
Abstract
Escherichia coli strains expressing the O-glucosyltransferases UGT73B3 or UGT84B1 were compared for the production of glucosides from quercetin supplied into a defined medium. The formation of quercetin-3-glucoside (Q3G) by UGT73B3 showed a maximum at 33 °C, while the formation of quercetin-7-glucoside by UGT84B1 increased with increasing temperature to 37 °C. The highest concentrations of Q3G were attained by strains having a deletion in the pgi gene-coding phosphoglucose isomerase, which effectively blocked the entry of glucose-6P into the Embden–Meyerhof–Parnas pathway. Formation of Q3G was improved in 1-L controlled bioreactors compared to shake flask cultures, a result attributed to the greater oxygen transfer rate in bioreactors. Under batch conditions with 30 g/L glucose as the sole carbon source, E. coli MEC367 (MG1655 pgi) expressing UGT73B3 generated 3.9 g/L Q3G in 56 h.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Flavonoids consist of a large group of natural polyphenols including quercetin and are widely found in cereals, fruits, and vegetables such as apples, onions, tea, and wine [1–4]. Like many flavonoids, quercetin possesses several beneficial biological activities including inhibition of cancer cells [5–7], anti-oxidative effects [8], anti-inflammatory activities [9], and vasodilation and reduction of blood pressure [10–12]. However, the low water solubility of quercetin limits its in vivo availability and effectiveness as a food additive and dietary supplement [13, 14].
Quercetin contains five hydroxyl groups, and O-glycosylation at any of these locations increases the water solubility by several folds [14], resulting in glucosides which exhibit greater bioavailability than the aglycone quercetin [15–19]. For example, as much as 5-fold greater concentrations of quercetin metabolites were found in organs of rats fed with quercetin-3-glucoside (Q3G) compared to rats whose diets were supplemented with quercetin [20]. In addition to greater bioavailability, quercetin glucosides benefit human health similar to the aglycone. For example, Q3G suppresses growth of various colon cancer cell lines (SW480, DLD-1, and HCT116) in vitro by targeting the Wnt/β-catenin signaling pathway but exerts no effect on non-tumor colon IEC-18 cells [21]. Other quercetin glucosides have similar biological activities [22–24].
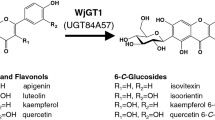
Although quercetin glucosides are widely distributed in nature, their concentrations in plant material are generally low, and therefore, extraction of quercetin glucosides directly from plants is rarely practical [25]. Direct biocatalytic formation of glucosides from quercetin using glucosyltransferases such as UGT73B3 and UGT84B1 requires equimolar quantities of expensive uridine diphosphate (UDP)-glucose (Fig. 1). Using whole cells may be a promising way to produce quercetin glucosides, because cell metabolism can provide a continual supply of UDP-glucose [26]. Previous research has established the effectiveness of using Escherichia coli as a cell factory for glucoside formation: expressing Medicago truncatula UGT71G1 Tyr202Ala mutant in a complex medium converted 34 mg/L quercetin into 20 mg/L Q3G in 48 h [27], expressing Oryza sativa glucosyltransferase cDNA RF5 converted 34 mg/L quercetin into 46 mg/L Q3G in 6 h [28], and expressing Arabidopsis thaliana UGT73B3 produced 9 mg/L Q3G in shake flasks using a complex medium and 99 mg/L Q3G in 20 h in a 2-L controlled bioreactor [29].

Quercetin glycosylation catalyzed by UDP-glucosyltransferases UGT73B3 and UGT84B1 [29]
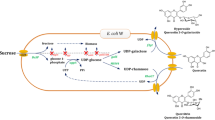
Intracellular accumulation of the sugar donor UDP-glucose in E. coli from glucose-6P (Fig. 2) is likely critical to improve the cellular production of quercetin glucosides although its intracellular concentration is usually only 1–2 mM [30]. Metabolic engineering directed at increasing the accumulation of glucose-6P, the precursor of UDP-glucose, could enhance the formation of the sugar donor and hence quercetin glucosides in a cell expressing glucosyltransferases. For example, E. coli with deletions in gene-coding phosphoglucose isomerase (pgi) or both pgi and glucose 6-phosphate dehydrogenase (zwf) showed elevated intracellular glucose-6P pools [31, 32], and thus, these mutants could elevate formation of products derived from glucose-6P. Of course, overexpressing enzymes involved in the conversion of glucose-6P to UDP-glucose may also improve UDP-glucose formation, and co-overexpressing phosphoglucomutase (pgm) and UDP-glucose pyrophosphorylase (galU) has previously increased the yield of UDP-sugar-derived disaccharides [30]. Previous studies, generally in shake flask culture, have focused both on flavanol synthesis from coumaric acid and glucosylation of the resulting flavanol [33, 34].

Hexose transformations in the upper metabolic pathways in Escherichia coli. The formation of quercetin glucosides from quercetin and metabolite UDP-glucose is mediated by glucosyltransferases
The objective of the current study is to engineer E. coli strains to generate high concentrations of quercetin glucosides from quercetin. In addition to optimizing the temperature for quercetin glycosylation using two glucosyltransferases, the pathway to UDP-glucose was engineered to understand the bottlenecks in quercetin glycosylation. Processes in shake flasks and in controlled bioreactor processes were compared to optimize the production of quercetin glucoside.
Materials and Methods
Strains
E. coli MG1655 (wild-type, F- λ- ilvG- rfb-50 rph-1) and its derivatives were used for this study (Table 1). Knockouts of zwf and pgi were constructed by transducing MG1655 with the corresponding Keio (FRT)Kan deletions [35] and, if necessary, to delete a second gene or transform with a plasmid, curing the Kan(R) using the pCP20 plasmid, which contains a temperature-inducible FLP recombinase as well as a temperature-sensitive replicon [36]. All strains were verified by PCR.
Construction of Plasmids
The UGT73B3 and UGT84B1 genes from A. thaliana were PCR cloned and digested with EcoRI and KpnI, then subsequently cloned into vector pTrc99A to yield plasmids pTrc99A-UGT73B3 and pTrc99A-UGT84B1. The UGT73B3 gene was PCR amplified with primers 5′-GGGAAAGAATTCATGAGTAGTGATCCTCATCGTAAGCTCCA-3′ (forward) and 5′-GGGAAAGGTACCTTACGAGGTAAACTCTTCTATGAAGCTGT-3′ (reverse), and the UGT84B1 gene was PCR amplified with primers 5′-GGGAAAGAATTCATGGGCAGTAGTGAGGGTCAAG-3′ (forward) and 5′-GGGAAAGGTACCTTAGGCGATTGTGATATCACTAATG-3′ (reverse). To construct the pCS27-pgm, pCS27-galU, and pCS27-glk plasmids, the pgm, galU, and glk genes were PCR amplified using E. coli BW25113 genomic DNA as the template. The PCR fragments were digested by KpnI and BamHI and ligated into the KpnI and BamHI sites of pCS27, generating the desired plasmids.
Growth Conditions
The defined medium used in all experiments contained (per L) the following: 13.3 g KH2PO4, 4.0 g (NH4)2HPO4, 1.2 g MgSO4·7H2O, 13.0 mg Zn(CH3COO)2·2H2O, 1.5 mg CuCl2·2H2O, 15.0 mg MnCl2·4H2O, 2.5 mg CoCl2·6H2O, 3.0 mg H3BO3, 2.5 mg Na2MoO4·2H2O, 100 mg Fe(III)citrate, 8.4 mg Na2EDTA·2H2O, 1.7 g citric acid, 4.5 mg thiamine·HCl, and carbon source (i.e., glucose, mannitol) as indicated. As appropriate for the strain, the medium contained 100 mg/L ampicillin and 50 mg/L kanamycin.
Strains transformed with pTrc99A-UGT73B3 or pTrc99A-UGT84B1 were first cultured in a 125-mL flask containing 20-mL medium with 8 g/L glucose. Strains having deletions in both the pgi and zwf genes (MEC374, MEC411) additionally contained 8 g/L mannitol. All shake flasks were incubated at the desired temperature and 250 rpm (19 mm pitch), and the pH adjusted to an initial value of 7.0 with 20% (w/v) NaOH. When the optical density (OD) reached 3, 2 mL was used to inoculate a 250-mL shake flask containing 50 mL of the identical medium. When the OD in this second flask reached 1, 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and 45 mg quercetin dissolved in 1.5 mL DMSO were added (resulting in a 900 mg/L effective quercetin concentration). The temperature range of 20–37 °C was first examined to establish the optimal temperature for glycosylation using MG1655. This same procedure was also used at one temperature to compare quercetin glycosylation in shake flasks among different strains. All shake flask experiments were conducted three to six times.
Controlled Batch Fermentations
For bioreactor experiments, a strain was first grown in a 250-mL shake flask containing 50 mL medium at a pH of 7.0 and incubated at 37 °C and 250 rpm. When the OD reached 3, the flask contents were used to inoculate a 2.5-L bioreactor (Bioflo 2000, New Brunswick Scientific Co., Edison, NJ) containing initially 1.0-L medium with 8 g/L glucose. Air was sparged at a flow rate of 1.0 L/min, and an agitation of 500 rpm ensured the dissolved oxygen remained greater than 70% saturation. The pH was controlled at 7.0 using 20% NaOH. When the OD reached 1, 0.5 mM IPTG and 2 g quercetin dissolved in 30 mL DMSO were added (resulting in a 2 g/L effective quercetin concentration). For microaerobic experiments, the strain was cultured under aerobic condition in the first 16 h, and then the dissolved oxygen level was decreased to 5–10% saturation by sparging a mixture of N2 and air. All bioreactor experiments were conducted in duplicate.
Analyses
The OD at 600 nm was used to monitor cell growth (DU-650 spectrophotometer, Beckman Instruments, San Jose, CA). Liquid chromatography was used to quantify glucose, mannitol, organic acids, and ethanol [37]. To analyze quercetin and quercetin glucosides, a 1-mL sample was centrifuged to obtain a supernatant and a cell pellet, the latter which was resuspended in 0.2 mL DMSO to extract the flavonoids. The combined supernatant and extracted pellet fractions were centrifuged again, and the supernatant was analyzed by HPLC at 370 nm using a 5-μm C18 column (250 × 4.6 mm, Whatman-Partisphere) and a linear gradient of 20 to 80% acetonitrile in H2O with 0.1% trifluoroacetic acid at 1 mL/min over 60 min [29].
Results
Effect of Temperature on Glucoside Formation
The glucosyltransferases UGT73B3 and UGT84B1 from A. thaliana mediate the biotransformation of quercetin to quercetin-3-glucoside (Q3G) and quercetin-7-glucoside (Q7G), respectively (Fig. 1). Previous research demonstrated the specificity of these enzymes at the low bacterial growth temperature of 20 °C but did not establish the optimal temperature for the whole cell glycosylation of quercetin by E. coli [29]. We therefore first completed a set of experiments over the temperature range of 20–37 °C expressing these genes individually in E. coli MG1655 (Fig. 3). The results demonstrated that the optimal temperature for Q3G formation using UGT73B3 was about 33 °C, while the formation of Q7G using UGT84B1 increased with increasing temperature over the entire experimental range (Fig. 3). While greater Q7G formation is possible above the range examined, E. coli degrades proteins at an increased rate above 37 °C [38], shows significant changes in protein expression above 40 °C [39] and does not grow in a defined medium above 44 °C [39]. With 8 g/L glucose as the sole carbon source and 900 mg/L quercetin as available precursor for glycosylation, the maximum glucoside concentration was 330 mg/L for Q3G and 95 mg/L for Q7G in the wild-type background E. coli. In all cases, 600 mg/L or more quercetin remained unreacted at the time of maximum glucoside formation. Typically, the maximum glucoside concentration was observed a few hours after glucose was depleted, although the difference between the concentration at the time of glucose exhaustion and the maximum was only about 10%. Note that the presence of sparingly soluble quercetin prevented the measurement of an accurate OD. Since UGT73B3 consistently generated more glucoside, all subsequent studies were conducted at 33 °C with UGT73B3.

Quercetin glycosylation in shake flasks at different temperatures. Generation of quercetin-3-glucoside (Q3G) by MG1655/pTrc99A-UGT73B3 (filled circle) and the generation of Q7G MG1655/pTrc99A-UGT84B1 (empty circle). Error bars indicate standard error from three to six experiments
Glucoside Formation with Engineered Strains in Shake Flask Culture
UGT73B3 requires UDP-glucose as the sugar donor for glucotransfer, and in E. coli, UDP-glucose is formed in two enzymatic steps from glucose-6P (Fig. 2), a metabolite that is normally directed into the Embden–Meyerhof–Parnas (EMP) pathway via glucose-6P isomerase (coded by the pgi gene) or into the pentose phosphate (PP) pathway via glucose-6P 1-dehydrogenase (zwf). In a defined medium with 5 g/L glucose as the sole carbon source, the growth rate of MG1655 was 0.73 h−1; the zwf deletion (MEC368) slightly reduced the growth rate to 0.69 h−1, whereas the deletion of pgi (MEC367) reduced the growth rate to 0.19 h−1.
We next investigated whether reducing bacterial metabolism through each of these pathways would affect the formation of Q3G. Specifically, we compared glucoside formation in MG1655/pTrc99A-UGT73B3, MEC367/pTrc99A-UGT73B3 (pgi deletion), and MEC368/pTrc99A-UGT73B3 (zwf deletion) in shake flask cultures with 8 g/L glucose and 900 mg/L quercetin. Both the wild-type expressing UGT73B3 and the zwf knockout expressing UGT73B3 accumulated 300–330 mg/L Q3G within 24 h, while the pgi knockout produced 560 mg/L Q3G, a 70% increase in Q3G concentration (Fig. 4).

Comparison of Q3G accumulation by different Escherichia coli strains in 250-mL baffled shake flasks. Each 50 mL culture contained 8 g/L glucose initially, and 900 mg/L quercetin was added when the OD reached 1.0. Escherichia coli strains with deletions in both the zwf and pgi genes additionally contained 8 g/L mannitol
The sugar donor, UDP-glucose, is generated in three steps from glucose (Fig. 2). Glucose is first transported into the cell and phosphorylated to glucose-6P by a phosphotransferase system (ptsG or manZ) or by glucokinase (glk). Glucose-6P is then isomerized to glucose-1P by phosphoglucomutase (pgm), and glucose-1P is converted to UDP-glucose by UDP-glucose pyrophosphorylase (galU). We next investigated whether the overexpression of glk, pgm, or galU would further enhance the production of Q3G in the pgi knockout. The plasmids pCS27-glk, pCS27-pgm, or pCS27-galU were individually transformed into MEC410 in addition to pTrc99A-UGT73B3. The glycosylations were again conducted in shake flasks with 8 g/L glucose and 900 mg/L quercetin. The strain overexpressing pgm gene increased Q3G production by 46%, while the overexpression of other two individual genes did not increase the formation of Q3G compared to MEC367/pTrc99A-UGT73B3 (Fig. 4).
We also were interested in forming Q3G in E. coli having knockouts in both the zwf and pgi genes, which together would effectively prevent glucose-6P from being metabolized through either the Embden–Meyerhof–Parnas or pentose phosphate pathways (Fig. 2). Of course, E. coli lacking both genes is unable to grow on glucose as the sole carbon source, and therefore, for these shake flask experiments, mannitol was added into the medium in addition to glucose. We also again examined the effect of expressing glucokinase, phosphoglucomutase, and UDP-glucose pyrophosphorylase individually on Q3G formation (Fig. 4). In all cases using the zwf pgi double knockout with 900 mg/L quercetin, 390–400 mg/L Q3G was formed, about 25% less than in MEC367 having the pgi knockout alone.
Glucoside Formation in Controlled Bioreactors
We next sought to investigate Q3G production in controlled bioreactors. These batch glycosylations were conducted with 8 g/L glucose and 2 g/L quercetin, and six strain constructs were examined: MG1655/pTrc99A-UGT73B3, MEC367/pTrc99A-UGT73B3 (pgi deletion), and MEC368/pTrc99A-UGT73B3 (zwf deletion) as well as the pgi knockout overexpressing one of the genes pgm, galU, or glk (Fig. 5). The wild-type expressing UGT73B3 accumulated 720 mg/L Q3G within 14 h. The zwf deletion did not alter Q3G formation, while the pgi deletion elevated the final concentration of Q3G to 1550 mg/L in 36 h, greater than twice the concentration generated by the wild-type (Fig. 5). Although the oxygen was not limited during any of these controlled cultures, the wild-type and the zwf knockout additionally generated 0.5–0.7 g/L acetate as a by-product, while the pgi knockout grew at about one third the rate and did not generate acetate. In contrast to the shake flask experiments, overexpression of any of the genes pgm, galU, or glk in the pgi knockout MEC410 did not increase the formation of Q3G compared to MEC367/pTrc99A-UGT73B3 (Fig. 5). Generally, batch cultures in a controlled bioreactor resulted in about a 2-fold increase in Q3G concentration compared to shake flask cultures with the corresponding strain. MEC410/pTrc99A-UGT73B3/pCS27-pgm was the only exception to this result, as this knockout expressing phosphoglucomutase accumulated about 800 mg/L Q3G in both shake flasks and bioreactors.

Comparison of Q3G accumulation by different Escherichia coli in controlled bioreactors with 1.0 L working volume. Each culture contained 8 g/L glucose initially, and 2 g/L quercetin was added when the OD reached 1.0
We suspected that dissolved oxygen level was likely the principal difference between shake flasks and controlled bioreactors. We therefore next conducted microaerobic experiments using MEC367/pTrc99A-UGT73B3 by maintaining the DO to less than 10% saturation after the first 16 h of growth in bioreactors. This controlled, oxygen-deprived culture generated 610 mg/L Q3G, 60% lower than the same strain in fully oxygenated conditions, and only 10% greater than the concentration observed when MEC367/pTrc99A-UGT73B3 was cultured in shake flasks (Fig. 4).
Finally, in order to determine if a greater concentration of quercetin glucoside could be generated in a batch culture, we conducted 1 L batch processes using 30 g/L glucose and 5 g/L quercetin. In this controlled experiment, 3.9 g/L Q3G was produced by MEC367/pTrc99A-UGT73B3 (pgi deletion) in 56 h (Fig. 6). Acetate, a typical product of aerobic E. coli processes, was not detected in the fermentation broth during growth of the pgi knockout strain (i.e., its concentration was less than 0.05 g/L).

Quercetin-3-glucoside (filled triangle) formation by Escherichia coli MEC367/pTrc99A-UGT73B3. The 1.0 L process was conducted in a controlled bioreactor with 30 g/L glucose (empty square ) initially, and 5 g/L quercetin was added when the OD reached 1.0 (at 9 h)
Discussion
In the current study, quercetin-3-glucoside (Q3G) and quercetin-7-glucoside (Q7G) were formed using glucosyltransferases UGT73B3 and UGT84B1, respectively, and adding the aglycone quercetin into the culture of growing E. coli cells. We first examined the glucoside production in shake flasks using wild-type E. coli expressing these enzymes over the range of 20 °C – 37 °C. Surprisingly, the maximal temperatures were significantly different for the two glycosylations, with UGT73B3 showing a distinct maximum in Q3G formation at about 33 °C while Q7G formation by UGT84B1 increased over the entire temperature range. A previous study used 20 °C for glucoside formation in shake flasks using a complex medium [29], and these conditions generated about 9 mg/L Q3G and 4.8 mg/L Q7G, respectively. Our shake flask studies using defined medium at this same temperature resulted in 32-fold greater Q3G (153 mg/L) and 3.5-fold greater Q7G (17 mg/L) than these previous results. Moreover, compared to results at 20 °C, we observed 2.2-fold greater Q3G at 33 °C (330 mg/L) and 5.6-fold greater Q7G at 37 °C (95 mg/L). Our results not only demonstrate the value of temperature optimization to improve glycosylation by whole-cell catalysis but also indicate that glucosyltransferases behave differently with temperature. We can rationalize the effect of temperature by noting that glucoside formation from glucose and quercetin will not only depend on the activity and denaturation of the specific glucosyltransferase relative to other enzymes competing with substrates in this pathway, but glucoside formation will also depend on the cell growth rate, on the availability of uridine-5′-triphosphate (UTP) to charge the sugar donor UDP-glucose, and on the equilibrium between glucose-6P and glucose-1P. Since the isomerization of glucose-6P to glucose-1P is thermodynamically unfavorable with ΔG°′ = 7.1 kJ mol−1 [40], yielding an equilibrium constant of 0.057 (at 25 °C), increasing temperature favors glucose-1P formation slightly.
The medium and carbon source likely also affects glucoside formation. Typically, lysogeny broth (“LB”) has been selected to investigate the performance of various enzymes on quercetin glycosylation (e.g., 29). By using a defined medium with glucose as sole carbon source, cells are able to generate glucose-6P directly. When E. coli grows on defined media with glucose as the carbon source, enzymes of the PTS (e.g., PtsHI, Crr, and ManX) and enzymes of the upper glycolytic pathway are upregulated compared to the cells growing in rich media such as LB or terrific broth [41].
Bioconversion of quercetin using various strains generally accumulated twice the amount of Q3G in controlled bioreactors as in shake flasks (Figs. 4 and 5). The availability of quercetin was not a limiting factor, as this substrate was always in excess for both the shake flask and bioreactor experiments, and residual quercetin always remained at the end of each culture. In shake flask cultures, the 900 mg/L quercetin present could theoretically generate 1380 mg/L Q3G, though most shake flask cultures attained less than 40% of this maximal concentration. We attribute the general improvement in Q3G yield in bioreactors to be due to improved oxygenation in these cultures. This conclusion is supported by results in which cultures were intentionally oxygen-deprived, resulting in Q3G concentrations close to those concentrations observed in shake flask cultures. Furthermore, MEC367/pCS27-pgm grew about 30% slower than MEC367, MEC367/pCS27-glk, and MEC367/pCS27-galU, while MEC367 grew about 75% slower than MG1655 and MEC368. Oxygen uptake rate is lower in slow growing strains, and therefore, the slow-growing MEC367/pCS27-pgm would encounter the least oxygen deprivation in shake flask cultures. Thus, the high glucoside concentration attained by MEC367/pTrc99A-UGT73B3/pCS27-pgm compared to other strains in shake flask culture could be partly attributed to greater oxygen availability during growth of this strain. In the controlled bioreactor in which all cultures were fully oxygenated, slow-growing MEC367/pTrc99A-UGT73B3/pCS27-pgm no longer outperformed the other strains. We speculate that accumulation of UDP-glucose, which serves as the sugar donor for glucoside formation, is coupled to UTP regeneration and limited by ATP availability. ATP synthesis is sensitive to oxygen supply, and aerobic growth of E. coli generates about 2.5 times more ATP than anaerobic growth [42].
The glucose donor UDP-glucose is derived from glucose via glucose-6P, and blocking glucose-6P metabolism through glycolysis or the PP pathway would benefit UDP-glucose accumulation. A pgi knockout blocks glycolysis between glucose-6P and fructose-6P, and with this knockout, the PP pathway becomes the primary pathway to metabolize glucose-6P (Fig. 2). A zwf knockout prevents glucose-6P from entering the PP pathway, and in this case, glucose-6P is only metabolized through the EMP pathway. We observed a significantly reduced growth rate in the pgi knockout, while the zwf knockout did not alter the specific growth rate, consistent with previous research results [43, 44]. Steady-state cultures of wild-type E. coli on a defined glucose medium have demonstrated a metabolic flux distribution of 80% glucose entering glycolysis and 20% entering the PP pathway, with the activity of phosphoglucose isomerase 3.6-fold higher than the activity of glucose 6-phosphate dehydrogenase [45]. In addition, NADPH competitively inhibits glucose 6-phosphate dehydrogenase [46]. Thus, the PP pathway likely has a limited capacity to metabolize glucose-6P, and the reduced glycolytic flux for the pgi mutant increases the glucose-6P pool, as reported by others [31, 32]. Decreased glucose uptake also increases phosphorylated EIIA (EIIA-P) and thus cAMP-Crp, which in turn activates TCA cycle [47], while decreased fructose-1,6P2 leads to activated glyoxylate pathway enzymes [48–51]. The absence of acetate in the pgi knockout can thus be attributed to these regulatory changes compared to the parent strain [47, 52–54]. Moreover, not only did the pgi deletion play a pivotal role in sugar donor accumulation by increasing the glucose-6P pool, this knockout could potentially improve heterologous protein expression by eliminating acetate formation and E. coli overflow metabolism [55].
Overexpression of phosphoglucomutase (pgm), UDP-glucose pyrophosphorylase (galU), or glucokinase (glk) individually did not improve the glucoside formation in the controlled bioreactor. The k CAT values of phosphoglucomutase for glucose-6P and UDP-glucose pyrophosphorylase for glucose-1P have not been reported. Overexpression of pgm alone might yield no benefit (other than reduced growth rate in shake flask described above) simply because glucose-6P and glucose-1P are already close to an (unfavorable) equilibrium in a pgi knockout growing on glucose. Moreover, glucose-1P may activate competing pathways such as its conversion to amylose and glycogen via glucose-1P adenylyltransferase and glycogen synthase. E. coli strains deficient in phosphoglucomutase synthesize significantly more amylose than wild-type when grown on galactose or maltose, due to accumulation of glucose-1P [56, 57]. Overexpression of glucokinase could increase the phosphorylation of glucose, but its overexpression might also accelerate ATP consumption, which affects UTP regeneration and coupled UDP-glucose formation. Like many prokaryotes and eukaryotes, E. coli regenerates UTP from UDP and ATP via nucleoside diphosphate kinase (Ndk) [58]. UTP regeneration may currently be the bottleneck for glucoside formation rather than the phosphorylation of glucose. Overexpression of Ndk has previously enhanced the formation of cyanidin 3-O-glucoside in E. coli [59], and strategies which elevate UTP regeneration are the next potential steps to further improve the production of quercetin glucosides.
Bacterial cultures growing on glucose for the production of glucosides have the significant advantage of continuously supplying sugar donors in vivo to the glucosyltransferase. In less than 60 h, E. coli with a single pgi knockout and expressing UGT73B3 generated nearly 4 g/L Q3G as the sole glucoside product observed in a defined medium.
References
Hertog, M. G. L., Hollman, P. C. H., & van de Putte, B. (1993). Content of potentially anticarcinogenic flavonoids in tea infusions, wines and fruit juices. Journal of Agricultural and Food Chemistry, 41, 1242–1246.
Picinelli, A., Sua, B., & Mangas, J. J. (1997). Analysis of polyphenols in apple products. Zeitschrift für Lebensmittel-Untersuchung und Forschung A, 204, 48–51.
Price, K. R., & Rhodes, M. J. C. (1997). Analysis of the major flavonol glycosides present in four varieties of onion (Allium cepa) and changes in composition resulting from autolysis. Journal of the Science of Food and Agriculture, 74, 331–339.
Neveu, V., Perez-Jiménez, J., Vos, F., Crespy, V., du Chaffaut, L., Mennen, L., Knox, C., Eisner, R., Cruz, J., Wishart, D., and Scalbert, A. (2010). Phenol-Explorer: an online comprehensive database on polyphenol contents in foods. Database.
Choi, E. J., Bae, S. M., & Ahn, W. S. (2008). Antiproliferative effects of quercetin through cell cycle arrest and apoptosis in human breast cancer MDA-MB-453 cells. Archives of Pharmacal Research, 31, 1281–1285.
Luo, H., Jiang, B. H., King, S. M., & Chen, Y. C. (2008). Inhibition of cell growth and VEGF expression in ovarian cancer cells by flavonoids. Nutrition and Cancer, 60, 800–809.
Jeong, J. H., An, J. Y., Kwon, Y. T., Rhee, J. G., & Lee, Y. J. (2009). Effects of low dose quercetin: cancer cell-specific inhibition of cell cycle progression. Journal of Cellular Biochemistry, 106, 73–82.
Robak, J., & Gryglewski, R. J. (1988). Flavonoids are scavengers of superoxide anions. Biochemical Pharmacology, 37, 837–841.
Inal, M. E., & Kahraman, A. (2000). The protective effect of flavonol quercetin against ultraviolet an induced oxidative stress in rats. Toxicology, 154, 21–29.
Duarte, J., Pérez-Palencia, R., Vargas, F., Ocete, M. A., Pérez-Vizcaíno, F., Zarzuelo, A., & Tamargo, J. (2001). Antihypertensive effects of the flavonoid quercetin in spontaneously hypertensive rats. British Journal of Pharmacology, 133, 117–124.
Sánchez, M., Galisteo, M., Vera, R., Villar, I. C., Zarzuelo, A., Tamargo, J., Pérez-Vizcaíno, F., & Duarte, J. (2006). Quercetin downregulates NADPH oxidase, increases eNOS activity and prevents endothelial dysfunction in spontaneously hypertensive rats. Journal of Hypertension, 24, 75–84.
Yamamoto, Y., & Oue, E. (2006). Antihypertensive effect of quercetin in rats fed with a high-fat high-sucrose diet. Bioscience, Biotechnology and Biochemistry, 70, 933–939.
Gugler, R., Leschik, M., & Dengler, H. J. (1975). Disposition of quercetin in man after single oral and intravenous doses. European Journal of Clinical Pharmacology, 9, 229–234.
Makino, T., Shimizu, R., Kanemaru, M., Suzuki, Y., Moriwaki, M., & Mizukami, H. (2009). Enzymatically modified isoquercitrin, α-oligoglucosyl quercetin 3-O-glucoside, is absorbed more easily than other quercetin glycosides or aglycone after oral administration in rats. Biological and Pharmaceutical Bulletin, 32, 2034–2040.
Hollman, P. C., de Vries, J., van Leeuwen, S. D., Mengelere, M. J., & Katan, M. B. (1995). Absorption of dietary quercetin glycosides and quercetin in healthy ileostomy volunteers. American Journal of Clinical Nutrition, 62, 1276–1282.
Hollman, P. C., van der Gaag, M., Mengelers, M. J., van Trijp, J. M., de Vries, J. H., & Katan, M. B. (1996). Absorption and disposition kinetics of the dietary antioxidant quercetin in man. Free Radical Biology and Medicine, 21, 703–707.
Hollman, P. C., van Trijp, J. M., Buysman, M. N., van der Gaag, M. S., Mengelers, M. J., de Vries, J. H., & Katan, M. B. (1997). Relative bioavailability of the antioxidant flavonoid quercetin from various foods in man. FEBS Letters, 418, 152–156.
Gee, J. M., DuPont, M. S., Day, A. J., Plumb, G. W., Willliamson, G., & Johnson, I. T. (2000). Intestinal transport of quercetin glycosides in rats involves both deglycosylation and interaction with the hexose transport pathway. Journal of Nutrition, 130, 2765–2771.
Crespy, V., Morand, C., Besson, C., Manach, C., Demigne, C., & Remesy, C. (2001). Comparison of the intestinal absorption of quercetin, phloretin and their glucosides in rats. Journal of Nutrition, 131, 2109–2114.
Paulke, A., Eckert, G. P., Schubert-Zsilavecz, M., & Wurglics, M. (2012). Isoquercitrin provides better bioavailability than quercetin: comparison of quercetin metabolites in body tissue and brain sections after six days administration of isoquercitrin and quercetin. Pharmazie, 67, 991–996.
Amado, N. G., Predes, D., Fonseca, B. F., Cerqueira, D. M., Reis, A. H., Dudenhoeffer, A. C., Borges, H. L., Mendes, F. A., & Abreu, J. G. (2014). Isoquercitrin suppresses colon cancer cell growth in vitro by targeting the Wnt/β-catenin signaling pathway. Journal of Biological Chemistry, 289, 35456–35467.
Day, A. J., Gee, J. M., DuPont, M. S., Johnson, I. T., & Williamson, G. (2003). Absorption of quercetin-3-glucoside and quercetin-4′-glycoside in the rat small intestine: the role of lactase phlorizin hydrolase and the sodium-dependent glucose transporter. Biochemical Pharmacology, 65, 1199–1206.
Cermak, R., Landgraf, S., & Wolffram, S. (2004). Quercetin glucosides inhibit glucose uptake into brush-border-membrane vesicles of porcine jejunum. British Journal of Nutrition, 91, 849–855.
Song, J. H., Park, K. S., Kwon, D. H., & Choi, H. J. (2013). Anti-human rhinovirus 2 activity and mode of action of quercetin-7-glucoside from Lagerstroemia speciosa. Journal of Medicinal Food, 16, 274–279.
Lu, Z., Wang, J., Lin, S., & Zhan, Y. (2013). Degradation of rutin into isoquercitrin by Bacillus litoralis strain C44. IOSR Journal of Engineering, 2, 1154–1161.
De Bruyn, F., Maertens, J., Beauprez, J., Soetaert, W., & De Me, M. (2015). Biotechnological advances in UDP-sugar based glycosylation of small molecules. Biotechnology Advances, 33, 288–302.
He, X. Z., Li, W. S., Blount, J. W., & Dixon, R. A. (2008). Regioselective synthesis of plant (iso)flavone glycosides in Escherichia coli. Applied Microbiology and Biotechnology, 80, 253–260.
Kim, J. H., Shin, K. H., Ko, J. H., & Ahn, J. H. (2006). Glucosylation of flavonols by Escherichia coli expressing glucosyltransferase from rice (Oryza sativa). Journal of Bioscience and Bioengineering, 102, 135–137.
Lim, E. K., Ashford, D. A., Hou, B., Jackson, R. G., & Bowles, D. J. (2004). Arabidopsis glycosyltransferases as biocatalysts in fermentation for regioselective synthesis of diverse quercetin glucosides. Biotechnology and Bioengineering, 87, 623–631.
Mao, Z., Shin, H. D., & Chen, R. R. (2006). Engineering the E. coli UDP-glucose synthesis pathway for oligosaccharide synthesis. Biotechnology Progress, 22, 369–374.
Lee, A. T., & Cerami, A. (1987). Elevated glucose 6-phosphate levels are associated with plasmid mutations in vivo. Proceedings of the National Academy of Sciences USA, 84, 8311–8314.
Morita, T., El-Kazzaz, W., Tanaka, Y., Inada, T., & Aiba, H. (2003). Accumulation of glucose 6-phosphate or fructose 6-phosphate is responsible for destabilization of glucose transporter mRNA in Escherichia coli. Journal of Biological Chemistry, 278, 15608–15614.
Yan, Y., Zhen, L., & Koffas, M. A. G. (2008). High-yield anthocyanin bioxynthesis in engineered Escherichia coli. Biotechnology and Bioengineering, 100, 126–140.
Lim, C. G., Wong, L., Bhan, N., Xu, P., Venkiteswaran, S., & Koffas, M. A. G. (2015). Development of a recombinant Escherichia coli strain for overproduction of the plant pigment anthocyanin. Applied and Environmental Microbiology, 81, 6276–6284.
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., Datsenko, K. A., Tomita, M., Wanner, B. L., & Mori, H. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular Systems Biology, 2, 1–11.
Datsenko, K. A., & Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences USA, 97, 6640–6645.
Eiteman, M. A., & Chastain, M. J. (1997). Optimization of the ion-exchange analysis of organic acids from fermentation. Analytica et Chemica Acta, 338, 69–75.
St. John, A. C., Conklin, K., Rosenthal, E., & Goldberg, A. L. (1978). Further evidence for the involvement of charged tRNA and guanosine tetraphosphate in the control of protein degradation in Escherichia coli. Journal of Biological Chemistry, 253, 3945–3951.
Herendeen, S. L., van Bogelen, R. A., & Neidhardt, F. C. (1979). Levels of major proteins of Escherichia coli during growth at different temperatures. Journal of Bacteriology, 139, 185–194.
Berg, J. M., Tymoczko, J. L., & Stryer, L. (2002). Biochemistry (5th ed.). New York: W H Freeman.
Li, Z., Nimtz, M., & Rinas, U. (2014). The metabolic potential of Escherichia coli BL21 in defined and rich medium. Microbial Cell Factories, 13, 1.
Chen, X., Alonso, A. P., Allen, D. K., Reed, J. L., & Shachar-Hill, Y. (2011). Synergy between 13C-metabolic flux analysis and flux balance analysis for understanding metabolic adaption to anaerobiosis in E. coli. Metabolic Engineering, 13, 38–48.
Canonaco, F., Hess, T. A., Heri, S., Wang, T., Szyperski, T., & Sauer, U. (2001). Metabolic flux response to phosphoglucose isomerase knockout in Escherichia coli and impact of overexpression of the soluble transhydrogenase UdhA. FEMS Microbiology Letters, 204, 247–252.
Fischer, E., & Sauer, U. (2003). Metabolic flux profiling of E. coli mutants in central carbon metabolism using GC-MS. European Journal of Biochemistry, 270, 880–891.
Zhao, J., Baba, T., Mori, H., & Shimizu, K. (2004). Effect of zwf gene knockout on the metabolism of Escherichia coli grown on glucose or acetate. Metabolic Engineering, 6, 164–174.
Olavarría, K., Valdés, D., & Cabrera, R. (2012). The cofactor preference of glucose-6-phosphate dehydrogenase from Escherichia coli—modeling the physiological production of reduced cofactors. FEBS Journal, 279, 2296–2309.
Yao, R., Hirose, Y., Sarkar, D., Nakahigashi, K., Ye, Q., & Shimizu, K. (2011). Catabolic regulation analysis of Escherichia coli and its crp, mlc, mgsA, pgi and ptsG mutants. Microbial Cell Factories, 10, 67.
Ramseier, T. M., Nègre, D., Cortay, J. C., Scarabel, M., Cozzone, A. J., & Saier, M. H. (1993). In vitro binding of the pleiotropic transcriptional regulatory protein, FruR, to the fru, pps, ace, pts and icd operons of Escherichia coli and Salmonella typhimurium. Journal of Molecular Biology, 234, 28–44.
Saier Jr., M. H., & Ramseier, T. M. (1996). The catabolite repressor/activator (Cra) protein of enteric bacteria. Journal of Bacteriology, 178, 3411–3417.
Shimada, T., Yamamoto, K., & Ishihama, A. (2011). Novel members of the Cra regulon involved in carbon metabolism in Escherichia coli. Journal of Bacteriology, 193, 649–659.
Kabir, M. M., & Shimizu, K. (2003). Gene expression patterns for metabolic pathway in pgi knockout Escherichia coli with and without phb genes based on RT-PCR. Journal of Biotechnology, 105, 11–31.
Hua, Q., Yang, C., Baba, T., Mori, H., & Shimizu, K. (2003). Responses of the central metabolism in Escherichia coli to phosphoglucose isomerase and glucose-6-phosphate dehydrogenase knockouts. Journal of Bacteriology, 185, 7053–7067.
Toya, Y., Ishii, N., Nakahigashi, K., Hirasawa, T., Soga, T., Tomita, M., & Shimizu, K. (2010). 13C-metabolic flux analysis for batch culture of Escherichia coli and its pyk and pgi gene knockout mutants based on mass isotopomer distribution of intracellular metabolites. Biotechnology Progress, 26, 975–992.
Usui, Y., Hirasawa, T., Furusawa, C., Shirai, T., Yamamoto, N., Mori, H., & Shimizu, H. (2012). Investigating the effects of perturbations to pgi and eno gene expression on central carbon metabolism in Escherichia coli using 13C metabolic flux analysis. Microbial Cell Factories, 11, 87.
Eiteman, M. A., & Altman, E. (2006). Overcoming acetate in Escherichia coli recombinant protein fermentations. Trends in Biotechnology, 24, 530–536.
Brautaset, T., Petersen, S. B., & Valla, S. (1998). An experimental study on carbon flow in Escherichia coli as a function of kinetic properties and expression levels of the enzyme phosphoglucomutase. Biotechnology and Bioengineering, 5, 299–302.
Brautaset, T., Petersen, S. B., & Valla, S. (2000). In vitro determined kinetic properties of mutant phosphoglucomutases and their effects on sugar catabolism in Escherichia coli. Metabolic Engineering, 2, 104–114.
Lu, Q., Zhang, X., Almaula, N., Mathews, C. K., & Inouye, M. (1995). The gene for nucleoside diphosphate kinase functions as a mutator gene in Escherichia coli. Journal of Molecular Biology, 254, 337–341.
Leonard, E., Yan, Y., Fowler, Z. L., Li, Z., Lim, C. G., Lim, K. H., & Koffas, M. A. (2008). Strain improvement of recombinant Escherichia coli for efficient production of plant flavonoids. Molecular Pharmaceutics, 5, 257–265.
Acknowledgements
The authors thank Sarah Lee, Li Wang, and Don Armento for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xia, T., Eiteman, M.A. Quercetin Glucoside Production by Engineered Escherichia coli . Appl Biochem Biotechnol 182, 1358–1370 (2017). https://doi.org/10.1007/s12010-017-2403-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-017-2403-x