
Abstract
Dipeptidyl aminopeptidases are enzymes involved in the posttranslational control of bioactive peptides. Here we identified the gene dapUm in Ustilago maydis by homology with other fungal dipeptidyl aminopeptidases. Analysis of the dapUm-deduced amino acid sequence indicated that it encodes for membrane-type serine protease with a characteristic prolyl oligopeptidase catalytic motif triad: Ser, Asp, His. In order to overexpress the DapUm, the gene encoding for it was cloned and transformed into Pichia. Using this system, we observed a ∼125-kDa recombinant protein with an optimal enzymatic activity at pH 6.0 and at 40 °C for the Ala-Pro-p-nitroanilide substrate and an experimental pH of 6.9. U. maydis DapUm was specifically inhibited by phenylmethylsulfonyl fluoride and Pefabloc, confirming the presence of a serine residue in the active site. To our knowledge, this study is the first report on the cloning and expression of a DPP IV dipeptidyl aminopeptidase from a basidiomycete organism. Moreover, the use of recombinant DapUm will allow us to further study and characterize this enzyme, in addition to testing chemical compounds for pharmaceutical purposes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The basidiomycete fungus Ustilago maydis, the causative agent of corn smut disease [1], has a high dipeptidyl aminopeptidase activity from intracellular extracts linked to the membrane fraction. This was observed when U. maydis was grown in broth media supplemented with proline or corn infusion as a nitrogen source during the late stationary and exponential growth phases, respectively [2].
Dipeptidyl aminopeptidases (DPPs, DAPs, or DPAPs) are proteases from the S9 family that are able to remove dipeptides from the N-terminus of proteins. DPPs are classified according to the dipeptide released from the substrate, for instance, DPPs with a type IV activity (DPP IV) remove N-terminal X-Pro and X-Ala dipeptides (X could be any amino acid residue in the peptide) [3]. Like many other proteases, DPP IV enzymes are very important in the posttranslational control and maturation of other peptides in the cell.
Particularly, DpapA or STE13 is a Golgi membrane-bound protease of Saccharomyces cerevisiae encoded by the STE13 gene that removes dipeptides X-Ala from the N-terminal of the alpha factor precursor. Non-mating (“sterile” or “ste”) alpha-cell mutants bearing defects in the STE13 gene do not produce normal alpha factor [4]. A second S. cerevisiae gene called DAP2 encodes a vacuolar membrane bound with DPP IV activity, DpapB (DAP2), whose function is unknown. However, when the DAP2 gene is overexpressed in STE13 mutants, DAP2 processes the pheromone precursor that complements the STE13 gene function [5].
Here we describe for the first time a DpapB-like enzyme from the basidiomycete U. maydis. We cloned and overexpressed it in a Pichia pastoris system and demonstrated its activity using multiple approaches. Given our experimental evidences, we conclude that U. maydis DapUm2 is a DPP type IV enzyme and propose that this protein can be used for further characterization of this and other DPPs.
Materials and Methods
Strains and Plasmids
The U. maydis FB1 a1b1 strain used in this study was kindly provided by Dr. Flora Banuett from UCSF. The Escherichia coli-P. pastoris shuttle vectors pPICZB and pPICZαB (Easy Select Pichia Expression Kit, Invitrogen, CA, USA) were used for cloning and expression of the dapUm gene from U. maydis. The P. pastoris strain X-33 was used for expression of the construct mentioned above, and the E. coli strain DH10b was used for plasmid construction and propagation.
Bioinformatic Analysis
The U. maydis dapUm nucleotide sequence was identified through a BLASTp analysis using deduced amino acid sequences from genes encoding for DPP IV in other yeasts and filamentous fungi [ID numbers: Cryptococcus neoformans (AAW47088), Aspergillus niger (An02g11420), Aspergillus fumigatus (Afu3g07850), S. cerevisiae (YHR028C), Magnaporthe grisea (EHA53149), and Candida albicans (CaO19.4322)]. BLASTp analysis was done using S. cerevisiae DAP2 in the MIPS U. maydis database (http://mips.gsf.de/genre/proj/ustilago). The structural analyses of the gene and the translated protein (i.e., ORF prediction, reverse complementary sequence, and restriction map) were performed with several software programs. The approximate molecular weight and the translation using an alternative yeast nuclear genetic code were determined by the DNAMAN v.3.0 software (developed by Lynnon BioSoft, 1994–1997). The approximate isoelectric point (pI), hydrophobicity, and helical membranous regions were searched with Antheprot 2000 v. 5.2 [6]. Prediction of molecular signatures and subcellular localization were performed using the ScanProsite database and PSORTII, respectively (http://www.expasy.org). The dap2 nucleotide sequence was aligned to those of other organisms at NCBI with the BLASTn program. Multiple alignments of nucleotide and protein sequences were performed with Clustal X v. 2 [7]. Protein similarity estimations were performed with MatGat software using the PAM 250 matrix [8].
Heterologous Expression of the U. maydis dapUm Gene in P. pastoris
The dapUm coding region was amplified by PCR with primers FwdEco 5′-TGGTCGAATTCATGGCGTCCAAGAAACGTGCG-3′ and NotRev 5′-GTTGCGGCCGCCTCTACGGAATTATGCAGCGC-3′, in which EcoRI and NotI restriction sites were included and are represented in italics, respectively. The amplicon was digested with EcoRI and NotI restriction enzymes and purified with QIAquick Gel Extraction kit (QIAGEN, USA) according to the manufacturer’s instructions. The purified product was cloned into the EcoRI and NotI sites of the pPICZB multiple cloning site to generate the plasmid pPICZADAPUm (Fig. 1b). P. pastoris was transformed with 10 μg of pPICZADAPUm previously linearized with PmeI. Transformant clones were selected according to the manufacturer’s instructions on YPDS zeocin medium (YPD, 100 μg/ml zeocin, and 1 M sorbitol). The P. pastoris transformants were confirmed by PCR amplification of the dapUm gene, using the same primers used to clone it and with the AOX forward and reverse primers supplied with the kit. P. pastoris X-33 and the Cα (carrying the empty plasmid) genomic DNAs were used as negative controls.

Schematic representation of the DapUm encoded by the dap2 gene from U. maydis and the strategy for its expression in P. pastoris. a Cartoon representing the amino acid sequence alignment of the DAP2/DAPB from different fungi: U. maydis (um05273), C. neoformans (AAW47088), A. niger (An02g11420), A. fumigatus (Afu3g07850), S. cerevisiae (YHR028C), M. grisea (EHA53149), and C. albicans (CaO19.4322). From left to right, boxes indicate the transmembranal domain (dark gray), the dipeptidyl peptidase IV N-terminal region (light gray), and the catalytic triad SDH (Ser920, Asp995, and His1028) (black box and lines). b Schematic representation of the pPICZBDapUm plasmid and the homologous recombination in P. pastoris to obtain a methanol-inducible recombinant protein
Partial Purification of the Recombinant DapUm
P. pastoris wild type and transformant strains were grown in 50 ml of glycerol-based yeast medium (1.34 % YNB without amino acids, 4 × 10−5 % biotin,10 mM potassium phosphate buffer, and 2 % glycerol, pH 6.0) at 28 °C in an agitated water bath until the DO600 reached ∼2.0. The cells were harvested and resuspended in 100 ml of the same medium supplemented with 0.5 % methanol (v/v) instead of glycerol and then incubated for 2 days. The cells were harvested and washed twice with cold distilled water and disrupted using glass beads (0.5-mm diameter) and lysis buffer (10 mM Tris-HCl, pH 7.5). The cellular extract was centrifuged for 10 min at 10,000×g, and the resulting supernatant was ultracentrifuged for 90 min at 100,000×g. The pellet was resuspended by gentle agitation in an ice bath using the same buffer supplemented with 1 % n-octyl-β-d-glucopyranoside, and the suspension was centrifuged again for 40 min at 12,000×g. Finally, the supernatant was recovered to detect enzymatic activity. The activities for the wild-type X-33 and the Cα strains were always measured as controls. To calculate protein molecular weight, we used Zwittergent 3-14 (BACHEM Bioscience Inc., USA) to dissolve membranes.
Enzymatic Assay
The enzymatic activity of the recombinant DapUm was determined as previously done in our lab [9] by using Ala-Pro-p-nitroanilide (pNA) as a substrate for DPP IV activity. The reaction mixture consisted of 450 μl of 50 mM Tris-HCl (pH 7.5), 25 μl of 10 mM Ala-Pro-pNA, and 25 μl of the crude extract with or without the enzyme. Absorbance at 405 nm was determined after 20 min of incubation at 37 °C. One unit of X-prolyl-dipeptidyl aminopeptidase was defined as the amount of enzyme that liberated 1 μmol of p-nitroaniline in the tested assay conditions [10].
Protein extracts were analyzed in a 10 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel as described by Laemmli [11]. Native polyacrylamide gels were stained with Ala-Pro-βNA (Bachem) and Fast Garnet (Sigma) to detect DPP IV activity [12]. The protein concentration was estimated according to a modified Lowry method [13]. The in situ assay was performed also with Ala-Pro-βNA and Fast Garnet in 10 mM Tris-HCl, pH 7.0, and 1 % agar. This mixture was used to cover colonies of 48 h in each medium [14].
Western blot assay was performed by transferring proteins from a 10 % SDS-PAGE gel to a PVDF membrane (Millipore), which was then blocked and then blotted with anti-His tag (C-term)-HRP antibody (Invitrogen, CA, USA) and stained with diaminobenzidine (Sigma) and 30 % hydrogen peroxide.
Isoelectric Point Determination and Effects of pH and Temperature on Recombinant DapUm DPP IV Activity
Isoelectric focusing was performed with a Rotofor system using Bio-Lyte 2 ampholytes (pH 3–10) (Bio-Rad Laboratories, Richmond, CA, USA). The optimal pH for protease activity was determined at 37 °C with different 50 mM buffers. For the pH ranges of 2.0–7.0, 7.0–10.0, and 9.0–11.0, McIlvaine, Tris-HCl, and glycine-NaOH buffers were used, respectively. The pH stability was determined by overnight preincubation of the purified enzyme in the appropriate buffer at different pH values ranging from 2 to 10 at 4 °C followed by a standard enzyme assay. Optimal temperature was examined between 5 and 80 °C by a standard enzyme assay. Thermal stability was evaluated by incubation of the enzyme solution at 5, 25, 30, 35, 40, 45, 50, 55, 60, 70, and 80 °C for 60 min, before performing the standard enzyme assay. Activity was always expressed as a percentage of the activity obtained at either the optimal pH or temperature.
Effects of Protease Inhibitors on Recombinant DapUm Activity
The effect of potential inhibitors, such as bestatin, pepstatin, leupeptin, Pefabloc, E-64, phenylmethylsulfonyl fluoride (PMSF), 1,10-phenanthroline, and EDTA-Na2 (Roche, Switzerland), was determined at two different concentrations (Table 2). Briefly, the purified enzyme was preincubated with the potential inhibitor for 30 min at 37 °C followed by the standard enzyme assay described above. Activity was expressed as a percentage where 100 % of enzymatic activity is considered to be the enzymatic activity obtained without the inhibitor.
Substrate Specificity
The relative activities of the recombinant DapUm against several dipeptide-pNA substrates, Gly-Arg and Gly-Phe (both substrates of the DPP I family), Lys-Ala (for the DPP II family), Arg-Arg (DPP III family), and Ala-Pro-pNA (DPP IV family), were determined by a standard activity assay with 10 mM of each substrate (BACHEM Bioscience Inc., USA). All determinations were performed at least twice with duplicates.
Results
Sequence Analysis of the U. maydis dapUm Gene
Using S. cerevisiae YHR028c sequence (which encodes for DAP2, DpapB, or DPP2), we identified an ORF with 39 % identity and 53 % similarity of 3,234 bp encoding for a 1,077-amino acid protein with a calculated molecular weight of approximately 117 kDa and a high similarity to DPP IV from other fungi (Fig. 1a). We were unable to detect introns in this gene, annotated as um05273 (with alternative entry: um10547) in the U. maydis genome project database. This protein is referred as MER107914 in the MEROPS database (http://merops.sanger.ac.uk/). It is an X-prolyl peptidase and can be considered as the ortholog of the S. cerevisiae DpapB (MER00405). Despite this, there is only one gene for DAP in U. maydis; hence, we decided to name it DapUm. The structural domains include the DWVYEEE (residues 432 to 438) and Gly-X-Ser-X-Gly (residues 918 to 922) consensus sequences that are characteristic of the S9B and α/β hydrolase families, respectively [15, 16]. The Ser residue usually is part of the putative catalytic amino acid triad as it is Ser920, Asp995, and His1028 in DapUm. Additionally, a hydrophobic region (residues 106 to128) was also identified at the N-terminal end corresponding to a putative transmembrane domain with a predicted topology of a type II membrane protein. Interestingly, the first 308 amino acid residues at the N-terminal end did not align with any of the other DAP2-like sequences (data not shown).
Considering this lack of alignment, and in an attempt to overexpress a recombinant protein, the dapUm gene was cloned starting from the amino acid residue at position 97, as a 2,946-bp fragment. This was done because at the time this work was planned, the available sequence in the U. maydis sequencing genome project website covered only to this position; several changes have been done since then. This section encodes a protein of 981 amino acids whose predicted molecular weight is 108 kDa. The corresponding DNA fragment was cloned into both pPICZB and pPICZαΑ expression vectors to generate plasmids pPICZBDapUm and pPICZαΑDapUm, the latter being in frame with the alpha pheromone secretion signal from S. cerevisiae. These plasmids were linearized and used to transform P. pastoris X-33 (wild type) (Fig. 1b). The insertion of the recombinant dapUm gene in the P. pastoris genome was confirmed by specific PCR amplification of the U. maydis dapUm using transformed P. pastoris total DNA. However, no DPP IV activity was detected in the culture medium from P. pastoris harboring the pPICZαADapUm plasmid (data not shown), and we decided to work only with the recombinant DapUm expressed intracellularly (this is the one encoded in pPICZBDapUm).
Partial Purification and Characterization of U. maydis DapUm
In order to test whether the dapUm gene encodes for an enzyme with an X-propyl peptidase activity, P. pastoris strains with or without plasmids pPICZB and pPICZBDapUm were grown in an agar medium with methanol as the only carbon source. We screened for several transformed P. pastoris clones and selected one clone harboring pPICZBDapUm (clone C41) after testing them for activity using Ala-Pro-βNA in agar (data not shown). Membrane and soluble fractions were obtained from wild-type (WT) and C41 strains, subjected to electrophoresis in a native PAGE and tested for X-propyl peptidase activity with Ala-Pro-βNA. Despite the fact that membrane fractions from both WT and C41 strains showed activity (as evidenced with a dark spot), it is clear that the C41 strain has more activity (Fig. 2a). P. pastoris C41 showed around a 7-fold increase in the DPP IV-specific activity in a cell-free extract as compared to P. pastoris X-33 (WT) (Fig. 2b), after 48 h of incubation in methanol-supplemented medium.

Recombinant U. maydis DapUm DPP IV activity in P. pastoris. Samples were obtained after growing P. pastoris clones for 48 h in induced (methanol-supplemented) or not medium. a Native acrylamide gel of the membrane (M) and soluble (S) fractions from the C41 (harboring pPICZBDapUm) and X-33 (WT) strains. The amount of loaded protein for each sample was previously normalized. Activity is shown as darker zone in the upper part of the gel after addition of substrate. b Specific DPP IV activity in a membrane extract of both C41 and X-33 strains measured by release of pNA. Strains were grown in inducing (M—methanol) and non-inducing (G—glycerol) conditions. Error bars indicate standard deviation
Taking advantage of a 6-histidine tag in both Pichia plasmids, the cell-free extracts of the DPP IV-overproducing P. pastoris clone C41 were passed through a Ni-NTA affinity chromatography column to purify this protein. However, we were not able to obtain any protein and only poor activity was detected in the eluted fractions (data not shown). Then, the membrane fractions of both strains were treated with different detergents. We observed a 4-fold increase in the enzymatic activity for the C41 strain after using Zwittergent 3-14 while the extract X-33 control extract did not show a change in the activity level. The extracts of both strains thus obtained were analyzed in an SDS-PAGE gel that showed a band of high molecular weight in the extract of the transformant C41 that was absent in the extract of the X-33 strain (Fig. 3a). From this, we estimated a molecular weight for the recombinant enzyme of 125 kDa. The Western blot assay showed also a band in this molecular weight (Fig. 3b).

Detection of recombinant DapUm. a Samples of the membrane fractions of the C41 strain expressing dap2 and the X-33 wild-type strain obtained with Zwittergent 3-14 detergent were subjected to SDS-PAGE; alcohol dehydrogenase is evident at 70 kDa in both samples. The arrow indicates that a ∼125-kDa band is evident only in the C41 strain. b A band of approximately120 kDa (arrow) is evidenced by Western blot using anti-His antibodies
Additionally, we found that this enzyme has a pH of 6.9 and optimal activity toward the Ala-Pro-pNA substrate in a narrow pH range around 7, which we consider as the optimum activity pH (Fig. 4a). Moreover, the enzyme was stable for 60 min in a range of 5 to 40 °C, with an optimal temperature of 35 °C (Fig. 4b).

Effect of a pH and b temperature on the activity of recombinant U. maydis DapUm. a For determination of activity at multiple pH, a McIlvaine (pH 3.0–7.0), b Tris-HCl (pH 8.0–9.0), and c glycine-NaOH (pH 9.0–10.0) buffers were used. Black curve indicates testing at indicated pH and gray curve indicates preincubation at pH 6.0 before addition of substrate and change to shown pH. b To determine optimum temperature for DPP IV activity (black curve), reactions were carried out at indicated temperatures. To determine stability (gray curve), reactions were preincubated at indicated temperatures prior to substrate addition and then changed to optimal temperature
There are five classes of DPP enzymes. In order to classify DapUm, we tested the concentrated fraction with substrates from each type group (Table 1). We observed that the concentrated extract was active against Ala-Pro-pNA but not against any of the other substrates, which demonstrates that is a DPP IV enzyme. We also tested the effect of a variety of inhibitors specific for DPP IV enzymes. Pefabloc and PMSF, which are serine protease inhibitors, specifically inhibited its activity (Table 2). No inhibitory effect was observed with the following cysteine and aspartic protease inhibitors: 1,10-phenanthroline, bestatin, leupeptin, pepstatin, E64, metalloprotease, or exopeptidase.
Discussion
Heterologous expression of industrially important enzymes, such as lipases and peroxidases, by employing biotechnology tools, has been of great relevance in recent years [17, 18]. In the present study, the dapUm gene of U. maydis was analyzed, cloned, and heterologously expressed in P. pastoris. A 2,943-bp amplicon corresponding to a portion of the dapUm gene (see “Results” section) was obtained and cloned in the expression vector pPICZB and transformed into P. pastoris. We determined that this gene was responsible for an increased DPP IV activity when P. pastoris was grown in induction conditions. The recombinant enzyme was partially characterized in terms of molecular weight and the capacity to remove X-Pro dipeptides preferentially at neutral pH which were similar to other known properties of DPP IV enzymes. For instance, DPP IV enzymes of Aspergillus oryzae, A. fumigatus, and Trichophyton rubrum, whose molecular weights are between 80 and 95 kDa, have an optimum pH activity between 7 and 9 [19–21]. The molecular weight of the DapUm protein was estimated to be 125 kDa, but the in silico analysis predicted a protein of at least 117 kDa. This difference can be explained by considering the multiple posttranslational sites that were also predicted, such as glycosylation, myristoylation, and phosphorylation sites. Similar to other DPP IV, it was inhibited by PMSF and Pefabloc, specific for serine proteases, which is in agreement with the presence of a predicted Gly-X-Ser-X-Gly consensus sequence and for the Ser-Asp-His catalytic triad.
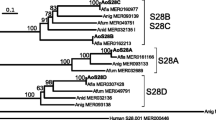
Contrary to other filamentous fungi and yeasts that harbor two DPP IV encoded by two paralogous genes, U. maydis seems to contain only one gene. This could be because the U. maydis genome is smaller than other plant pathogenic fungi genomes [21], and in general, the secreted hydrolases of U. maydis are different than their phylogenetically related parents Malassezia spp. and C. neoformans (Fig. S1) [21, 22]. Moreover, extracellular DPP IV activity was not detected in either U. maydis FB1 or FB2 haploid cells in different growth conditions nor during the dimorphic events induced by pH [2]. It is known that many biologically active peptides are protected from general proteolytic degradation and activation by the structural features imposed by proline residues [23]. Thus, the biological importance of prolyl-specific peptidases is the high potential for drug discovery for this family of enzymes [24].
Different studies have shown that the U. maydis fungus and humans share disease-related proteins that are not found in S. cerevisiae [25]. Human DPP IV, which is a member of the S9B serine protease family, cleaves a number of regulatory factors, including chemokines and growth factors. DPP IV inhibitors have recently emerged as an effective treatment option for type 2 diabetes [26], and it has recently been suggested that DPP IV inhibitors could be used as novel agents in cases of inflammatory disease [27]. Although human DAP IV recombinant protein has recently been purified [28], its acquisition for in vitro studies in many countries outside the USA is expensive and poses regulatory challenges. Given this and from our findings, we propose that DapUm from U. maydis could be used as an alternative model for the study of inhibitors with therapeutic purposes.
References
Banuett, F. (1995). Annual Review Genetics Review, 29, 179–208.
Mercado-Flores, Y., Hernández-Rodríguez, C., Ruiz-Herrera, J., & Villa-Tanaca, L. (2003). Mycologia, 95, 327–339.
Ogasawara, W., Ochiai, K., Ando, K., Yano, K., Yamasaki, M., Okada, H., & Morikawa, Y. (1996). Journal of Bacteriology, 178, 1283–1288.
Julius, D. B., Brake, L., Sprague, G., & Thorner, J. (1983). Cell, 32, 839–852.
Roberts, C. J., Pholig, J. H., Rothman, J. H., & Stevens, T. H. (1989). Journal of Cell Biology, 108, 1363–1373.
Deléage, G., Combet, C., Blanchet, C., & Geourjon, C. (2001). Computers in Biology and Medicine, 31, 259–267.
Larkin, M. A., Blackshields, G., Brown, N. P., et al. (2007). Bioinformatics, 23, 2947–2948.
Campanella, J. J., Bitincka, L., & Smalley, J. (2003). BMC Bioinformatics, 4, 29.
Suárez-Rendueles, M. P., Schwenke, J., García-Alvarez, N., & Gascón, S. (1981). FEBS Letters, 131, 296–300.
Bautista-Muñoz, C., Hernández-Rodríguez, C., & Villa-Tanaca, L. (2005). Analysis and expression of STE13ca gene encoding a putative X-prolyl dipeptidyl aminopeptidase from Candida albicans. FEMS Immunology and Medical Microbiology, 45, 459–469.
Laemmli, U. K. (1970). Nature, 227, 351–355.
García-Alvarez, N., Bordallo, C., Gascón, S., & Suárez-Rendueles, P. (1985). Biochemical and Biophysical Acta, 832, 119–125.
Lowry, O. H., Rosebrough, N. J., Farr, A. L., & Randall, R. J. (1951). Journal of Biological Chemistry, 193, 265–275.
Villa, L., & Suárez-Rendueles, P. (1994). FEMS Microbiology Letters, 120, 211–216.
Ogata, S., Misumi, Y., Tsuji, E., Takami, N., Oda, K., & Ikehara, Y. (1992). Biochemistry, 31, 2582–2587.
Abbott, C. A., McCaughan, G. W., & Gorell, M. D. (1999). FEBS Letters, 458, 278–284.
Ayala, M., Pickard, M. A., & Vazquez-Duhalt, R. (2008). Journal of Molecular Microbiology and Biotechnology, 15, 172–180.
Zheng, J., Liu, L., Liu, C., & Jin, Q. (2012). Journal of Molecular Microbiology and Biotechnology, 22, 300–311.
Doumas, A., van den Broek, P., Affolter, M., & Monod, M. (1998). Applied and Environmental Microbiology, 64, 4809–4815.
Monod, M., Lechenne, B., Jousson, O., Grand, D., Zaugg, C., Stocklin, R., & Grouzmann, E. (2005). Microbiology, 151, 145–155.
Kämper, J., Kahmann, R., Bolker, M., et al. (2006). Nature, 444, 97–101.
Xu, J., Saunders, C. W., Hu, P., et al. (2007). Proceedings of the National Academy of Sciences of the United States of America, 104, 18730–18735.
Cunningham, D. F., & O’Connor, B. (1997). Biochimica et Biophysica Acta, 1343, 160–186.
Juillerat-Jeanneret, L. (2009). Current Chemical Biology, 2, 97–109.
Münsterkötter, M., & Steinberg, G. (2007). Molecular Plant-Microbe Interactions, 21, 110–121.
Palalau, A. I., Tahrani, A. A., Piya, M. K., & Barnett, A. H. (2009). Postgraduate Medicine, 121, 70–100.
Yazbeck, R., Howarth, G. S., & Abbott, C. A. (2009). Trends in Pharmacological Sciences, 30, 600–607.
Hu, C. X., Huang, H., Zhang, L., Huang, Y., Sheng, Z. F., Cheng, K. D., Du, G. H., & Zhu, P. (2009). Biotechnological Letters, 31, 979–984.
Acknowledgments
Authors would like to thank Martha Thayer for reviewing this manuscript thoroughly. MJM was a recipient of fellowships from CONACyT, ICyT-DF, and PIFI-IPN; LVT and CHHR received support from COFAA-IPN and EDI-IPN, as well as grants from ICyT-DF (PICSO10-95, Convenio 254/10) and CONACyT (208247, SIP-IPN 20120750, and 20131171). JAI was hired through the “Programa Institucional de Contratación de Personal Académico de Excelencia IPN.”
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Fig. S1
Neighbor-joining phylogenetic tree of intracellular and extracellular dipeptidyl aminopeptidases of multiple fungi. Dipeptidyl aminopeptidases from mammals and bacteria were used as external group (JPEG 62 kb)
Rights and permissions
About this article
Cite this article
Juárez-Montiel, M., Ibarra, J.A., Chávez-Camarillo, G. et al. Molecular Cloning and Heterologous Expression in Pichia pastoris of X-Prolyl-dipeptidyl Aminopeptidase from Basidiomycete Ustilago maydis . Appl Biochem Biotechnol 172, 2530–2539 (2014). https://doi.org/10.1007/s12010-013-0682-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-013-0682-4