/Trsp fl/fl mice and did not survive beyond P13. More interestingly, Tα1-Cre/Gpx4 fl/fl mice phenocopied the cerebellar phenotype with respect to cerebellar hypoplasia and stunted growth of Purkinje cell dendrites or Purkinje cell loss (Fig. 3a). Purkinje cell loss again occurred along parasagittal stripes in Tα1-Cre/Gpx4 fl/fl mice (Fig. 3b). Since parvalbumin-positive GABAergic interneurons were the most affected cortical neurons, we asked whether interneurons in the cerebellum are also affected in our mutants. Purkinje cells (which are also GABAergic) express both calbindin and parvalbumin. Co-labeling of calbindin (red) and parvalbumin (green) in fluorescence microscopy therefore allows visualizing parvalbumin-positive interneurons (green) on a yellow background of Purkinje cells. As shown in Fig. 3c, parvalbumin-positive interneurons, presumably basket and/or stellate cells, are completely absent in Tα1-Cre/Trsp fl/fl mice, while they partly remain in Tα1-Cre/Gpx4 fl/fl mice.

Conditional inactivation of Gpx4 mimics Trsp deficiency. a Cerebellar hypoplasia and Purkinje cell loss in Gpx4-mutant mice on P12. Cresyl violet stain (blue) and α-calbindin (brown). Scale bars = 200 μm (upper panel) and 50 μm (lower panel). b Stripe-like loss (arrows) of Purkinje cells on P8. α-Calbindin (brown). c Few parvalbumin + cerebellar interneurons remain in Gpx4-mutant mice on P12. Purkinje cells are labeled yellow (parvalbumin is green, and calbindin is red), while interneurons express only parvalbumin (green)
Bergmann Glia
Staining of radial glia with GFAP as a marker did not reveal a reduction of Bergmann fiber density or radial glia morphology. Even in areas where Purkinje cell dendrites have degenerated, there is no indication of structural changes of Bergmann glial fibers (Fig. 4).

The density and orientation of radial glial fibers are not disrupted in Trsp-mutant cerebella on P12. a Staining of radial glia with an antibody against GFAP reveals intact pial contacts of radial glial fibers. b Double immunohistochemistry of calbindin and GFAP reveals that the radial glial network is not disrupted at sites of Purkinje cell loss
Discussion
Initial observations in SePP −/− mice and later in Apoer2 −/− mice established that selenoprotein expression in the mammalian brain is essential [4–7, 23–25]. Among the neurological phenotypes of affected mice were epilepsy/hyperexcitability and movement disorders. However, initial studies of SePP −/− mice did not demonstrate a clear pathology of the cerebellum, despite an abnormal gait of the mice [21, 23, 24]. In order to elicit the maximal phenotype associated with a lack of selenoprotein expression, we abrogated selenoprotein biosynthesis by inactivating Trsp, specifically in neurons using pan-neuronal Tα1-Cre (including cerebellum) and CamK-Cre (forebrain-specific, excluding cerebellum). In the cerebral cortex and hippocampus, parvalbumin-expressing interneurons did not develop at all, while parvalbumin-expressing cells in the globus pallidus were still detectable [12]. Most importantly, neuron-specific CamK-Cre/Trsp fl/fl mice exhibited hyperexcitability. Here, we describe that Tα1-Cre/Trsp fl/fl mice, in which cerebellar neurons are affected, show cerebellar hypoplasia. Epilepsy and cerebellar hypoplasia are among the phenotypes of children afflicted with mutations in PCCA due to mutations in the SEPSECS gene [16]. Therefore, the essential role of selenoproteins in the mammalian brain can now be considered firmly established.
What could be the specific roles of selenoproteins in the brain? We show here that inactivation of Gpx4 alone already mimics a loss of all selenoproteins when cerebellar hypoplasia and Purkinje cell death are chosen as parameters. This finding is reminiscent of findings in forebrain interneurons and cultivated cortical neurons (mostly glutamatergic neurons) [12]. However, when comparing Tα1-Cre/Trsp fl/fl mice and Tα1-Cre/Gpx4 fl/fl mice, it was noticed that loss of Gpx4 yields a slightly milder phenotype, indicating that besides Gpx4, a yet to be identified selenoprotein may contribute to the more severe pathology as observed in Tα1-Cre/Trsp fl/fl mice.
Thioredoxin reductases are essential selenoproteins, but their inactivation in neurons using Tα1-Cre was shown not to cause to a cerebellar phenotype [15].
Cerebellar development represents a special paradigm since a secondary proliferating neuroepithelium is established in the course of its development. The first neurons to develop in the primary neuroepithelium of the cerebellar primordium are principal neurons of the deep nuclei and, most importantly, the Purkinje cells [26]. In a second step, neuronal precursors migrate through the Purkinje cell layer and form the external germinal layer where the precursors proliferate postnatally. The newly developed neurons, granule cells, then migrate along Bergmann glia processes inward through the Purkinje cell layer and establish the internal granule cell layer. Cerebellar hypoplasia is usually associated with a failure of the secondary neuroepithelium to produce the huge numbers of granule cells, which make up most of the cerebellar volume. Finally, GABAergic interneuron precursors are born in the primary neuroepithelium and migrate outwards where basket cells mature close to Purkinje cells and stellate cells migrate into the molecular layer and reside among the Purkinje cell dendrites. In our Tα1-Cre/Trsp fl/fl mouse model, Purkinje cell death was observed at a stage when the Purkinje cell layer is already formed; hence, we find gaps in the layer. The Tα1-Cre transgene is only expressed in neurons, not neuronal precursors consistent with the loss of postmitotic Purkinje cells. Why Purkinje cell degeneration occurs often along parasagittal stripes (e.g., preferentially in aldolase C-negative cells in our model) is not known but may be associated with the functional patterning of the cerebellum [22]. Purkinje cells secrete along their dendrite factors stimulating granule cell proliferation, including sonic hedgehog and others [27]. A reduced number of Purkinje cells may thus entail reduced promitotic stimulation of granule cell precursors in the external germinal layer. Migration of granule cells along Bergmann fibers may occur normally, since we did not find evidence of radial glia defects. Targeting neural precursors and Bergmann fibers, nestin-Cre; Txnrd1 fl/fl mice also suffer cerebellar hypoplasia associated with defective foliation/migration disorganized Bergmann fibers [15]. This may bear some relevance in the case of global selenoprotein deficiency as in PCCA. Of note, our Tα1-Cre transgene targets only postmitotic neurons and not glia. We find, however, TUNEL-positive cells mostly in the internal granule cell layer, suggesting that death of granule cells contributes to the observed cerebellar hypoplasia. Whether granule cells degenerate in a cell autonomous process or because their synaptic targets, Purkinje cells, degenerate is not known. We observed that GABAergic interneurons, which express parvalbumin once they reach their destination in the cerebellar cortex, are virtually absent in Tα1-Cre/Trsp fl/fl mice. Their numbers are also reduced in Gpx4 mutants, but occasionally, we observed few of these neurons in the Purkinje cell layer.
It is interesting to note that the neurons most severely affected by the lack of selenoprotein expression (or Gpx4 alone) are GABAergic neurons that also express the calcium-binding protein parvalbumin: cortical interneurons, cerebellar interneurons, and Purkinje cells. Cortical GABAergic neurons expressing calbindin or calretinin are less affected [12]. However, neurons in the globus pallidus are also GABAergic and express parvalbumin and are not affected, or at least, they do not degenerate up to the latest time points we have studied them. Therefore, further studies are needed to define the mechanism of how neurons are damaged by the lack of selenoproteins. While “oxidative stress” is often invoked and massive oxidation of cell constituents is assumed, we speculate that specific developmental mechanisms are targeted. Dysregulations of redox-sensitive pathways are our prime suspects for further study, because oxidative stress may not be very cell-specific at all. For example, signaling by receptor tyrosine kinases often entails transient oxidation of protein phosphatases. It has been shown that lipid hydroperoxides generated enzymatically by 12/15-lipoxygenase efficiently inactivate kinases [28]. Targeting of Gpx4, a lipid hydroperoxide-specific peroxidase, recapitulates the cerebellar phenotype, and we speculate that selenoprotein deficiency may act along such a mechanism.
References
Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN (2003) Characterization of mammalian selenoproteomes. Science 300(5624):1439–1443. doi:10.1126/science.1083516
Zhang Y, Zhou Y, Schweizer U, Savaskan NE, Hua D, Kipnis J, Hatfield DL, Gladyshev VN (2008) Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. J Biol Chem 283(4):2427–2438. doi:10.1074/jbc.M707951200
Schweizer U, Bräuer AU, Köhrle J, Nitsch R, Savaskan NE (2004) Selenium and brain function: a poorly recognized liaison. Brain Res Brain Res Rev 45(3):164–178. doi:10.1016/j.brainresrev.2004.03.004
Hill KE, Zhou J, McMahan WJ, Motley AK, Atkins JF, Gesteland RF, Burk RF (2003) Deletion of selenoprotein P alters distribution of selenium in the mouse. J Biol Chem 278(16):13640–13646
Schomburg L, Schweizer U, Holtmann B, Flohé L, Sendtner M, Köhrle J (2003) Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem J 370(Pt 2):397–402. doi:10.1042/BJ20021853
Burk RF, Hill KE, Olson GE, Weeber EJ, Motley AK, Winfrey VP, Austin LM (2007) Deletion of apolipoprotein E receptor-2 in mice lowers brain selenium and causes severe neurological dysfunction and death when a low-selenium diet is fed. J Neurosci 27(23):6207–6211
Valentine WM, Abel TW, Hill KE, Austin LM, Burk RF (2008) Neurodegeneration in mice resulting from loss of functional selenoprotein P or its receptor apolipoprotein E receptor 2. J Neuropathol Exp Neurol 67(1):68–77
Seeher S, Mahdi Y, Schweizer U (2012) Post-transcriptional control of selenoprotein biosynthesis. Curr Protein Pept Sci 13:337–346
Kumaraswamy E, Carlson BA, Morgan F, Miyoshi K, Robinson GW, Su D, Wang S, Southon E, Tessarollo L, Lee BJ, Gladyshev VN, Hennighausen L, Hatfield DL (2003) Selective removal of the selenocysteine tRNA[Ser]Sec gene (Trsp) in mouse mammary epithelium. Mol Cell Biol 23(5):1477–1488
Carlson BA, Novoselov SV, Kumaraswamy E, Lee BJ, Anver MR, Gladyshev VN, Hatfield DL (2004) Specific excision of the selenocysteine tRNA[Ser]Sec (Trsp) gene in mouse liver demonstrates an essential role of selenoproteins in liver function. J Biol Chem 279(9):8011–8017. doi:10.1074/jbc.M310470200
Schweizer U, Streckfuss F, Pelt P, Carlson BA, Hatfield DL, Köhrle J, Schomburg L (2005) Hepatically derived selenoprotein P is a key factor for kidney but not for brain selenium supply. Biochem J 386(Pt 2):221–226. doi:10.1042/BJ20041973
Wirth EK, Conrad M, Winterer J, Wozny C, Carlson BA, Roth S, Schmitz D, Bornkamm GW, Coppola V, Tessarollo L, Schomburg L, Köhrle J, Hatfield DL, Schweizer U (2010) Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. FASEB J 24(3):844–852. doi:10.1096/fj.09-143974
Roth S, Zhang S, Chiu J, Wirth EK, Schweizer U (2010) Development of a serum-free supplement for primary neuron culture reveals the interplay of selenium and vitamin E in neuronal survival. J Trace Elem Med Biol 24(2):130–137. doi:10.1016/j.jtemb.2010.01.007
Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M (2008) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8(3):237–248. doi:10.1016/j.cmet.2008.07.005
Soerensen J, Jakupoglu C, Beck H, Forster H, Schmidt J, Schmahl W, Schweizer U, Conrad M, Brielmeier M (2008) The role of thioredoxin reductases in brain development. PLoS One 3(3):e1813. doi:10.1371/journal.pone.0001813
Agamy O, Ben Zeev B, Lev D, Marcus B, Fine D, Su D, Narkis G, Ofir R, Hoffmann C, Leshinsky-Silver E, Flusser H, Sivan S, Soll D, Lerman-Sagie T, Birk OS (2010) Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am J Hum Genet 87(4):538–544
Ben Zeev B, Hoffman C, Lev D, Watemberg N, Malinger G, Brand N, Lerman-Sagie T (2003) Progressive cerebellocerebral atrophy: a new syndrome with microcephaly, mental retardation, and spastic quadriplegia. J Med Genet 40(8):e96
Coppola V, Barrick CA, Southon EA, Celeste A, Wang K, Chen B, Haddad E, Yin J, Nussenzweig A, Subramaniam A, Tessarollo L (2004) Ablation of TrkA function in the immune system causes B cell abnormalities. Development 131(20):5185–5195
Sillitoe RV, Hawkes R (2002) Whole-mount immunohistochemistry: a high-throughput screen for patterning defects in the mouse cerebellum. J Histochem Cytochem 50(2):235–244
Conrad M, Jakupoglu C, Moreno SG, Lippl S, Banjac A, Schneider M, Beck H, Hatzopoulos AK, Just U, Sinowatz F, Schmahl W, Chien KR, Wurst W, Bornkamm GW, Brielmeier M (2004) Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol Cell Biol 24(21):9414–9423
Schomburg L, Riese C, Michaelis M, Griebert E, Klein MO, Sapin R, Schweizer U, Köhrle J (2006) Synthesis and metabolism of thyroid hormones is preferentially maintained in selenium-deficient transgenic mice. Endocrinology 147(3):1306–1313. doi:10.1210/en.2005-1089
Sarna JR, Hawkes R (2003) Patterned Purkinje cell death in the cerebellum. Prog Neurobiol 70(6):473–507
Schweizer U, Michaelis M, Köhrle J, Schomburg L (2004) Efficient selenium transfer from mother to offspring in selenoprotein-P-deficient mice enables dose-dependent rescue of phenotypes associated with selenium deficiency. Biochem J 378(Pt 1):21–26. doi:10.1042/BJ20031795
Hill KE, Zhou J, McMahan WJ, Motley AK, Burk RF (2004) Neurological dysfunction occurs in mice with targeted deletion of the selenoprotein P gene. J Nutr 134(1):157–161
Renko K, Werner M, Renner-Muller I, Cooper TG, Yeung CH, Hollenbach B, Scharpf M, Köhrle J, Schomburg L, Schweizer U (2008) Hepatic selenoprotein P (SePP) expression restores selenium transport and prevents infertility and motor-incoordination in Sepp-knockout mice. Biochem J 409(3):741–749. doi:10.1042/BJ20071172
Altman J, Bayer SA (1985) Embryonic development of the rat cerebellum. III. Regional differences in the time of origin, migration, and settling of Purkinje cells. J Comp Neurol 231(1):42–65. doi:10.1002/cne.902310105
Corrales JD, Blaess S, Mahoney EM, Joyner AL (2006) The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development 133(9):1811–1821
Conrad M, Sandin A, Forster H, Seiler A, Frijhoff J, Dagnell M, Bornkamm GW, Radmark O, Hooft van Huijsduijnen R, Aspenstrom P, Bohmer F, Ostman A (2010) 12/15-lipoxygenase-derived lipid peroxides control receptor tyrosine kinase signaling through oxidation of protein tyrosine phosphatases. Proc Natl Acad Sci U S A 107(36):15774–15779. doi:10.1073/pnas.1007909107
Acknowledgments
The authors thank Silke Kappler for technical assistance and Dr. Richard Hawkes for zebrin II antibody. The study was funded by Deutsche Forschungsgemeinschaft DFG Schw914/2-1 and Charité-Universitätsmedizin Berlin.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Wirth, E.K., Bharathi, B.S., Hatfield, D. et al. Cerebellar Hypoplasia in Mice Lacking Selenoprotein Biosynthesis in Neurons. Biol Trace Elem Res 158, 203–210 (2014). https://doi.org/10.1007/s12011-014-9920-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-014-9920-z