
Abstract
Detection of species-specific sarcoplasmic peptides can be used as proteomic markers for fish food authentication and identification of species of origin in processed products. In the present study, proteomics technology was employed for differential characterization of sarcoplasmic peptides of two closely related fish species, Sperata seenghala and Sperata aor. Species-specific peptides were searched in white muscle extracts of the two species for identification of unique peptides that might aid in differentiation of the species, under two-dimensional gel electrophoresis platform. A total of 19 proteins were identified by combined matrix-assisted laser desorption ionization time-of-flight mass spectrometry and liquid chromatography-tandem mass spectrometry, of which nine and two proteins were found to be unique to S. seenghala and S. aor, respectively. One of the proteins, triosephosphate isomerase (TPI) was found to have three isoforms, out of which two were specific to S. aor, and one was specific to S. seenghala. All the three isoforms of TPI were present in the mixed samples of raw protein extracts of S. seenghala and S. aor, an observation that can be exploited to differentiate between the species and detection of deceptive practices of fraudulent substitution of commercially valuable fish species with inferior ones and differential characterization between closely related fish species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Food safety and quality is demanding greater attention by the food processing industries and consumers. One of the relevant points concerning food quality is the authentication of food components. Food products may be adulterated, and highly valuable species may be substituted, partially or entirely, by closely related but cheaper varieties. Food authentication is a major concern due to the global commercial demands concerning labeling and traceability for the prevention of commercial fraud and also for the assessment of safety risks derived from the advertent introduction of food ingredients which might be harmful for human health [1]. In addition to the negative effects that adulteration can cause to the food industry and the consumers and to the safety risks derived, this practice can also reduce the effectiveness of conservation management programs that help to protect different important fish species [2].
Fish food products include an extensive variety of species widely used for human nutrition, having a significant impact in food industry. As fishery products are among the most traded food commodities internationally [2], species identification of fish food products is important for the implementation of the labeling regulations as set by many countries [1] to assure complete and correct information, guaranteeing market transparency. The identification of species in fish food products has traditionally relied on morphological features, which are particularly difficult to use in species differentiation, because of their phenotypic similarities and as their external carapace is often removed during processing. Therefore, unintentional fraud may occur due to interspecies phenotypic similarities, which may lead to inadvertent adulteration and mislabeling of products [2].
Proteomics offers a comprehensive approach to study biochemical systems by expanding the investigation from single proteins to a wide range of proteins. Proteomics tools have been applied for the characterization and identification of species in fish products [2], for studying postmortem changes [3] and monitoring the effect of additives during the processing of fishes [3]. Different techniques, viz., sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) [4–7], native or urea isoelectric focusing electrophoresis (IEF) [8, 9], two-dimensional electrophoresis (2-DE) [10, 11], high-performance liquid chromatography [12], and capillary electrophoresis [13], have been employed for the purpose of fish species differentiation. Similarly, DNA-based techniques, such as the DNA bar coding [14], the single-strand conformation polymorphism [15], and the sequence analysis of the part of species-specific gene [16], provided discriminatory power for identification of processed fish products.
Sperata species (family: Bagridae) are commercially important species available in tropical rivers and enjoy high consumer preference [17]. Sperata seenghala and Sperata aor are two important species under the genus Sperata, and phenotypically, they look very similar. Some features like the length of barbel and distance between the dorsal and pelvic fin are used to differentiate these two species; however, in processed form, it is not possible to differentiate them. In the present study, by employing proteomics technology, we identified some species-specific sarcoplasmic peptides which could be used as markers for identification of the species.
Materials and Methods
Test Animals
S. seenghala and S. aor were caught from the River Ganga, India, and the whole fishes were identified based on their external anatomical and morphological features. The weight and length of the specimens studied were in the range of 700–800 g and 51–52 cm, respectively.
Preparation of Muscle Extracts
Axial white skeletal muscle from midway down the body of each fish (under the dorsal fin and above the lateral line) was swiftly dissected out from fishes euthanized with MS 222 (>100–200 mg/l). For protein extraction, white muscle tissues collected from different animals were pooled and mechanically homogenized in ice-cold PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4·7H2O, 1.4 mM KH2PO4), pH 7.3, containing protease inhibitor cocktail (Sigma P8340) [18]. To minimize protein modification or degradation, all dissections and sample processing were performed on ice. The homogenates were centrifuged in a high-speed refrigerated centrifuge (Biofuge Fresco, Heraeus) at 10,000 rpm at 4 °C for 10 min, and supernatants (representing the soluble protein extracts) were aspirated out. For mixed protein samples, similar quantity of protein extract from S. seenghala and S. aor was mixed. The samples were stored at −40 °C, as aliquots, until further use.
Protein Quantification
Protein concentration of the muscle protein extracts were determined by the Bradford assay [19] using BSA as the standard.
SDS-PAGE
The soluble tissue proteins were separated by SDS-PAGE through 12 % (w/v) polyacrylamide gel with a 5 % (w/v) stacking gel at a constant potential of 200 V on the Mini-PROTEAN 3 electrophoresis cell. Samples were boiled at 100 °C for 5 min in Laemmli final sample buffer [20], and 20 μg of protein was loaded in each lane. Protein molecular weight markers (S8445 Sigma) were co-run with samples for determination of molecular weight. Gels were stained with Coomassie Brilliant Blue R250 (CBB) for visualization of the protein bands. The gels were scanned, and the images were acquired by ImageScanner III LabScan 6.0 (GE Healthcare Biosciences), following a standard protocol as given in manufacturer’s instructions.
Two-Dimensional Gel Electrophoresis
The first dimension isoelectric focusing was performed using the PROTEAN IEF Cell (Bio-Rad) with 11-cm immobilized pH gradient (IPG) strips (pH 5–8; Sigma) following a standard protocol [21]. Briefly, the protein sample (150 μg) was premixed with “rehydration buffer” (8 M urea, 2 M thiourea, 2 % CHAPS, 50 mM dithiothreitol (DTT), 0.2 % Biolyte, 5/8 ampholyte, and 0.001 % bromophenol blue), and rehydration of the IPG strips was carried out for 12 h. The rehydrated strips were isoelectrofocussed at a current of 50 μA/strip at the stated voltage gradient as follows: for 11-cm IPG strips, 250 V for 20 min, 500 V for 30 min, 1,000 V for 15 min, 2,000 V for 15 min, 4,000 V for 15 min, 8,000 V for 2 h 30 min, and at 8,000 V for 20,000 V-h with an end voltage of ∼30,000 V-h, using the PROTEAN IEF Cell (Bio-Rad). After the IEF run, the focused strips were equilibrated with the “equilibration buffers” I and II [equilibration buffer 1 (reducing buffer), 0.375 M Tris–HCl at pH 8.8, 6 M urea, 20 % v/v glycerol, 2 % SDS, 130 mM DTT; equilibration buffer II (alkylating buffer), 0.375 M Tris–HCl at pH 8.8, 6 M urea, 20 % v/v glycerol, 2 % SDS, 135 mM iodoacetamide] and then placed on SDS polyacrylamide slab gels for second dimension run. The second dimension SDS-PAGE was performed using 12 % separating gels with 5 % (w/v) stacking gel (ATTO). The gels were stained with Coomassie Brilliant Blue.
Protein Identification by Mass Spectrometry
Matrix-Assisted Laser Desorption Ionization Tandem Time-of-Flight Mass Spectrometry
Protein spots of interest were cut from the gel, destained in methanol and ammonium bicarbonate buffer, and digested overnight with trypsin. The resulting peptides were extracted following standard techniques [22] by two 20-min incubations with 10–20 μL acetonitrile (ACN) containing 1 % TFA, depending on the size of the gel piece. The resulting tryptic peptide extract was dried by rotary evaporation and stored at −20 °C for further analysis by MS. The peptides were analyzed by matrix-assisted laser desorption ionization tandem time-of-flight (MALDI-TOF/TOF) mass spectrometry using the 5800 Proteomics Analyzer (AB Sciex).
For protein identification, peptide masses from trypsin digests derived using the MALDI-TOF/TOF mass spectrometry (MS) were used to search against Ludwig NR database and taxonomy set to Actinopterygii (ray-finned fishes) using the MASCOT sequence matching software (Matrix Science, www.matrixscience.com).
The MASCOT search parameters were as follows: peptide mass accuracy was 100 ppm, and protein modifications were cysteine as S-carbamidomethyl derivative and oxidation of methionine allowed. The default search parameters used were enzyme, trypsin; max. missed cleavages, 1; fixed modifications, carbamidomethyl (C); variable modifications, oxidation (M); peptide tolerance, +0.4 Da; fragment mass tolerance, +0.4 Da; protein mass, unrestricted; and instrument, default.
Liquid Chromatography-Tandem Mass Spectrometry
Protein spots of interest were cut from the gel, destained in methanol and ammonium bicarbonate buffer, and digested overnight with trypsin. Selected protein spots were also analyzed following standard techniques [23] by liquid chromatography-tandem mass spectrometry (LC-MS/MS) on the HCT Ultra PTM Discovery System (Bruker Daltonics, Coventry, UK) coupled to the UltiMate 3000 LC System (Dionex, Camberley, Surrey, UK). Peptides were separated on the Monolithic Capillary Column (200 mm id, 65 cm; Dionex part no. 161409). For the LC fractionation of the peptides, eluent A was 3 % ACN in water containing 0.05 % formic acid, and eluent B, 80 % ACN in water containing 0.04 % formic acid with a gradient of 3–45 % B in 12 min at a flow rate of 2.5 mL/min. Peptide fragment mass spectra were acquired in data-dependent AutoMS (2) mode with a scan range of 300–1,500 m/z; three averages and up to three precursor ions were selected from the MS scan (100–2,200 m/z). Precursors were actively excluded within a 1-min window, and all singly charged ions were excluded.
Results
SDS-PAGE
Representative SDS-PAGE profiles of the muscle protein extract of S. seenghala, S. aor, and protein mix of both the species are shown in Fig. 1. CBB-stained 12 % SDS-polyacrylamide gels separated the protein extracts into 29, 27, and 29 protein bands in the 14 to >205 kDa range in case of S. seenghala, S. aor, and mixed muscle protein extracts, respectively. Two protein bands, 29 and 52 kDa, which are present in S. seenghala, and the mixed protein extract were found to be absent in S. aor (Fig. 1).

SDS-PAGE of white muscle of S. seenghala (lane 1), S. aor (lane 2), and mixed muscle extract (S. seenghala and S. aor) (lane 3). Arrow indicates bands unique to S. seenghala
Two-Dimensional Gel Electrophoresis
CBB-stained 2-D polyacrylamide gels separated the white muscle protein extract of S. seenghala, S. aor, and mixed raw muscle protein samples into 180, 150, and 150 spots, respectively (Fig. 2). Protein spots were distributed over the molecular weight range of 14 to 200 kDa and pI range of 5–8; however, majority of constituent proteins separated at the basic zone, between pI 6.5 and 8. A closer examination of the muscle proteomes showed that nine spots were present exclusively in the muscle extract of S. seenghala (shown by arrows, Fig. 2a) and were absent in S. aor; similarly, two spots (shown by curved arrows, Fig. 2b) were present exclusively in S. aor muscle proteome. These spots were selected for identification by mass spectrometry for the identification/differentiation of species. Besides these 11 spots, another eight spots which were common to both the species were also selected for proteomic analysis (Fig. 2a).

Two-dimensional gel profiles of raw white muscle protein of S. seenghala (a), S. aor (b), and mixed (c) samples. Separation was performed on an 11-cm IPG strip with pH gradients of 5–8. Gels were Coomassie stained for visualization of proteins. Protein spots indicated by arrows were excised for identification by MALDI-TOF MS and LC-MS/MS. Proteins unique to S. seenghala and S. aor raw sample are indicated by arrows and curved arrows in case of S. seenghala and S. aor, respectively. Eight proteins (SS7, SS8, SS9, SS16, SS17, SS22, and SS23) marked by circles are common to both the species. Arrows indicated protein spots in the 2-D GE of mixed sample, which are unique to S. seenghala and S. aor. TPI triosephosphate isomerase, CK creatine kinase, RERGL Ras-related and estrogen-regulated growth inhibitor-like protein, PP protein phosphatase, ADK adenylate kinase, Pax7a paired box protein 7a, Hb hemoglobin embryonic-α, GP glycogen phosphorylase, ENO3 β-enolase-like isoform 1
MALDI-TOF MS
A total of 12 spots (12/19) were identified by MALDI-TOF MS analysis (Table 1), out of which four spots (SS13, SS19, SS20, and SS23) were identified as creatine kinase (CK) or CK isoforms, and three spots (SS5, SA1, and SA3) were identified as triosephosphate isomerase (TPI) (Fig. 2a, b). Another five spots (SS8, SS9, SS14, SS15, and SS22) were identified as PAX-7a (paired box protein 7a), hemoglobin embryonic-α, Ras-related and estrogen-regulated growth inhibitor-like protein (RERGL), protein phosphatase, and adenylate kinase isozyme 1, respectively. Out of the above 12 spots, five spots (SS5, SS13, SS14, SS15, and SS19; Fig. 2a) were S. seenghala specific, and two protein spots were S. aor specific (SA1 and SA3; Fig. 2b).
LC-MS/MS
Seven protein spots from 2-D gels of S. seenghala muscle, whose identity could not be ascertained by MALDI-TOF MS, were identified by LC-MS/MS (Table 2). Out of these, three spots (S1, S2, and S6) were identified as CK (Fig. 2a). Another two spots (SS17 and SS18) were identified as β-enolase-like isoforms (Fig. 2a), of which SS18 is present only in S. seenghala, whereas SS17 was found in both the species. Other two spots, SS16 and S7, were identified as glycogen phosphorylase and uncharacterized protein, respectively (Fig. 2a).
Discussion
A great number of species of fish traded and the diversity of processed sea products justify the development of suitable methods of identification to verify their authenticity. Classical methods, such as SDS-PAGE [6, 9], and isoelectric focusing [7, 9] have been used traditionally for such purposes as these are faster to perform and demand less sophisticated technology. However, increase in fraudulent practices involving large varieties of species available for substitution with commercially important fish species has made it imperative to find out more efficient and robust technology. Recently, the proteomics tools have been widely applied for the characterization and identification of species in fish products [10, 11, 24]. Analysis of parvalbumin fractions allowed the differential classification of ten closely related species of the family Merlucciidae [25] in an earlier study. Characterization and partial sequencing of species-specific sarcoplasmic peptides from commercial hake species led to the identification of five commercially important hake species, viz., Merluccius merluccius, Merluccius australis, Merluccius hubbsi, Merluccius gayi, and Merluccius capensis [11]. The objective of the present study was to identify species-specific peptides, employing proteomics tools, which could be used to distinguish between S. seenghala and S. aor.
As shown in Fig. 1, SDS-PAGE profiles of the two species indicated differences in some protein bands. However, SDS-PAGE has its limitations; protein isoforms and proteins which are very close in molecular weight cannot be clearly distinguished by this method. The combination of IEF (first dimension) and SDS-PAGE (second dimension) forms the classical separation technique in proteomics. IEF separates the proteins on a basis of their charge, whereas SDS-PAGE separates proteins on a basis of their molecular weight. Therefore, the classical proteomics technology, which combines these two techniques, generates an array of proteins where it becomes easier to identify subtle changes. Therefore, we analyzed the muscle proteins of these two closely related species by 2-D gel electrophoresis (GE). Visual comparison of both the 2-D GE profiles revealed differences that were investigated for species differentiation. Protein spots unique to S. seenghala and S. aor, nine in the former and two spots in the later, were subjected to proteomic analysis. Besides, some additional spots common to both the species were excised and identified by MALDI-TOF MS and/or LC-MS/MS.
The protein spots SS1, SS2, SS6, SS13, and SS19 were identified as positional variants of CK and were found to be specific to S. seenghala (Tables 1 and 2, Fig. 2a). CK catalyzes the transphosphorylation between phosphocreatine and ADP and is central to the regulation of muscle bioenergetics. CK isoenzymes are present in all vertebrates. Two protein spots, SS17 and SS18, were identified as β-enolase-like isoform 1, of which SS18 is S. seenghala specific. It has been shown earlier that human muscle-specific β-enolase can be used as a marker for species identification, which is very effective in forensic medicine [26].
Protein spot SS22 was identified as adenylate kinase (ADK), which is a phosphotransferase enzyme catalyzing the interconversion of adenine nucleotides and plays an important role in cellular energy homeostasis. SS8 which was identified as Pax-7a (paired box protein 7a) is encoded by PAX7 gene in the human that belongs to the family of paired box transcription factors. These genes play critical roles during fetal development (neural crest development and gastrulation) and cancer growth [27]. Another three spots (SS9, SS14, and SS16) were identified as hemoglobin-α, RERGL, and glycogen phosphorylase, respectively.
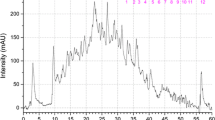
TPI is an important protein that is identified in this study. TPI (EC 5.3.1.1) is involved in carbohydrate metabolism and catalyzes the interconversion of the three-carbon sugars glyceraldehyde 3-phosphate and dihydroxyacetone phosphate. At least three spots were identified as TPI; one positional variant (SS5) of TPI was found to be species specific and was present only in muscle proteome of S. seenghala (Fig. 2a). Two positional variants of TPI (SA1 and SA3) were also found specifically in S. aor (Fig. 2b). Peptide mass fingerprint (PMF) of this protein exhibited specific peaks (Fig. 3d) for S. seenghala (m/z 954.5, Fig. 3a) and S. aor (m/z 2197.0, Fig. 3b, c), which could be useful as species-specific polypeptides for differentiation of these two commercially important species. Fingerprints of TPI in these two species also exhibited a common peak at m/z 1656.8 which can possibly be utilized for identification of the genus Sperata. PMF of sarcoplasmic arginine kinase have been successfully employed earlier for differentiation of commercially important shrimp and prawn species [28, 29].

Triosephosphate isomerase PMF for protein spots from S. seenghala [SS5] (a) and S. aor [SA1 and SA3] (b, c). Genus (Sperata)-specific peptides (filled circle), S. seenghala (asterisk), and S. aor (filled triangle)-specific peptides are highlighted. d Flow diagram showing the systematic identification of Sperata species using TPI. Y denotes the presence, and N, the absence of a particular peptide peak
To conclude, in the present study, we have generated reference muscle proteome mass for the riverine catfish S. seenghala and S. aor, for the first time. We could find typical mass spectra profile of TPI in sarcoplasmic peptides of S. seenghala and S. aor which are useful for differentiation of these two closely related species. The present study and earlier studies in this line reaffirm the fact that proteomic technology can be suitably used for identification of specific biomarkers for commercially important species. Such work could be useful to design fast and handy analysis kits for the rapid detection of fraudulent practice of substitution of low quality meat with commercially important species.
References
Mermelstein, M. H. (1993). Food Technology, 47, 81–96.
Ortea, I., Pascoal, A., Canas, B., Gallardo, J. M., Barros-Velazquez, J., & Calo-Mata, P. (2012). Electrophoresis, 33, 2201–2211.
Martinez, J., & Friis, T. J. (2004). Proteomics, 4, 347–354.
Chen, T. Y., & Hwang, D. F. (2002). Journal of Food Science, 67, 936–942.
Etienne, M., Jerome, M., Fleurence, J., Rehbein, H., Kundiger, R., Mendes, R., Costa, H., & Martinez, I. (2001). Food Chemistry, 72, 105–112.
Pineiro, C., Barros-Velazquez, J., Perez-Martin, R., Martinez, I., Jacobsen, T., Rehbein, H., Kundiger, R., Mendes, R., Etienne, M., Jerome, M., Craig, A., Mackie, I., & Jessen, F. (1999). Electrophoresis, 20, 1425–1432.
Etienne, M., Jerome, M., Fleurence, J., Rehbein, H., Kundiger, R., Mendes, R., Costa, H., Perez-Martin, R., Pineiro-Gonzalez, C., Craig, A., Mackie, I., Malmheden-Yman, I., Ferm, M., Martinez, I., Jessen, F., Smelt, A., & Luten, J. (2000). Journal of Agricultural and Food Chemistry, 48, 2653–2658.
Etienne, M., Jerome, M., Fleurence, J., Rehbein, H., Kundiger, R., Yman, I. M., Ferm, M., Craig, A., Mackie, I., Jessen, F., Smelt, A., & Luten, J. (1999). Electrophoresis, 20, 1923–1933.
Mackie, I., Craig, A., Etienne, M., Jerome, M., Fleurence, J., Jessen, F., Smelt, A., Kruijt, A., Malmheden-Yman, I., Ferm, M., Martinez, I., Perez-Martin, R., Pineiro, C., Rehbein, H., & Kundiger, R. (2000). Food Chemistry, 71, 1–7.
Pineiro, C., Barros-Velazquez, J., Sotelo, C. G., Perez-Martin, R. I., & Gallardo, J. M. (1998). Journal of Agricultural and Food Chemistry, 46, 3991–3997.
Pineiro, C., Vazquez, J., Marina, A., Barros-Velazquez, J., & Gallardo, J. M. (2001). Electrophoresis, 22, 1545–1552.
Armstrong, S. G., Leach, D. N., & Wyllie, S. G. (1992). Food Chemistry, 44, 147–155.
Gallardo, J. M., Sotelo, C. G., Pineiro, C., & Perez-Martin, R. J. (1995). Journal of Agricultural and Food Chemistry, 43, 1238–1244.
Wong, L. L., Peatman, E., Lu, J., Kucuktas, H., He, S., Zhou, C., Na-nakorn, U., & Liu, Z. (2011). PLoS One, 6, e17812.
Mackie, I., Pryde, S. E., Sotelo, C. G., Medina, I., Perez-Martin, R., Quinteiro, J., Rey-Mendez, M., & Rehbein, H. (1999). Trends in Food Science and Technology, 10, 9–14.
Cheng, C. A., Hsieh, Y. W., Noguchi, T., Arakawa, O., & Hwang, D. F. (2001). Journal of Food and Drug Analysis, 9, 232–237.
Mohanty, B. P., Paria, P., Das, D., Ganguly, S., Mitra, P., Verma, A., Sahoo, S., Mahanty, A., Aftabuddin, M., Behera, B. K., Sankar, T. V., & Sharma, A. P. (2012). National Academy Science Letters, 35, 155–161.
Mohanty, B. P., Bhattacharjee, S., Mondal, K., & Das, M. K. (2010). National Academy Science Letters, 33, 177–182.
Bradford, M. M. (1976). Analytical Biochemistry, 72, 248–254.
Laemmli, U. K. (1970). Nature, 227, 680–685.
O’Farrell, P. H. (1975). Journal of Biological Chemistry, 250, 4007–4027.
Bringans Dr, S., Eriksen, S., Kendrick, T., Gopalakrishnakone, P., Livk, A., Lock, R., & Lipscombe, R. (2008). Proteomics, 8, 1081–1096.
Martin, S. A. M., Mohanty, B. P., Cash, P., Houlihan, D. F., & Secombes, C. J. (2007). Proteomics, 7, 2275–2286.
Chen, T. Y., Shiau, C. Y., Wei, C. I., & Hwang, D. F. (2004). Journal of Agricultural and Food Chemistry, 52, 2236–2241.
Carrera, M., Canas, B., Pineiro, C., Vazquez, J., & Gallardo, J. M. (2006). Proteomics, 6, 5278–5287.
Matsuda, H., Seo, Y., & Takahama, K. (2000). Legal Medicine, 2, 42–45.
Eccles, M. R., He, S., Legge, M., Kumar, R., Fox, J., Zhou, C., French, M., & Tsai, R. W. S. (2002). International Journal of Developmental Biology, 46, 535–544.
Ortea, I., Barros, L., & Gallardo, J. M. (2009). Journal of Aquatic Food Product Technology, 18, 146–155.
Ortea, I., Canas, B., Calo-Mata, P., Barros-Velazquez, J., & Gallardo, J. M. (2009). Journal of Agricultural and Food Chemistry, 57, 5665–5672.
Acknowledgments
This work was supported by the CIFRI Core project no. FHE/ER/07/06/005 (BPM) and ICAR NFBSFARA project no. AS: 2001 (SM and BPM). The authors are thankful to Professor A. P. Sharma, Director, CIFRI, for the facilities and encouragement.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Barik, S.K., Banerjee, S., Bhattacharjee, S. et al. Proteomic Analysis of Sarcoplasmic Peptides of Two Related Fish Species for Food Authentication. Appl Biochem Biotechnol 171, 1011–1021 (2013). https://doi.org/10.1007/s12010-013-0384-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-013-0384-y