
Abstract
Bacillus amyloliquefaciens LL3 is a glutamate-independent poly-γ-glutamic acid (γ-PGA) producing strain which consists of a circular chromosome (3,995,227 bp) and an endogenous plasmid pMC1 (6,758 bp). The study of the function of native plasmid and the genome-size reduction of the B. amyloliquefaciens LL3 strain requires elimination of the endogenous plasmid. Traditional plasmid-curing procedures using sodium dodecyl sulfate (SDS) or acridine orange combined with heat treatment have been shown to be ineffective in this strain. Plasmid incompatibility is an effective method for curing which has been studied before. In our research, the hypothetical Rep protein gene and the origin of replication of the endogenous plasmid were cloned into the temperature-sensitive vector yielding the incompatible plasmid pKSV7-rep-ori. This plasmid was transformed into LL3 by electroporation. The analysis of the strain bearing incompatible plasmids after incubation at 30 °C for 30 generations showed the production of plasmid cured strains. High frequency of elimination was achieved with more than 93 % of detected strains showing to be plasmid-cured. This is the first report describing plasmid cured in a γ-PGA producing strain using this method. The plasmid-cured strains showed an increase of γ-PGA production by 6 % and led to a yield of 4.159 g/l, compared to 3.918 g/l in control and cell growth increased during the early stages of the exponential phase. Gel permeation chromatography (GPC) characterization revealed that the γ-PGA produced by plasmid-cured strains and the wild strains were identical in terms of molecular weight. What is more, the further study of plasmid function showed that curing of the endogenous plasmid did not affect its sporulation efficiency.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Poly-γ-glutamic acid (γ-PGA) is an unusual anionic polypeptide which consists of D-glutamate and L-glutamate monomers connected by amide linkage between the γ-carboxyl and α-amino groups [1]. γ-PGA is water-soluble, biodegradable, edible, and nontoxic towards human and it is widely applied in food, cosmetics, medical industries, and agriculture [2]. All the γ-PGA producing strains can be classified into two categories: glutamate-dependent strains and glutamate-independent strains. The yield of γ-PGA in glutamate-dependent strains is significantly higher than that in glutamate-independent strains. Most of the γ-PGA strains used in the industry are glutamate-independent strains, since the former microorganisms need a large amount of exogenous L-glutamate which increases the cost of γ-PGA production. Bacillus amyloliquefaciens LL3 is a glutamate-independent γ-PGA producing strain [3], which can produce γ-PGA directly from sucrose, and it has been used in industrial production. The whole genome sequence of B. amyloliquefaciens LL3 (GenBank ID: CP002634) consists of a 3,995,227-bp circular chromosome and a 6,758-bp endogenous plasmid pMC1 (GenBank ID: CP002635) [4]. In this study, we aimed to increase the γ-PGA production and obtain a better insight of the function of its endogenous plasmid.
Plasmids are species of extrachromosomal DNA that replicate autonomously in bacterial cells. These elements often contain important genes involved in metabolism and symbiosis in some strains [5, 6]. Rap proteins which are shown to be response regulator aspartate phosphatases have been mostly studied in Bacillus subtilis [7]. A subset of proteins consisting of RapA, RapB, RapE, and RapH can dephosphorylate Spo0F and consequently reduce the level of phosphorylated Spo0A in the cell, thus inhibiting sporulation [8, 9].These phosphatases act as negative regulators of the developmental process and deletion of rap genes may result in the increasing of sporulation efficiency [10]. A hypothetical Rap protein gene has been found on the LL3 native plasmid, but its effects on the sporulation are still unknown. In some reports, the plasmidless derivatives were observed to grow at a faster rate compared to the wild-type strain [11]. Elimination of the plasmid is an important step for their functional analysis; moreover, it may induce a faster growth of cell and increase the production.
SDSFootnote 1 [12] and acridine orange [13] have been used for elimination of the native plasmid and the sub-inhibitory concentrations of these compounds have been shown to play an important role in plasmid curing [14]. Heat-shock treatment is also an efficient method, which is usually used in combination with SDS, acridine orange, or other curing agents [15, 16]. Although widely used, these procedures have been reported to be ineffective in certain cases [17]. Plasmid incompatibility, which is defined as unstable inheritance of two co-resident plasmids in the absence of external pressure [18], has been used for plasmid curing in some species [19]. The sharing of elements for plasmid replication or partitioning system results in plasmid incompatibility [18].
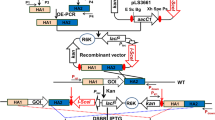
Previous studies have shown that the origin of replication of the plasmid and the rep protein were the main elements in its plasmid replication [20]. In this work, the origin fragment (ori) and the replication protein gene (rep) from plasmid pMC1 were cloned into a temperature-sensitive shuttle plasmid, pKSV7 [21]. The pKSV7 plasmid, derived from pE194ts, contains a conditional defective replication origin, which can replicate at a temperature as low as 30 °C, but it is unstable at a high temperature like 42 °C. The constructed incompatible plasmid pKSV7-rep-ori was then used for curing. The function of the endogenous plasmid and its effect on cell growth and γ-PGA production of the strain were also investigated.
Materials and Methods
Bacterial Strains, Plasmid, and Cultivation
The strains and plasmids used in this study are listed in Table 1. All the Escherichia coli strains were cultured in liquid Luria–Bertani medium (LB) or on LB agar. The B. amyloliquefaciens strains LL3-zx and LL3 were cultured in γ-PGA fermentation medium, which contains: 50 g/l sucrose, 6 g/l (NH4)2SO4, 0.6 g/l MgSO4, 6 g/l KH2PO4, and 14 g/l K2HPO4. E. coli strains were grown at 37 °C; the wild-type B. amyloliquefaciens LL3 strain was grown at 37 °C and the LL3 strain containing the incompatibility plasmid was cultivated at 30 °C. In order to cure the incompatibility plasmid, the pMC1 cured and bearing the incompatibility plasmid LL3 strains were grown at 42 °C. The following antibiotic concentrations were used for selection: chloramphenicol (Cm) 5 μg/ml for LL3 and ampicillin (Amp) 100 μg/ml for E. coli. The primers used in this study are listed in Table 2.
Reagents
SDS and acridine orange were purchased from Ding Guo Biotechnology (Tianjin, China) and Sigma, respectively.
The enzymes used for polymerase chain reaction (PCR) and DNA ligase and restriction enzymes were purchased from TaKaRa Biotechnology (Dalian, China). The pMD19-T simple vector was also purchased from TaKaRa Biotechnology. BamHI methyltransferase was bought from New England Biolabs.
Incompatible Plasmid Construction
The fragments rep and ori of plasmid pMC1 were amplified by PCR using primers rep F/R and ori F/R, respectively. The PCR assay was performed in a final volume of 50 μl containing 5 μl 10× Ex Taq buffer, 4 μl dNTP mixture, 2 μl of each primer, 1 μl template, 0.6 μl Ex Taq DNA polymerase, and 35.4 μl double distilled water. The touchdown PCR protocol was used in the assay with the following thermal cycling parameters: predenaturation at 94 °C for 10 min, followed by 20 cycles of 94 °C for 40 s, an initial annealing temperature of 68 °C (with a temperature decrement of 0.5 °C following each cycle) for 30 s, 72 °C for 2 min and 10 cycles of 94 °C for 40 s, 58 °C for 30 s, 72 °C for 2 min, and a final elongation at 72 °C for 10 min. The purified amplicons were ligated to the pMD19-T simple vector for sequencing. The 1.5-kb PstI-HindIII rep fragment was introduced into PstI-HindIII cleaved pKSV7 vector to obtain the plasmid, pKSV7-rep. The 1.8-kb KpnI-XbaI ori fragment was introduced into the KpnI-XbaI cleaved pKSV7-rep vector to generate the incompatible plasmid pKSV7-rep-ori. The generated plasmids were introduced into E. coli DH5α by calcium chloride transformation.
Introduction of Incompatible Plasmid into B. amyloliquefaciens
The incompatible plasmid was isolated from E. coli DH5α and introduced into E. coli GM2163 for demethylation. The plasmids isolated from E. coli GM2163 were first treated with BamHI methyltransferase and then introduced into B. amyloliquefaciens LL3 by electroporation (36 μF, 2.3 kV). After pulsing, the cells were grown at 30 °C (180 rpm) for 3 h and then plated onto the Cm-selective agar plates. Colonies were selected and incubated in LB medium with chloramphenicol at 30 °C for 18 h. Strains containing incompatible plasmids were identified by PCR using primers M13 F/R.
Traditional Treatment for Plasmid Curing
SDS and acridine orange combined with heat treatment were used for plasmid curing. An overnight culture of B. amyloliquefaciens LL3 in LB medium was diluted 10 times and a 50-μl aliquot was sub-cultured in 5 ml fresh broth with SDS and acridine orange at sub-inhibitory concentrations (0.005 %, 24 μg/ml, respectively). After inoculation, the culture was incubated at 42 °C (180 rpm) for 12 h. The treatment was repeated 30 times and plasmid-cured strains were verified by PCR using primers JC F/R.
Curing Endogenous Plasmid by Plasmid Incompatibility
After introducing the incompatible plasmid into B. amyloliquefaciens LL3, the strain was cultured at 30 °C for 12 h and sub-cultured 30 generations with chloramphenicol. The treated culture was diluted 105 times, spread on agar plates containing Cm and cultured at 30 °C for 18 h. Single colonies were picked out and primers JC F/R were used to select positive colonies. The primers JC F/R amplified a 600-bp fragment present in the endogenous plasmid but not in the incompatible plasmid. Therefore, colonies whose amplification of this fragment was negative represent the target colonies.
Curing of the Incompatible Plasmid
The strain cured of endogenous plasmid was cultivated in fresh LB medium at 42 °C for 24 h in the absence of antibiotic. The culture was then spread on LB agar plates for 12 h. Single colonies were picked and cultured on plates with and without Cm. Cm-sensitive colonies were identified as positive strains and verified by PCR using the primers rep F/R, ori F/R. Colonies in which no fragments were amplified represent the strains cured of the endogenous plasmid and the incompatible plasmid.
Detection of Spore Formation
The B. amyloliquefaciens LL3 wild-type strain and LL3-zx plasmid-cured derivatives were cultured in LB medium and sporulated by exhaustion in the Difco sporulation medium (DSM) [22]. Starter culture (1 ml) was transferred into 100 ml DSM and incubated at 37 °C (180 rpm) for 48 h. The cultures were adjusted at the same cell concentration and then treated at 80 °C for 20 min before 1 ml treated culture was applied to LB agar plates and cultured at 37 °C for 12 h to detect spore growth and obtain the number of CFU (colony-forming units) [23]. All the experiments were performed in triplicate.
Influence of Plasmid Curing
One milliliter of LL3-zx and LL3 seed cultures was incubated in 500 ml flasks filled with 100 ml LB medium, shaking the culture at 37 °C for 48 h. All the experiments were carried out in triplicate. The optical density (OD600) was monitored to differentiate cell growth of the two strains.
The fermentation product, γ-PGA, was purified according to a previously reported method [24]. The molecular weight was measured by gel permeation chromatography. An Alltech system controller (Alltech Associates Inc., US) fitted with a Shodex KW804 column (Showa Denko KK, Japan) and Schambeck SFD RI detector (Germany) were used for γ-PGA analysis. Samples were eluted with 0.25 N NaNO3. The flow rate was set at 0.6 ml/min. The Shodex Pullulan-82 standards were used to construct the calibration curve.
Results
Incompatible Plasmid Construction
An incompatible plasmid (pKSV7-rep-ori) was constructed based on pKSV7, which carries the replication gene (rep, 1,500 bp) and the origin of replication fragment ori (1,800 bp) from the endogenous plasmid pMC1. Digestion of the plasmid with the corresponding restriction endonucleases confirmed that the plasmid was successfully constructed.
Curing the Endogenous Plasmid by Plasmid Incompatibility
The methylated incompatible plasmid pKSV7-rep-ori was introduced into B. amyloliquefaciens LL3. The transformed cells were grown at 30 °C in the presence of Cm to maintain the dominant position of the incompatible plasmid. After growth of the transformed cells for 30 generations, single colonies were selected and these colonies devoid of the endogenous plasmid were verified by PCR with the primers JC F/R. A 600-bp amplified fragment represents the existence of endogenous plasmid. Curing experiment using SDS, acridine orange and heat revealed that these treatments did not eliminate the plasmid, while the experiment using plasmid incompatibility showed high frequency of plasmid elimination. More than 93 % (14/15) of the selected strains showed the absence of endogenous plasmid, with the exception of the strain shown in lane 10 (Fig. 1).

Identification of the plasmid-cured strains by PCR. Lane M DNA marker III, lanes 1 to 15 PCR product from independent isolates following the incompatibility curing procedure, and 16 PCR positive control from wild-type strain LL3
Curing of the Incompatible Plasmid, pKSV7-rep-ori
A strain bearing pKSV7-rep-ori was cultured at 42 °C for 12 h, and the Cm-sensitive clones were selected for further detection. Agarose gel electrophoresis of PCR amplification products obtained using the primers rep F/R, ori F/R demonstrated the absence of the pKSV7-rep-ori, and also confirmed the loss of endogenous plasmid (Fig. 2). LL3 strain and its derived Cm-sensitive clones were subjected to a plasmid extraction procedure, using a commercial plasmid extraction kit (Tiangen Biotech, Beijing, China), and the result of the extraction was analyzed by agarose gel electrophoresis. As expected, LL3 strain produced a band of the size represents the endogenous plasmid pMC1, whereas no plasmids were found in Cm-sensitive strains LL3-zx1 and LL3-zx2 (Fig. 3). These observations confirmed the curing of incompatible plasmid and native plasmid in LL3 strain transformed with pKSV7-rep-ori plasmid.

Confirmation of the loss of plasmid pKSV7-rep-ori by PCR. Lane M DNA marker III, 1 to 4 the results of strains that amplified with primers ori F/R; 1–3 chloramphenicol-sensitive strains, 4 wild-type strain LL3. 5–8 The results of strains that amplified with primers rep F/R; 5–7 three previous chloramphenicol-sensitive strains, 8 wild-type strain LL3

Agarose gel electrophoresis of plasmids extracted from plasmid-cured strain and wild-type strain LL3. Lane M DNA marker III, lanes zx-1 and zx-2 results of plasmids extracted from plasmid-cured strains, and lane wild result of plasmid extracted from wild-type strain LL3
Effect of Endogenous Plasmid Loss in B. amyloliquefaciens LL3 Strain
One milliliter of LL3, LL3-zx, and E. coli DH5α heat treated cultures was spread on the LB plates and the results were shown in Fig. 4. Cell growth was detected on the LL3 plate (Fig. 4a) and LL3-zx plate (Fig. 4b) while no cell growth was detected on the control plate (Fig. 4c) spreading E. coli DH5α. The average colony-forming units (CFU) of LL3 and LL3-zx were 146 and 128, respectively (Table 4). It was confirmed that no increase of sporulation efficiency was identified for LL3-zx. Thus the hypothetical response regulator aspartate phosphatase gene on the plasmid does not affect sporulation efficiency. The existence of other response regulator aspartate phosphatase family genes in the chromosome serves the same function, or this type of Rap protein does not influence its sporulation.

Identification of sporogenesis; 1 ml heat-treated culture spread on the plate, a LL3, b LL3-zx, and c E. coli DH5α
As shown in Table 3, the growth rates of plasmid-cured strain and LL3 wild-type strain were the same at the early stage of the exponential phase (0-4 h); then the plasmid-cured strain exhibited a significantly higher growth rate than the LL3 wild-type strain during the middle stage (4-12 h after incubation) with a highest significance of 68.2 % at 8 h; after that, the growth rates of the two strains were found to be comparable (data not shown). The level of γ-PGA production was observed between the two strains after they were cultured at 37 °C for 48 h (Table 4). LL3-zx showed a 6 % increase at 4.159 g/l compared to LL3, 3.918 g/l. Their molecular weight carried out that LL3 (449,424) and LL3-zx (458,889) are comparable.
Discussion
Numerous methods have been described for plasmid curing. The conventional methods involve chemical and physical treatments. A variety of compounds have been reported as curing agents [25, 26]. SDS and acridine orange are the most common agents for this purpose [15, 27]. Heat treatment is also an effective physical treatment for elimination of plasmids, which is always used in combination with other methods [15, 16]. The mechanisms of plasmid curing in bacteria by these conventional methods had been studied previously and they were mostly involved in two possible target sites during plasmid replication. One was membrane binding sites and the other was plasmid replication [14]. SDS can damage the cell membrane sites while acridine orange and high temperature can block the replication of plasmid, thus leading to the curing of plasmid [14]. However, some plasmids cannot be cured by conventional methods and the effectiveness of these methods varies according to the strains involved. B. amyloliquefaciens LL3 has a very stable plasmid. We chose SDS and acridine orange as the curing agents which were used combining with heat treatment at a high temperature (42 °C). However, it was proved ineffective in this strain. Plasmid incompatibility is an efficient method for elimination which has been widely used for plasmid curing [28]. In this study, we constructed a novel curing plasmid, pKSV7-rep-ori, which contains the origin of replication region of the native plasmid and the replication protein gene in order to replace the native plasmid due to incompatibility. The use of the plasmid encoding the replication protein gene alone was not sufficient to achieve plasmid curing strains (data not shown), while the incompatible plasmid, pKSV7-rep-ori, showed high efficiency for plasmid elimination and more than 93 % of the detected strains were plasmid-cured derivatives. This confirmed that simply using the single replication protein gene for plasmid curing was not enough. Furthermore, this method is recommended as a much safer technique for plasmid curing as it is not associated with gene mutation of the host chromosome [19]. For the principles of this method are clear and high efficiency of curing is achieved, it is recommended to use this method for elimination in those strains whose plasmid sequences are clear.
Following the curing process, the plasmid influence on the cured derivatives and those of their respective wild-type strains were compared. A gene on the plasmid encoding a hypothetical response regulator aspartate phosphatase and some of these members have been reported to play important regulatory roles in the initiation of sporulation in B. subtilis and the deletion of the gene may result in the increase of sporulation efficiency [7, 10]. Other homologous genes have also been detected in plasmids in some strains [29, 30], though their functions were not defined. In this work, we found that both LL3 and LL3-zx strains could form spores and their CFU were comparable. The plasmid did not affect the sporulation as expected. The reason might be existence of other intracellular proteins of this family performing the same function, or the hypothetical protein was not classified in the members influencing sporulation. We also compared cell growth and γ-PGA production between the two strains. Plasmid-cured strains grew at a faster rate compared to the wild-type strains during the early stages of the exponential phase. The yield of γ-PGA of the LL3-zx strains slightly increased compared to that of the wild-type LL3 strains and the γ-PGA molecular weights of the two strains were equivalent. The possible reasons were as follows: (a) genome-size reduction may decrease the redundant metabolic networks and provide a more efficient system than the wild-type strain. The construction of the reduced-genome of industrial microbiology strains not only increases cell growth but also improves the production of relevant product [31–33]; (b) there is no gene in the endogenous plasmid involving the γ-PGA synthesis and the deletion fragment is not large enough to influence the γ-PGA production significantly. Although curing the endogenous plasmid does not markedly increase the γ-PGA yield, it shows a prospect that the genome-size reduction of industrial bacteria do not just remove the by-product, but also increase the production of target products.
Curing of the plasmid is an important step for the research on its function. Although the conventional chemical treatments are the most widely-used methods, they are unsafe and inefficient. Moreover, it cannot be used in certain conditions since its curing mechanisms remain unclear. Plasmid incompatibility is an efficient and safe method for its curing mechanism is distinct and it can be used in almost all the target strains. In this study, we successfully cured the endogenous plasmid of a γ-PGA producing B. amyloliquefaciens LL3 using this efficient method. Overall, plasmid incompatibility is a recommended choice for curing plasmid with a clear background in those strains.
Notes
SDS: sodium dodecyl sulfate
References
Ashiuchi, M., & Misono, H. (2002). Biochemistry and molecular genetics of poly-γ-glutamate synthesis. Applied Microbiology and Biotechnology, 59, 9–14.
Shih, I. L., & Van, Y. T. (2001). The production of poly-(γ-glutamic acid) from microorganisms and its various applications. Bioresource Technology, 79, 207–225.
Cao, M. F., Geng, W. T., Liu, L., Song, C. J., Xie, H., Guo, W. B., Jin, Y. H., & Wang, S. F. (2011). Glutamic acid independent production of poly-γ-glutamic acid by Bacillus amyloliquefaciens LL3 and cloning of pgsBCA genes. Bioresource Technology, 102, 4251–4257.
Geng, W. T., Cao, M. F., Song, C. J., Xie, H., Liu, L., Yang, C., Feng, J., Zhang, W., Jin, Y. H., Du, Y., & Wang, S. F. (2011). Complete genome sequence of Bacillus amyloliquefaciens LL3, which exhibits glutamic acid-independent production of poly-γ-glutamic acid. Journal of Bacteriology, 193, 3393–3394.
Nojiri, H., Shintani, M., & Mori, T. (2004). Divergence of mobile genetic elements involved in the distribution of xenobiotic-catabolic capacity. Applied Microbiology and Biotechnology, 64, 154–174.
Rotger, R., & Casadesus, J. (1999). The virulence plasmids of Salmonella. International Microbiology, 2, 177–184.
Perego, M., Hanstein, C., Welsh, K. M., Djavakhishvili, T., Glaser, P., & Hoch, J. A. (1994). Multiple protein-aspartate phosphatases provide a mechanism for the integration of diverse signals in the control of development in B. subtilis. Cell, 79, 1047–1055.
Jiang, M., Grau, R., & Perego, M. (2000). Differential processing of propeptide inhibitors of Rap phosphatases in Bacillus subtilis. Journal of Bacteriology, 182, 303–310.
Smits, W. K., Bongiorni, C., Veening, J. W., Hamoen, L. W., Kuipers, O. P., & Perego, M. (2007). Temporal separation of distinct differentiation pathways by a dual specificity Rap-Phr system in Bacillus subtilis. Molecular Microbiology, 65, 103–120.
Perego, M. (2001). A new family of aspartyl phosphate phosphatases targeting the sporulation transcription factor Spo0A of Bacillus subtilis. Molecular Microbiology, 42, 133–143.
Ghosh, S., Mahapatra, N. R., Ramamurthy, T., & Banerjee, P. C. (2000). Plasmid curing from an acidophilic bacterium of the genus Acidocella. FEMS Microbiology Letters, 183, 271–274.
EI-Mansi, M., Anderson, K. J., Inche, C. A., Knowles, L. K., & Platt, D. J. (2000). Isolation and curing of the Klebsiella pneumonia large indigenous plasmid using sodium dodecyl sulphate. Research in Microbiology, 151, 201–208.
Hara, T., Aumayr, A., Fujio, Y., & Ueda, S. (1982). Elimination of plasmid-linked polyglutamate production by Bacillus subtilis (natto) with acridine orange. Applied and Environment Microbiology, 44, 1456–1458.
Spengler, G., Molnár, A., Schelz, Z., Amaral, L., Sharples, D., & Molnár, J. (2006). The mechanism of plasmid curing in bacteria. Current Drug Targets, 7, 823–841.
Kulkarni, R. S., & Kanekar, P. P. (1998). Effects of some curing agents on phenotypic stability in Pseudomonas putida degrading ε-caprolactam. World Journal of Microbiology and Biotechnology, 14, 255–257.
Imre, A., Olasz, F., Kiss, J., & Nagy, B. (2006). A novel transposon-based method for elimination of large bacterial plasmids. Plasmid, 55, 235–241.
Rajini Rani, D. B., & Mahadevan, A. (1992). Plasmid mediated metal and antibiotic resistance in marine pseudomonas. Biometals, 5, 73–80.
Novick, R. P. (1987). Plasmid incompatibility. Microbiology and Molecualr Biology Reviews, 51, 381–395.
Uraji, M., Suzuki, K., & Yoshida, K. (2002). A novel plasmid curing method using incompatibility of plant pathogenic Ti plasmids in Agrobacterium tumefaciens. Genes & Genetic Systems, 77, 1–9.
Baker, T. A., & Bell, S. P. (1998). Polymerases and the replisome: machines within machines. Cell, 92, 295–305.
Smith, K., & Youngman, P. (1992). Use a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spoIIM gene. Biochimie, 74, 705–711.
Cliff, J. B., Jarman, K. H., Valentine, N. B., Golledge, S. L., Gaspar, D. J., Wunschel, D. S., & Wahl, K. L. (2005). Differentiation of spores of Bacillus subtilis grown in different media by elemental characterization using Time-of-Flight secondary ion mass spectrometry. Appllied and Environment Microbiology, 71, 6524–6530.
Fay, A., & Dworkin, J. (2009). Bacillus subtilis homologs of MviN (MurJ), the putative Escherichia coli lipid II flippase, are not essential for growth. Journal of Bacteriology, 191, 6020–6028.
Kubota, H., Matsunobu, T., Uotani, K., Takebe, H., Satoh, A., Tanaka, T., & Taniguchi, M. (1993). Production of poly (γ-glutamic acid) by Bacillus subtilis F-2-01. Bioscience, Biotechnology, and Biochemistry, 57, 1212–1213.
Bouanchaud, D. H., Scavizzi, M. R., & Chabbert, Y. A. (1969). Elimination of ethidium bromide of antibiotic resistance in Enterobacteria and Staphylococci. Journal of General Microbiology, 54, 417–425.
Mchuge, G. L., & Swartz, M. N. (1977). Elimination of plasmids from several bacterial species by novobiocin. Antimicrobial Agents and Chemotherapy, 12, 423–426.
Pickett, M. A., Everson, J. S., Pead, P. J., & Clarke, I. N. (2005). The plasmids of Chlamydia trachomatis and Chlamydophila pneumoniae (N16): accurate determination of copy number and the paradoxical effect of plasmid-curing agents. Microbiology, 151, 893–903.
Posno, M., Leer, R. J., van Luijk, N., van Giezen, M. J., Heuvelmans, P. T. H. M., Lokman, B. C., & Pouwels, P. H. (1991). Incompatibility of Lactobacillus vectors with replicons derived from small cryptic Lactobacillus plasmids and segregational instability of the introduced vectors. Applied and Environment Microbiology, 57, 1822–1828.
Meijer, W. J., de Boer, A. J., van Tongeren, S., Venema, G., & Bron, S. (1995). Nucleic Acids Research, 23, 3214–3223.
Mueller, J. P., Bukusoglu, G., & Sonenshein, A. L. (1992). Transcriptional regulation of Bacillus subtilis glucose starvation-inducible genes: control of gsiA by the ComP-ComA signal transduction system. Journal of Bacteriology, 174, 4361–4373.
Mizoguchi, H., Mori, H., & Fujio, T. (2007). Biotechnology and Applied Biochemistry, 46, 157–167.
Mizoguchi, H., Sawano, Y., Kato, J., & Mori, H. (2008). Superpositioning of deletions promotes growth of Escherichia coli with a reduced genome. DNA Research, 15, 277–284.
Morimoto, T., Kadoya, R., Endo, K., Tohata, M., Sawada, K., Liu, S. G., Ozawa, T., Kodama, T., Kakeshida, H., Kageyama, Y., Manabe, K., Kanaya, K., Ara, K., Ozaki, K., & Ogasawara, N. (2008). Enhanced recombinant protein productivity by genome reduction in Bacillus subtilis. DNA Research, 15, 73–81.
Acknowledgments
This study was financially supported by National key 296 Basic Research Program of China (“973”-Program) 2012CB725204, National High Technology Research and Development Program of China (“863”-Program) 2012AA021505, Natural Science Foundation of China Grant Nos. 31070039, 31170030, and 51073081, Project of Tianjin, China (11JCYBJC09500).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Jun Feng and Yanyan Gu contributed equally to this work.
Rights and permissions
About this article
Cite this article
Feng, J., Gu, Y., Wang, J. et al. Curing the Plasmid pMC1 from the Poly (γ-glutamic Acid) Producing Bacillus amyloliquefaciens LL3 Strain Using Plasmid Incompatibility. Appl Biochem Biotechnol 171, 532–542 (2013). https://doi.org/10.1007/s12010-013-0382-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-013-0382-0