
Abstract
Glycoside hydrolases (GHs) are enzymes found in all living kingdoms that are involved in multiple physiological functions. Due to their multiple enzymatic activities, GHs are broadly applied in bioethanol, food, and paper industry. In order to increase the productivity of these industrial processes, a constant search for novel and efficient enzymes has been proved to be necessary. In this context, metagenomics is a powerful approach to achieve this demand. In the current study, we describe the discovery and characterization of a novel member of GH16 family derived from the sugarcane soil metagenome. The enzyme, named SCLam, has 286 amino acid residues and displays sequence homology and activity properties that resemble known laminarases. SCLam is active against barley beta-glucan, laminarin, and lichenan (72, 33, and 10 U mg−1, respectively). The optimal reaction conditions were identified as 40 °C and pH 6.5. The low-resolution structure was determined using the small-angle X-ray scattering technique, revealing that SCLam is a monomer in solution with a radius of gyration equal to 19.6 Å. To the best of our knowledge, SCLam is the first nonspecific (1,3/1,3:1,4)-β-d-glucan endohydrolase (EC 3.2.1.6) recovered by metagenomic approach to be characterized.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metagenomics has become a powerful tool to access the genetic diversity of microbial world, given a fact that the majority of the microorganisms from different environments cannot be cultured using standard laboratory techniques [1]. Basically, metagenomic analysis involves isolation of DNA from an environmental sample, followed by development of function-driven and/or sequence-driven approaches in order to extract biological information from the genetic material [2, 3]. Collectively, these approaches have resulted in the identification of novel metabolites [4–6] and biocatalysts, including several novel glycoside hydrolases (GHs) belonging to different GH families [7–12].
The GHs are a group of enzymes of a great importance in carbohydrate metabolism [13], and therefore, they play key roles in biotechnological processes, such as the production of biofuels from plant feedstocks [14], pulp and paper industry [15], and food quality improvement [16].
Among the GHs, GH16 family is composed of enzymes that act on the hydrolysis of a wide range of polysaccharides, including 1,3-β-glucans, present in fungal cell walls [17]; mixed (1,3:1,4)-linkage β-glucans, widely distributed in plant family Poaceae, which includes species of cereals and grasses [18]; lichenan, a mixed-linkage β-glucan commonly found in lichens [19]; and laminarin, an energy storage glucose polysaccharide found in brown algae [20]. Members of GH16 family are distributed among the three domains of life, and many of them had already been characterized [21–24].
In 2004, Strohmeier et al. [25] proposed a classification of GH16 glycoside hydrolases into five subgroups based on their substrate specificities. The first subgroup is composed of (1,3)-β-galactan endohydrolases that are able to hydrolyze (1,3)-β-galactosyl linkages such as the one found in agarose; the second subgroup is composed of specific enzymes to (1,4)-β-galactosyl linkages. The nonspecific (1,3/1,3:1,4)-β-d-glucan endohydrolases belong to the third group, and they are able to cleave both (1,3) and (1,4) linkages adjacent to a (1,3)-β-d-glucosyl residue on the nonreducing end of hydrolyzed linkage [26]. The fourth group is composed of (1,3:1,4)-β-d-glucan endohydrolases that specifically cleave (1,4)-β-d-glucosyl linkage adjacent to (1,3)-β-d-glucosyl residue in the direction of the nonreducing end. The fifth group is composed of xyloglucan-modifying enzymes.
The β-1,3-glucanases have potential application in biotechnological process, since they act in a variety of natural polysaccharides containing 1,3-β linkages, resulting in the release of oligosaccharides with bioactive properties [27, 28]. Another potential area of β-1,3-glucanases application is in the food industry, particularly in the production of beverages such as beers and wine where, by avoiding premature filter fouling, they facilitate the filtration [29, 30].
To the best of our knowledge, our work describes the first characterization of a GH16 family member-nonspecific (1,3/1,3:1,4)-β-d-glucan endohydrolase derived from a metagenomic approach. Herein, we discuss the biochemical and biophysical properties of SCLam in order to evaluate the potential application of this enzyme for conversion of natural polysaccharides into bio-based products.
Materials and Methods
Identification and Sequence Analysis of the SCLam Gene
The coding gene for SCLam was identified after functional screening of a metagenomic library derived from sugarcane soil, as described previously [9]. The plasmid from the clone retrieved from metagenomic library was extracted and had its DNA insert sequenced on an ABI Prism 377 Genetic Analyzer (Applied Biosystems, USA) at the Brazilian Bioethanol Science and Technology Laboratory. The generated sequence reads were assembled using Geneious Pro 4.8.5, which was also applied for open reading frame (ORF) identification. Based on the preliminary sequence, the identified ORF was compared to a collection of public databases at National Center for Biotechnology Information (NCBI) using the BLASTX tool. Prediction of signal peptide was performed using SignalP 4.1 [31].
The nucleotide sequence of the SCLam gene was deposited at NCBI GenBank under accession number KP207684. The ProtParam tool from ExPASy (http://web.expasy.org/protparam/) [32] was used to predict physical and chemical parameter of SCLam. Construction of the phylogenetic tree was performed in Mega 6 [33], using the neighbor-joining method [34]. In relation to the phylogenetic tree, selected sequences were members of GH16 family (http://www.cazy.org/) [35] that presented experimental evidences at protein level.
Expression and Purification of SCLam
The coding sequence for SCLam was amplified by PCR using the forward and reverse primers 5′ TATATATACATATGCAGCAGCCCGAGGGTGTG 3′ and 5′ TATATATACTCGAGTCAGCGCTTCTGATACAC, containing NdeI and XhoI restriction sites (underlined in the primer sequences), respectively. The amplified sequence was cloned into pET28a (Novagen). The recombinant plasmid pET28a(+)-SCLam was transformed in the Escherichia coli Rosetta 2 strain (Novagen) for protein production. The expression was performed by cultivation at 37 °C and 200 rpm in LB broth containing 50 μg mL−1 of kanamycin until A 600 reached 1.0 followed by induction with 1 mM IPTG for approximately 16 h at 18 °C. Afterwards, the cells were harvested by centrifugation and resuspended in 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 10 mM imidazole, and 5 % glycerol. The cells were sonicated after treatment with 1 mg/mL lysozyme and submitted to centrifugation at 10,000g for 1 h. The recovered supernatant was incubated with TALON resin (Clontech), and the retained proteins were eluted with 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 300 mM imidazole, and 5 % glycerol. The protein was further purified on a Superdex 75 16/60 prep grade column, equilibrated with 20 mM sodium phosphate (pH 7.4) and 50 mM NaCl-containing buffer. The purity of the protein sample obtained was confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) [36] under reducing conditions. Protein concentration was assessed by absorbance at 280 nm.
Enzymatic Characterization
The hydrolytic activity of SCLam was determined by the amount of the reducing sugars released from different polysaccharides (purchased from Megazyme, Ireland, and Sigma-Aldrich, USA), using the DNS method [37], after incubation at 37 °C for 30 min using 100 mM sodium acetate buffer (pH 5.5) following Alvarez et al. [9]. One unit (U) was defined as the quantity of enzyme able to release reducing sugars at a rate of 1 μmol min−1. To determine the optimum pH, enzymatic reactions were carried in solutions with different pH levels using 300 mM citrate-phosphate-glycine buffer system, containing barley β-glucan 0.5 % (w/v), for 30 min at 37 °C. For optimal temperature experiments, the reactions were conducted at optimum pH and in the same buffer maintaining incubation temperatures in the range of 20–60 °C for 30 min. Thermal stability of SCLam was evaluated by incubation of the protein in a range of temperatures and removal of aliquots in defined time intervals followed by measurement of residual activity at the optimal pH and temperature.
For the determination of kinetic parameters, K m , V max, k cat, and k cat / K m , reaction time and enzymatic units were adjusted to allow the estimation of initial velocities, using a substrate range from 0.4 to 5.4 mg mL−1. Assays were performed in quintuplicate, using 100 mM sodium acetate buffer in optimal pH and temperature for 30 min. The mathematical calculations were made using the software GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA; www.graphpad.com).
Analysis of Hydrolysis Products
In order to evaluate the cleavage pattern of SCLam, the product of enzymatic hydrolysis of laminarin, barley β-glucan, laminarihexaose (L6), and cellohexaose (C6) were evaluated by capillary electrophoresis. The enzymatic hydrolysis reactions were performed by overnight incubation at 40 °C of 6 ng μL−1 and 0.2 mg mL−1 of SCLam with polysaccharides (2 g L−1) and oligosaccharides (L6 and C6, 4.5 mM), respectively. Following incubation, samples were derivatized by reductive amination with 8-aminopyrene-1,3,6-trisulfonic (APTS) [38] and evaluated by capillary electrophoresis (P/ACE MDQ system; Beckman Coulter) equipped with a laser-induced fluorescence detector. Separation was performed in a neutral capillary (Beckman Coulter) of 50 μm in internal diameter and 50 cm in length at 20 kV/70–100 μA in 40 mM potassium phosphate buffer (pH 2.5), with the cathode in the inlet. The molecules labeled with APTS were excited at 488 nm, and emissions were collected in a 520-nm bandpass filter. The comparison of separate electrophoresis runs showed a slight difference in retention times due to the small volumes of capillary electrophoresis and variations in buffer strength. Information of electrophoretic behavior combined to coelectrophoresis with oligosaccharide standards was used for identification of hydrolysis products.
Circular Dichroism and Thermal Unfolding Studies
The far-UV spectra (200–260 nm) were recorded on a Jasco J-815 spectropolarimeter (Jasco Inc., Tokyo, Japan) coupled to a Peltier temperature controller using a 0.1-cm path quartz cuvette. Protein was used at a concentration of 20 μM in 20 mM phosphate buffer (pH 7.4) and 50 mM NaCl. Data collection parameters were set to a scan rate of 50 nm min−1, response time of 4 s, sensitivity of 100 mdeg, and accumulation of 20. Results were expressed as mean residue ellipticity (deg cm2 dmol−1).
For evaluation of thermal stability, circular dichroism (CD) spectra were analyzed at different temperatures, ranging from 20 to 80 °C, at a heating rate of 1 °C min−1. Thermal unfolding was monitored by measuring the ellipticity change at 217 nm.
Small-Angle X-ray Scattering
Small-angle X-ray scattering (SAXS) data for SCLam were collected on the SAXS2 beamline at the Brazilian Synchrotron Light Laboratory at concentrations of 1 and 5.1 mg mL−1 and using the monochromatic X-rays with the wavelength (λ) of 1.55 Å. X-ray scattering patterns were recorded using a two-dimensional detector (MarResearch, USA). The sample-to-detector distance was set to 1000 mm to give a range of the scattering vectors (q) from 0.016 to 0.33 Å−1, where q is the magnitude of the q vector defined by the following equation: q = 4π sin θ/λ (2θ is the scattering angle). The SAXS patterns were integrated using Fit2D software, and the curves were scaled using the protein concentration [39].
Radius of gyration (R g) was computed from Guinier equation [40] and using an indirect Fourier transform method using GNOM package [41]. The distance distribution function (p(r)) also was calculated using GNOM package, and the maximum diameter (D max) was obtained. SAXS-based estimate of protein molecular weight was computed using the procedure implemented in the Web tool SAXS MoW [42]. Dummy residue (DR) models were calculated from the SAXS curves of the SCLam using the Gasbor package [43]. Damaver was used for automated analysis and averaging of multiple reconstructions, permitting both to analyze the stability of the reconstruction convergence and to yield the most probable averaged particle model [44]. Crysol 2.7 [45] was used to generate the simulated scattering curve from DR and the high-resolution crystallographic structure of laminarase from Rhodothermus marinus, RMLam (PDB id: 3ILN) [46], and to evaluate their R g and D max. GNOM was used to obtain the distance distribution function (p(r)) from a crystallographic model. Supcomb [43] was used to superimpose the low-resolution SCLam DR and the high-resolution structure of the R. marinus laminarase [46].
Results and Discussion
Phylogenetic Analysis of SCLam Amino Acid Sequence
After functional screening of a metagenomic library, a clone harboring an insert of approximately 3.5 kb was isolated. After sequencing the insert, we identified an ORF encoding a protein with 286 amino acid residues containing a putative N-terminal signal peptide followed by a conserved domain of GH16 family. The sequence showed considerable similarity to glycoside hydrolase from Sphingomonas (GenBank: WP_025560764.1) and Sphingomonas sp. JGI 0001003-D23 (GenBank: WP_029724034.1) with amino acid sequence identity of 74 %, to glycoside hydrolase from Sphingomonas elodea (GenBank: WP_010544485.1; 68 % of sequence identity) and to a hypothetical protein from Oxalobacteraceae bacterium JGI 0001004-K23 (GenBank: WP_018058119.1; sequence identity of 59 %). With respect to the previously characterized laminarases, SCLam displayed 48 and 39 % amino acid sequence identity to the laminarase from R. marinus (GenBank: AAC69707.1) and Thermotoga petrophila (GenBank: ABQ46917.1), respectively. The predicted molecular weight and isoelectric point for the mature SCLam were estimated as 30 kDa and 5.6, respectively. The predicted molecular weight of SCLam was confirmed by SDS-PAGE analyses of purified samples (Figure S1).
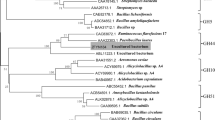
Accordingly to Strohmeier et al. [25], GH16 is classified into five subgroups based on enzymatic activity: (1,3)-β-d-galactanases (which includes agarases), (1,4)-β-d-galactanases/k-carrageenases, nonspecific (1,3/1,3:1,4)-β-d-glucanases (which includes laminarases), (1,3:1,4)-β-d-glucanases (which includes lichenases), and xyloglucan transglycosylases/hydrolases (XTHs). In order to analyze which subgroup SCLam would be related based on amino acid sequence, sequences from all subgroups, except XTHs, were retrieved from the CAZy database. XTHs’ sequences were not retrieved since none of them is classified as characterized in the CAZy database. The phylogenetic tree evidenced the distribution of four clades (Fig. 1a), in accordance to the subgroups described above. In addition, it was possible to observe a close association of SCLam sequence with sequences of known laminarases, which indicated that SCLam is expected to be a nonspecific (1,3/1,3:1,4)-β-d-glucanase.

a Phylogenetic analysis of SCLam with members of GH16 family based on amino acid sequences. b Substrate specificity for SCLam
Optimum pH, Temperature, and Thermal Stability of the Enzyme
The experiments conducted with barley β-glucan revealed an optimum value for enzymatic activity at pH 6.5 (Fig. 2a). SCLam was able to retain approximately 60 % of its maximum activity at pH 9.0. Similar values of optimum pH was observed for an endo-1,3-β-glucanase from Arthrobacter sp. [47] (pH 6.5) and for TpLam from T. petrophila (around pH 6.2) [48].

a, b Influence of pH and temperature on SCLam activity, respectively. c Thermal stability of SCLam evaluated through circular dichroism
The highest value of enzymatic activity was observed at 40 °C (Fig. 2b). This is the same value observed for the catalytic domain of an endo-1,3-β-glucanase from Paenibacillus sp. [49], but it is lower in comparison to TpLam and 1,3-β-glucanase from Streptomyces sioyaensis, which have the optimum temperatures for enzymatic activity at 91 and 75 °C, respectively [48, 50]. In addition, SCLam exhibited considerable thermal stability at 30 and 40 °C for up to 18 h, while at the temperatures above 40 °C, SCLam was very unstable (data not shown).
These data are consistent with the denaturation curve of SCLam generated using CD spectroscopy. In order to evaluate the enzyme thermostability, its CD spectra were recorded at different temperatures and the variation in molar ellipticity at 217 nm indicated a melting temperature (T m) of 41.3 °C (Fig. 2c).
Substrate Cleavage Analysis
The enzymatic activity of SCLam was evaluated using a set of different polysaccharides. The enzyme has a highest specific activity for barley β-glucan (72.26 ± 3.19 μmol min−1 mg−1), followed by laminarin (33.24 ± 1.24 μmol min−1 mg−1) and lichenan (10.19 ± 0.9 μmol min−1 mg−1) (Fig. 1b). These results indicated that SCLam preferentially hydrolyzes β-glucan, a mixed β-1,3-1,4-glucan, with the 1,4-to-1,3-β-d ratio varying from 2:1 to 3:1 [51]. The second preferred substrate is laminarin, a branched (β-1,6)-β-1,3-glucan chain, followed by lichenan, which is also a mixed β-1,3-1,4-glucan with the 1,4-to-1,3-β-d ratio varying from 2:1 to 3:1, thus with a higher proportion of 1,3 to 1,4-β-d linkages in comparison to β-glucan [52]. These results agree with our phylogenetic analysis, which showed SCLam closely associated to laminarases that are classified within the subgroup of nonspecific (1,3/1,3:1,4)-β-d-glucanases [25]. Important differences in substrate specificity were observed among the characterized enzymes from the phylogenetic tree (Fig. 1a). The endo-β-1,3-glucanase from Pyrococcus furiosus (AAC25554.2), TpLam from T. petrophila (ABQ46917.1), Curd1 from S. sioyaensis (AAF31438.1), and rPglA from Paenibacillus sp. S09 (AFO67889.1) exhibit preference for 1,3-β-homoglucans in comparison to mixed β-1,3-1,4-glucan, while SCLam, LamR from R. marinus (AAC69707.1), and BglF from Nocardiopsis sp. strain F96 (BAE54302.1) were more active on mixed β-1,3-1,4-glucan in comparison to 1,3-β-homoglucans [21, 24, 48, 50, 53, 54].
The apparent kinetic parameters were calculated from the initial rates of hydrolysis in variable concentrations of barley β-glucan (data not shown). Values of K m and V max were 1.09 ± 0.07 mg mL−1 and 32.44 ± 1.1 μmol min−1 mg−1, respectively. The calculated values for k cat and k cat / K m were 16.22 s−1 and 14.83 s−1 mg−1 mL−1, respectively.
The hydrolysis product of L6, C6 (both in the presence of 0.2 mg mL−1 of SCLam), barley β-glucan, and laminarin (both in the presence of 6 ng μL−1 of SCLam) was analyzed by capillary electrophoresis (Fig. 3). The SCLam was able to convert L6 molecules mostly into glucose (G), but smaller amounts of laminaribiose (L2) were also accumulated (Fig. 3c). In relation to polysaccharides, degradation of laminarin (Fig. 3b) revealed a similar pattern, resulting in the formation of mainly glucose and disaccharide (DP2), followed by a small quantity of trisaccharide (DP3). Apart from DP1, DP2, and DP3, hydrolysis of β-glucan (Fig. 3a) also resulted in accumulation of oligosaccharides with a higher degree of polymerization, such as DP5 and DP4. Collectively, these results suggest that SCLam is able to cleave both β-1,3 and β-1,4 linkages in β-glucan, leading to formation of glucose as a final product. This was confirmed by the ability of the enzyme to hydrolyze C6 and produce cellopentaose (C5), cellotetraose (C4), and glucose as hydrolysis product (Fig. 3d). Since C6 is an oligosaccharide composed only by β(1,4) linkages, this result contrasts with the definition of nonspecific (1,3/1,3:1,4)-β-d-glucan endohydrolases, which states that hydrolysis of (1,4) linkages occurs when it is adjacent to a (1,3)-β-d-glucosyl residue on the nonreducing end of hydrolyzed linkage [25, 26]. However, as shown in Fig. 3d, SCLam was not able to completely convert C6, while the degradation of L6 resulted in the total conversion of the substrate into glucose and DP2, suggesting lower affinity of SCLam for C6. The laminarase LamR from R. marinus exhibited a similar cleavage pattern both in polysaccharides (β-glucan and laminarin) and 1,3-β-oligosaccharides. Interestingly, LamR was also able to convert cello-oligosaccharides (from D2 to D5), resulting in the formation of mainly DP2 and glucose [54].

Analysis of hydrolysis products released by action of SCLam after overnight incubation in a β-glucan, b laminarin, c laminarihexaose (L6), and d cellohexaose (C6). C5 cellopentaose, C4 cellotetraose, G glucose, L2 laminaribiose. DP5, DP4, DP3, and DP2 stands for the degree of polymerization
Small-Angle X-ray Scattering
Since SCLam does not have high degree of identity with other enzymes whose structure was solved, we used SAXS to gain insight into structural properties in solution as size, shape, and oligomerization state. The crystallographic structure of RMLam, which has the highest sequence identity (48 %) with SCLam, was selected for comparison. Once its crystallographic structure is available as a dimer in asymmetric unit, SAXS-simulated curves were obtained for the monomeric and dimeric forms.
The SAXS analyses were performed from samples at 1 mg mL−1, due to a compromise between scattering detection quality and reduced aggregation effect. Applying the Guinier approximation to the scattering curve (qR g <1.3), we obtained a radius of gyration of SCLam equal to 19.6 Å (Fig. 4a, insert). This value is in close agreement with those obtained by the GNOM (19.40 ± 0.024) Å. Using a SAXS MoW server [42], SAXS-based estimate of the SCLam molecular mass was 27.70 kDa, with an error of 7.7 % relative to the theoretical molecular mass.

a Experimental SAXS curve of SCLam and fitting procedure with simulated curves. Experimental data 1 mg mL−1 (open circles), PDB: 3ILN monomer (continuous line), PDB: 3ILN dimer (dashed line). Insert contains Guinier analysis. b p(r), the same symbols are used. c Superposition of SCLam ab initio DAM and the GH16 monomeric crystal structure (PDB: 3ILN). The center model is rotated 90° relative to the x-axis and the model on the right is rotated 90° to the y-axis
The ab initio DR model was performed with a Gasbor package [43] without symmetry constraint. The DR model resolution, calculated using the equation Resolution = 2π/q max, was determined as 19.1 Å. The superposition of SCLam DR model and monomeric crystallographic structure of R. marinus laminarase is shown in Fig. 4c, evidencing an excellent fit between the models. The calculated parameters are given in Table 1.
Crysol package was used to simulate SAXS curves from ab initio DR model and from the RMLam X-ray structure (PDB: 3ILN) (Fig. 4a). As expected, the RMLam monomer has a much smaller χ parameter (1.9), indicating a better fit to the experimental data than the dimer (χ = 24.6). The distance distribution function (p(r)) of SCLam is well centered and fits better the monomeric laminarinase structure (Fig. 4b). The above information clearly indicates the monomeric state of the enzyme in solution and an overall shape similar to RMLam.
In conclusion, we have described herein the isolation and successful recombinant production of a novel GH16 family member isolated using a metagenomic approach. We evaluated the biochemical properties and the mode of operation and built the low-resolution model of this novel enzyme. This study provides a biochemical and structural basis for further studies on GH16 family members.
References
Amann, R. I., Ludwig, W., & Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiological Reviews, 59, 143–169.
Handelsman, J. (2004). Metagenomics: application of genomics to uncultured microorganisms. Microbiology and Molecular Biology Reviews, 68, 669–685.
Schloss, P. D., & Handelsman, J. (2003). Biotechnological prospects from metagenomics. Current Opinion in Biotechnology, 14, 303–310.
Kallifidas, D., Kang, H.-S., & Brady, S. F. (2012). Tetarimycin A, an MRSA-active antibiotic identified through induced expression of environmental DNA gene clusters. Journal of the American Chemical Society, 134, 19552–19555.
MacNeil, I. A., Tiong, C. L., Minor, C., August, P. R., Grossman, T. H., Loiacono, K. A., Lynch, B. A., Phillips, T., Narula, S., Sundaramoorthi, R., Tyler, A., Aldredge, T., Long, H., Gilman, M., Holt, D., & Osburne, M. S. (2001). Expression and isolation of antimicrobial small molecules from soil DNA libraries. Journal of Molecular Microbiology and Biotechnology, 3, 301–308.
Wang, G. Y., Graziani, E., Waters, B., Pan, W., Li, X., McDermott, J., Meurer, G., Saxena, G., Andersen, R. J., & Davies, J. (2000). Novel natural products from soil DNA libraries in a streptomycete host. Organic Letters, 2, 2401–2404.
Yun, J., Kang, S., Park, S., Yoon, H., Kim, M.-J., Heu, S., & Ryu, S. (2004). Characterization of a novel amylolytic enzyme encoded by a gene from a soil-derived metagenomic library. Applied and Environmental Microbiology, 70, 7229–7235.
Jiang, C., Li, S.-X., Luo, F.-F., Jin, K., Wang, Q., Hao, Z.-Y., Wu, L.-L., Zhao, G.-C., Ma, G.-F., Shen, P.-H., Tang, X.-L., & Wu, B. (2011). Biochemical characterization of two novel β-glucosidase genes by metagenome expression cloning. Bioresource Technology, 102, 3272–3278.
Alvarez, T. M., Goldbeck, R., dos Santos, C. R., Paixão, D. A. A., Gonçalves, T. A., Franco Cairo, J. P. L., Almeida, R. F., de Oliveira Pereira, I., Jackson, G., Cota, J., Büchli, F., Citadini, A. P., Ruller, R., Polo, C. C., de Oliveira Neto, M., Murakami, M. T., & Squina, F. M. (2013). Development and biotechnological application of a novel endoxylanase family GH10 identified from sugarcane soil metagenome. PLoS One, 8, e70014.
Liu, J., Liu, W.-D., Zhao, X.-L., Shen, W.-J., Cao, H., & Cui, Z.-L. (2011). Cloning and functional characterization of a novel endo-β-1,4-glucanase gene from a soil-derived metagenomic library. Applied Microbiology and Biotechnology, 89, 1083–1092.
Brennan, Y., Callen, W. N., Christoffersen, L., Dupree, P., Goubet, F., Healey, S., Hernández, M., Keller, M., Li, K., Palackal, N., Sittenfeld, A., Tamayo, G., Wells, S., Hazlewood, G. P., Mathur, E. J., Short, J. M., Robertson, D. E., & Steer, B. A. (2004). Unusual microbial xylanases from insect guts. Applied and Environmental Microbiology, 70, 3609–3617.
Alvarez, T. M., Paiva, J. H., Ruiz, D. M., Cairo, J. P. L. F., Pereira, I. O., Paixão, D. A., de Almeida, R. F., Tonoli, C. C., Ruller, R., Santos, C. R., Squina, F. M., & Murakami, M. T. (2013). Structure and function of a novel cellulase 5 from sugarcane soil metagenome. PLoS One, 8, e83635.
Henrissat, B. (1991). A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochemistry Journal, 280, 309–316.
Mamo, G., Faryar, R., & Karlsson, E. N. (2013). Microbial glycoside hydrolases for biomass utilization in biofuels applications. In V. K. Gupta & M. G. Tuhoy (Eds.), Biofuel technologies: Recent developments (pp. 171–188). Heidelberg: Springer.
Juturu, V., & Wu, J. C. (2012). Microbial xylanases: engineering, production and industrial applications. Biotechnology Advances, 30, 1219–1227.
Zheng, H., Guo, B., Chen, X.-L., Fan, S.-J., & Zhang, Y.-Z. (2011). Improvement of the quality of wheat bread by addition of glycoside hydrolase family 10 xylanases. Applied Microbiology and Biotechnology, 90, 509–515.
Peberdy, J. F. (1990). Fungal cell walls. A review. In P. J. Kuhn, A. P. J. Trinci, M. J. Jung, & M. W. Goosey (Eds.), Biochemistry of cell walls and membranes in fungi (pp. 5–30). New York: Springer.
Burton, R. A., & Fincher, G. B. (2009). (1,3;1,4)-beta-D-glucans in cell walls of the poaceae, lower plants, and fungi: a tale of two linkages. Molecular Plant, 2, 873–882.
Olafsdottir, E. S., & Ingólfsdottir, K. (2001). Polysaccharides from lichens: structural characteristics and biological activity. Planta Medica, 67, 199–208.
Davis, T. A., Volesky, B., & Mucci, A. (2003). A review of the biochemistry of heavy metal biosorption by brown algae. Water Research, 37, 4311–4330.
Gueguen, Y., Voorhorst, W. G., van der Oost, J., & de Vos, W. M. (1997). Molecular and biochemical characterization of an endo-beta-1,3-glucanase of the hyperthermophilic archaeon Pyrococcus furiosus. Journal of Biological Chemistry, 272, 31258–31264.
Kawai, R., Igarashi, K., Yoshida, M., Kitaoka, M., & Samejima, M. (2006). Hydrolysis of beta-1,3/1,6-glucan by glycoside hydrolase family 16 endo-1,3(4)-beta-glucanase from the basidiomycete Phanerochaete chrysosporium. Applied Microbiology and Biotechnology, 71, 898–906.
Iakiviak, M., Mackie, R. I., & Cann, I. K. (2011). Functional analyses of multiple lichenin-degrading enzymes from the rumen bacterium Ruminococcus albus 8. Applied and Environmental Microbiology, 77, 7541–7550.
Masuda, S., Endo, K., Koizumi, N., Hayami, T., Fukazawa, T., Yatsunami, R., Fukui, T., & Nakamura, S. (2006). Molecular identification of a novel beta-1,3-glucanase from alkaliphilic Nocardiopsis sp. strain F96. Extremophiles, 10, 251–255.
Strohmeier, M., Hrmova, M., Fischer, M., Harvey, A. J., Fincher, G. B., & Pleiss, J. (2004). Molecular modeling of family GH16 glycoside hydrolases: potential roles for xyloglucan transglucosylases/hydrolases in cell wall modification in the poaceae. Protein Science, 13, 3200–3213.
Parrish, F. W., Perlin, A. S., & Resse, E. T. (1960). Selective enzymolysis of poly-J3-D-glucans, and the structure of the polymers. Canadian Journal of Chemistry, 38, 2094–2104.
Pang, Z., Otaka, K., Maoka, T., Hidaka, K., Ishijima, S., Oda, M., & Ohnishi, M. (2005). Structure of beta-glucan oligomer from laminarin and its effect on human monocytes to inhibit the proliferation of U937 cells. Bioscience Biotechnology and Biochemistry, 69, 553–558.
Zhan, X.-B., Lin, C.-C., & Zhang, H.-T. (2012). Recent advances in curdlan biosynthesis, biotechnological production, and applications. Applied Microbiology and Biotechnology, 93, 525–531.
Bamforth, C. W. (2009). Current perspectives on the role of enzymes in brewing. Journal of Cereal Science, 50, 353–357.
Humbert-Goffard, A., Saucier, C., Moine-Ledoux, V., Canal-Llaubères, R.-M., Dubourdieu, D., & Glories, Y. (2004). An assay for glucanase activity in wine. Enzyme and Microbial Technology, 34, 537–543.
Petersen, T. N., Brunak, S., von Heijne, G., & Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature Methods, 8, 785–786.
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D., & Bairoch, A. (2005). Protein identification and analysis tool on the ExPASy server. In J. M. Walker (Ed.), The proteomics protocols handbook (pp. 571–607). Totowa: Humana.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., & Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729.
Saitou, N., & Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406–425.
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., & Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Research, 42, D490–D495.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685.
Miller, G. L. (1959). Use of dinitrosalicylic acid reagent for determination of reducing sugar. Analytical Chemistry, 31, 426–428.
Evangelista, R. A., Liu, M.-S., & Chen, F.-T. A. (1995). Characterization of 9-aminopyrene-1,4,6-trisulfonate derivatized sugars by capillary electrophoresis with laser-induced fluorescence detection. Analytical Chemistry, 67, 2239–2245.
Hammersley, A. P., Svensson, S. O., Hanfland, M., Fitch, A. N., & Hausermann, D. (1996). Two-dimensional detector software: from real detector to idealised image or two-theta scan. High Pressure Research, 14, 235–248.
Guinier, A., Fournet, G., Walker, C., & Yudowitch, K. (1955). Small angle scattering of X-rays. New York: Wiley.
Svergun, D. I. (1992). Determination of the regularization parameter in indirect-transform methods using perceptual criteria. Journal of Applied Crystallography, 25, 495–503.
Fischer, H., de Oliveira Neto, M., Napolitano, H. B., Polikarpov, I., & Craievich, A. F. (2009). Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. Journal of Applied Crystallography, 43, 101–109.
Svergun, D. I., Petoukhov, M. V., & Koch, M. H. (2001). Determination of domain structure of proteins from X-ray solution scattering. Biophysical Journal, 80, 2946–2953.
Volkov, V. V., & Svergun, D. I. (2003). Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography, 36, 860–864.
Svergun, D., Barberato, C., & Koch, M. H. J. (1995). CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. Journal of Applied Crystallography, 28, 768–773.
Bleicher, L., Prates, E. T., Gomes, T. C. F., Silveira, R. L., Nascimento, A. S., Rojas, A. L., Golubev, A., Martínez, L., Skaf, M. S., & Polikarpov, I. (2011). Molecular basis of the thermostability and thermophilicity of laminarinases: X-ray structure of the hyperthermostable laminarinase from Rhodothermus marinus and molecular dynamics simulations. Journal of Physical Chemistry B, 115, 7940–7949.
Pang, Z., Otaka, K., Suzuki, Y., Goto, K., & Ohnishi, M. (2004). Purification and characterization of an endo-1,3-β-glucanase from Arthrobacter sp. Journal of Biological Macromolecules, 4, 57–66.
Cota, J., Alvarez, T. M., Citadini, A. P., Santos, C. R., de Oliveira Neto, M., Oliveira, R. R., Pastore, G. M., Ruller, R., Prade, R. A., Murakami, M. T., & Squina, F. M. (2011). Mode of operation and low-resolution structure of a multi-domain and hyperthermophilic endo-β-1,3-glucanase from Thermotoga petrophila. Biochemical and Biophysical Research Communications, 406, 590–594.
Cheng, Y.-M., Hong, T.-Y., Liu, C.-C., & Meng, M. (2009). Cloning and functional characterization of a complex endo-beta-1,3-glucanase from Paenibacillus sp. Applied Microbiology and Biotechnology, 81, 1051–1061.
Hong, T. Y., Cheng, C. W., Huang, J. W., & Meng, M. (2002). Isolation and biochemical characterization of an endo-1,3-beta-glucanase from Streptomyces sioyaensis containing a C-terminal family 6 carbohydrate-binding module that binds to 1,3-beta-glucan. Microbiology, 148, 1151–1159.
Bacic, A., Fincher, G. B., & Stone, B. A. (2009). Chemistry, biochemistry, and biology of 1-3 beta glucans and related polysaccharides. St. Louis: Elsevier.
Wood, P. J., Weisz, J., & Blackwell, B. A. (1994). Structural studies of (1-3)(1-4)-beta-D-glucans by 13C-nuclear magnetic resonance spectroscopy and by rapid analysis of cellulose-like regions using high-performance anion-exchange chromatography of oligosaccharides released by lichenase. Cereal Chemistry, 7, 301–307.
Cheng, R., Chen, J., Yu, X., Wang, Y., Wang, S., & Zhang, J. (2013). Recombinant production and characterization of full-length and truncated β-1,3-glucanase PglA from Paenibacillus sp. S09. BMC Biotechnology, 13, 105.
Krah, M., Misselwitz, R., Politz, O., Thomsen, K. K., Welfle, H., & Borriss, R. (1998). The laminarinase from thermophilic eubacterium Rhodothermus marinus-conformation, stability, and identification of active site carboxylic residues by site-direct mutagenesis. European Journal of Biochemistry, 257, 101–111.
Acknowledgments
The authors would like to acknowledge FAPESP for financial support to FMS (08/58037-9) and TMA (2010/11469-1); CNPq for financial support to FMS (442333/2014-5 and 310186/2014-5), MVL (158882/2014-8), and MON (478900/2012-0); and SAXS2 (LNLS/CNPEM) and CTBE/CNPEM for technical support.
Conflict of Interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
SDS–PAGE analysis of SCLam purification. A) Affinity chromatography. Lane MW – molecular weight marker (labelled in kDa). Lane 1 – soluble fraction. Lane 2 – Flow-through. Lane 3 -Wash. Lanes 4 to 8 – Elutions. B) Size-exclusion chromatography. Lane MW - molecular weight marker (labelled in kDa). Lanes 1 to 3 - elution fractions from size-exclusion chromatography, in 20 mM sodium phosphate pH 7.4 containing 50 mM NaCl buffer. (JPEG 268 kb)
Rights and permissions
About this article

Cite this article
Alvarez, T.M., Liberato, M.V., Cairo, J.P.L.F. et al. A Novel Member of GH16 Family Derived from Sugarcane Soil Metagenome. Appl Biochem Biotechnol 177, 304–317 (2015). https://doi.org/10.1007/s12010-015-1743-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-015-1743-7