
Abstract
MicroRNAs (miRNAs) are small, noncoding RNAs that play key roles in regulating gene expression in animals, plants, and viruses, which involves in biological processes including development, cancer, immunity, and host–microorganism interactions. In this present study, we have used the computational approach to identify potent miRNAs involved in Anopheles gambiae immune response. Analysis of 217,261 A. gambiae ESTs and further study of RNA folding revealed six new miRNAs. The minimum free energy of the predicted miRNAs ranged from −27.2 to −62.63 kcal/mol with an average of −49.38 kcal/mol. While its A + U % ranges from 50 to 65 % with an average value of 57.37 %. Phylogenetic analysis of the predicted miRNAs revealed that aga-miR-277 was evolutionary highly conserved with more similarity with other mosquito species. Observing further the target identification of the predicted miRNA, it was noticed that the aga-miR-2304 and aga-miR-2390 are involved in modulation of immune response by targeting the gene encoding suppressin and protein prophenoloxidase. Further detailed studies of these miRNAs will help in revealing its function in modulation of A. gambiae immune response with respect to its parasite.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Small RNAs are nonconventional, noncoding, functional, or regulatory RNAs that directly function at RNA level in cells, but they are not messenger, transfer, or ribosomal RNAs. RNA-based regulation of gene expression is an evolutionarily conserved mechanism regulated by two kinds of small RNAs [1]. Small interfering RNAs, termed as siRNA, are produced by cleavage of double stranded RNA, which gives rise to two classes of siRNA based on their length (20 to 22 and 24 to 26 mers). The other type of small RNA, termed as MicroRNAs (miRNAs), are endogenous∼22 nucleotide (nt) RNAs, some of which are known to play important regulatory roles by serving as guide RNAs for the posttranscriptional repression of protein coding genes [2]. As the first step miRNA genes are transcribed as long pre-miRNAs, RNase III enzyme Drosha processes them to ∼70 nt precursors (pre-miRNA) with stem-loop. Then ∼22 nt mature miRNAs from pre-miRNA are released by another RNase III enzyme Dicer [3, 4]. Finally, RNA-induced silencing complexes are formed to regulate the expression of target genes via complementary base pair interactions [5].
Direct cloning, forward genetics, and computational approaches using bioinformatics tools are the three main approaches globally used in detection of miRNA. In direct cloning, small RNA were isolated from biological samples and cloned to make a cDNA library for small endogenous RNAs [6]. In direct cloning approach, low expressed miRNAs are hard to clone and are limited to highly-expressed miRNAs. Forward genetics approach involves identification of mutation responsible for a certain phenotype. But this approach has not been used very much to detect miRNA families due to certain limitations like small size of miRNA and the “seed” sequence, which doesn’t mutate easily [7]. Bioinformatics approaches and genome sequence analysis could be potentially used to overcome the limitations of the above two methods. Bioinformatics approach is based on the information about certain miRNA features generated by cloning approach. Computational approach in identification of miRNAs is widely used because they are evolutionary conserved. Bioinformatics predictions with experimental validation indicate that the total number of miRNAs is significantly higher than previously estimated [8].
Mosquitoes of the genera Aedes, Anopheles, and Culex transmit serious human diseases such as malaria, yellow fever, West Nile fever, and chikungunya. Every year, 500 million people are affected by malaria, transmitted by Anopheles sp. with above 2 million death cases. In the absence of an effective control strategy, these diseases have a major impact on the public health, environmental safety, and the socioeconomic development of a country [9]. Discovery of miRNAs lin-4 and let-7 started in Caenorhabditis elegans [10] and later, several hundred miRNAs with posttranscriptional regulatory functions have been reported from viruses, plants, and animals. The recent availability of the complete genome and development of large scale expressed sequence tag (EST) of the malaria mosquito A. gambiae [11] has enabled the identification of miRNAs from its genome. In spite this vast study about miRNA, research in the area of insect host–microorganism interactions has been limited, accumulating evidences in vertebrates suggests that miRNAs play major roles, including antipathogen responses targeting the microorganism directly or altering the expression of host genes that are beneficial to the microorganism [12–14]. Pathogens are also able to manipulate host miRNA to facilitate its replication [15] and the manipulation will be very parasite-species-specific as one miRNA might be downregulated by one parasite while upregulated by another closely related species [16].
In the present study, we have utilized all miRNAs reported from different species for the identification of its conserved miRNA homologues from A. gambiae ESTs. Here, we reported six new miRNAs from A. gambiae EST, identified its targets, and discussed in detail. We believe that the approach employed here will help in the prediction of miRNA in distantly-related species as well.
Materials and methods
Collection of reference miRNA, nucleotide, and EST sequences
We used EST analysis method for the identification of miRNAs from A. gambiae. All known pre-miRNA (18,226 as of June 2012) and mature miRNA (21,623 as of June 2012) were retrieved from the miRBase (http://www.mirbase.org/). After elimination of homolog, rests of the miRNAs were used for searching A. gambiae miRNA. EST sequences (217,261 as of June 2012) of A. gambiae were downloaded from the Vectorbase EST database (http://www.agambiae.vectorbase.org/). Local nucleotide database was created after eliminating redundant and poor quality sequences.
Identification of miRNAs and their precursor sequences
Instead of mature miRNA sequences of A. gambiae, pre-miRNAs were used as reference material for the search of its homolog in A. gambiae transcriptome. Reference miRNA sequences were used as a query for homology search against our local A. gambiae nucleotide sequence database at e-value threshold <0.01 using BLAST 2.2.22+ program with all other parameters as default [17]. The FASTA formats of all the candidate sequences were saved. The repeated ESTs from the same gene were removed by BLAST against the A. gambiae ESTs Database using blastn with default parameters and single tone EST was created for each miRNA. Reference miRNA sequence was aligned against the corresponding singleton ESTs using ClustalW [18] multiple sequence alignment tool and initial miRNA candidate was created. Target sequences with not more than four mismatches were validated for their nonprotein encoding phenomenon using BLAST against protein database at NCBI using blastx with default parameter [19].
Validation of precursor candidate miRNAs
The candidate pre-miRNA satisfying the above conditions were considered for prediction and validation of secondary structure using Mfold v 3.2 (http://www.mfold.rna.albary.edu/).The precursor sequences were selected from 100 nucleotides upstream or downstream from the location of mature miRNAs. While selecting a RNA sequence as a candidate miRNA precursor, the following criteria were used according to Zhang et al. [20]: (a) RNA sequence is able to fold into an appropriate stem-loop hairpin secondary structure, (b) mature miRNA sequence site in one arm of the hairpin structure, (c) miRNAs should have less than seven mismatches with the opposite miRNA* sequence in the other arm, (d) loop or break in miRNA sequences should not be noticed, (e) had less than seven mismatches with the opposite miRNA sequence in the other arm, (4) no loop or break in miRNA sequences, (5) predicted secondary structures had higher negative energy MFEs (≤ −18 kcal/mol) and 40–70 % A + U contents, (6) predicted secondary structures should have higher negative energy MFEs (≤ −18 kcal/mol) and 40–70 % A + U contents.
Prediction of A. gambiae miRNAs targets
The identified A. gambiae miRNA query sequences were used for their target prediction using NCBI blastn program [17]. The parameters such as Database (Nucleotide collection), organism (A. gambiae—taxid: 3328) and program selection (highly similar sequences—megablast) were selected for analysis. The sequences showing 60 % query coverage were selected and subjected to RNA-hybrid, a miRNA target prediction tool (http://www.bibiserv.techfak.uni-bielefeld.de/rnahybrid/).
Nomenclature of predicted miRNAs
Names for the predicted miRNAs were assigned as per the pattern of miRBase Griffiths-Jones et al., [21]. The mature sequences were designated “miR” and the precursor hairpins were labeled as “mir” with the prefix “aga” for A. gambiae.
Phylogenetic analysis
Phylogenic relationship of A. gambiae miRNA conservation with their orthologues was analyzed. Candidate A. gambiae miRNAs was searched for their homolog against all miRNAs using NCBI standalone BLAST [17] allowing a maximum of three mismatches and e-value <0.001. The corresponding precursor sequences of homolog miRNA’s were identified, collected, and aligned with homolog A. gambiae miRNA using ClustalW [18]. Phylogenetic analysis of the aligned miRNA sequences of the selected was performed with Mega5. The evolutionary distances were computed using the maximum composite likelihood method [22] and are in the units of the number of base substitutions per site. Finally, the evolutionary analyses were conducted in MEGA5 [23].
Results and discussion
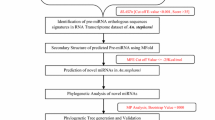
Here in this study, we employed computational prediction of A. gambiae miRNA from its EST resource (Fig. 1). miRNAs have been shown to regulate diverse and important processes such as B-cell differentiation [24], adipocyte differentiation [25], cardiogenesis [26], insulin secretion [27], antiviral defense [28], and the development of cancer [29]. A total of 217,261 A. gambiae EST sequences were retrieved from the Vectorbase database and used for prediction of miRNAs from A. gambiae genome. From the present study on in silico search for novel miRNAs, we have noticed that out of the whole EST sequences, only 105 sequences were recorded with less than four mismatches. Further analysis of the sequence for their protein encoding nature using BLAST against protein database at NCBI using blastx revealed the presence of 63 nonprotein encoding ESTs. Six new A. gambiae miRNAs was reported after a careful evaluation of the secondary structure analysis. Though miRNA research in the area of insect host–microorganism interactions has been limited, accumulating evidences in vertebrates suggests that miRNAs play major roles, including antipathogen responses targeting the microorganism directly or altering the expression of host genes that are beneficial to the microorganism [13, 14]. For the predicted miRNAs, Table 1 provides details such as source sequences, length of precursor sequences, and their minimum folding free energies and A + U content. Though Chatterjee and Chaudhuri, [30] had reported 41 A. gambiae using Drosophila melanogaster miRNA dataset, they failed to predict the miRNA which are absent in D. melanogaster.

A schematic illustration of different steps involved in miRNA and their target prediction
Among the predicted miRNAs from A. gambiae EST, only two were noticed in direct strand and the rest of the predictions were observed in indirect strand. The predicted miRNAs had minimum free energy ranging from −27.2 to −62.63 kcal/mol with an average of −49.38 kcal/mol. While considering the A + U percentage in the predicted miRNAs, it ranges from 50 to 65 % with an average value of 57.37 % (Table 1). Hackl et al. [31] extracted pre-miRNA from reference genomes folded in silico to verify correct structures from Chinese hamster ovary cell lines. Among the miRNAs predicted, only two (aga-miR-277 and aga-miR-466i) sequences were matched 100 % with their homologue in miRNA database. Analysis of RNA folding using Mfold revealed that, all the mature A. gambiae miRNAs that were found in the stem portion of the hairpin structures are broken or loopless inside the sequences (Fig. 2 a-f) and contains less than seven mismatches in the other arm.

Hairpin structure of predicted Anopheles gambiae miRNA. a aga-miR-277, b aga-miR-466b, c aga-miR-466i, d aga-miR-1638, e aga-miR-2304, and f aga-miR-2390
Phylogenetic analysis of the predicted miRNAs revealed miRNA (aga-miR-277) was highly conserved among the insect species, with more similarity with other mosquito species. Conservation of miRNA sequence was observed among the mosquito A. gambiae and Anopheles stephensi species [32]. Phylogenetic tree revealed their evolutionary development among the insect species (Fig. 3 a-b). For the six predicted miRNAs, we have identified 20 potent target genes. Among the miRNAs, a maximum of six targets were identified for the aga-miR-466b.

a Phylogenetic tree showing evolutionary conservation of aga-miR-277 among insect species b Multiple sequence alignment of aga-miR277 with its related species showing conserved region
Genes encoding Netrin-B, starry night, and visceral mesodermal armadillo-repeats were predicted as targets of aga-miR-277. Thus, the predicted aga-miR-277 could modulate the expression pattern of genes involved in biological process such as axon guidance and rhabdomere development. The aga-miR-466b are able to target the expression of genes encoding proteins such as N-acetyl transferase, N-formylmethionylaminoacyl-tRNA deformylase, rho guanine dissociation factor, Lipase, rap GTPase-activating protein, and innexin. Thus, aga-miR-277 could be involved in modulation of biological process such as cell proliferation, lipid metabolism, signal transduction, and response to light stimulus. Genes encoding protein like ceramidase, glial maturation factor, G-protein coupled receptor, and cytoplasmic dynein intermediate chain are targeted by aga-miR-466i and modulates the hatching behavior, gliogenesis, G-protein coupled receptor-signaling pathway, and microtubule cytoskeleton organization biological process (Table 2). The other miRNAs, such as aga-miR-1638, 2304, and 2390, are involved in biological process such as transcription, protein phosphorylation, immune response, and signaling cascade by targeting the genes encoding protein phosphatase, cyclin-dependent kinase, suppressin, monooxygenase, and protein prophenoloxidase. Pathogens are also able to manipulate host miRNA to facilitate its replication [9] and the manipulation will be very parasite-species-specific as one miRNA might be downregulated by one parasite while upregulated by another closely related species [16]. Recent report of Hussain et al., [33] demonstrated that Wolbachia could use host miRNA to manipulate host gene expression for its colonization in dengue vector Aedes aegypti. Differential expression of miRNA in response to the malaria parasite Plasmodium berghei was reported from the midgut of the mosquito following parasite invasion [34]. Results from the study of Winter et al., [34] suggested that host miRNAs might play key roles in antiparasitic responses and resistance of A. gambiae to P. berghei, perhaps by regulating defense-related host genes. Though Skalsky et al. [35] discovered that only small portion of miRNAs was found to show variation in Culex quinquefasciatus mosquitoes infected with the West Nile virus.
As conclusion, the present study reported six new miRNA candidates from A. gambiae using EST resource of the target organism. Further analysis of the miRNA targets revealed that biological process such as immune response, signaling cascade, cell proliferation, and post translation modification are controlled by the identified candidate miRNAs. Further future studies are to be carried out with wet lab to unravel the importance of the identified miRNA in response to the host–parasite interaction and developmental pattern of the vector mosquito.
References
Reinhart, B. J., Weinstein, E. G., Rhoades, M. W., Bartel, B., & Bartel, D. P. (2002). MicroRNAs in plants. Genes and Development, 16(13), 1616–1626.
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell, 116, 281–297.
Lee, Y., Jeon, K., Lee, J. T., Kim, S., & Kim, V. N. (2002). MicroRNA maturation: stepwise processing and subcellular. EMBO Journal, 21(17), 4663–4670.
Lee, Y., Ahn, C., Han, J., Choi, H., Kim, J., Yim, J., et al. (2003). The nuclear RNase III Drosha initiates microRNA processing. Nature, 425(6956), 415–419.
Hammond, S. C., Bernstein, E., Beach, D., & Hannon, G. J. (2000). An RNA-directed nuclease mediates post-transcriptional gene silencing in drosophila cells. Nature, 404(6775), 293–296.
Ambros, V., & Lee, R. C. (2004). Identification of microRNAs and other tiny noncoding RNAs by cDNA cloning. Methods Mol Biol, 265, 131–158.
Berezikov, E., Cuppen, E., & Plasterk, R. H. (2006). Approaches to microRNA discovery. Nat Genet, 38, S2–S7.
Xie, J., Techritz, S., Haebel, S., Horn, A., Neitzel, H., Klose, J., et al. (2005). A two-dimensional electrophoretic map of human mitochondrial proteins from immortalized lymphoblastoid cell lines: a prerequisite to study mitochondrial disorders in patients. Proteomics, 5(11), 2981–2999.
Manguin, S., Bangs, M. J., Pothikasikorn, J., & Chareonviriyaphap, T. (2010). Review on global co-transmission of human Plasmodium species and Wuchereria bancrofti by Anopheles mosquitoes. Infection, Genetics and Evolution, 10(2), 159–177.
Lee, R. C., Feinbaum, R. L., & Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 75(5), 843–854.
Holt, R. A., Subramanian, G. M., Halpern, A., Sutton, G. G., Charlab, R., Nusskern, D. R., et al. (2002). The genome sequence of the malaria mosquito Anopheles gambiae. Science, 298(5591), 129–149.
Cameron, J. E., Yin, Q., Fewell, C., Lacey, M., Mcbride, J., Wang, X., et al. (2008). The Epsteinn–Barr virus latent membrane protein 1 (LMP1) induces cellular miRNA-146a, a modulator of lymphocyte signaling pathways. Journal of Virology, 82(4), 1946–1958.
Pedersen, I. M., Cheng, G., Wieland, S., Volinia, S., Croce, C. M., Chisari, F. V., et al. (2007). Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature, 449(7164), 919–922.
Xiao, C., & Rajewsky, K. (2009). MicroRNA control in the immune system: basic principles. Cell, 136(1), 26–36.
Lei, X., Bai, Z., Ye, F., Xie, J., Kim, C. G., Huang, Y., et al. (2010). Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol, 12(2), 193–199.
Zeiner, G. M., Norman, K. L., Thomson, J. M., Hammond, S. M., & Boothroyd, J. C. (2010). Toxoplasma gondii infection specifically increases the levels of key host microRNAs. PLoS ONE, 5(1), e8742.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., & Lipman, D. J. (1990). Basic local alignment search tool. J Mol Biol, 215(3), 403–410.
Thompson, J. D., Higgins, D. G., & Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties, and weight matrix choice. Nucleic Acids Res, 22(22), 4673–4680.
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res, 25(17), 3389–3402.
Zhang, B., Pan, X., & Anderson, T. A. (2006). Identification of 188 conserved maize microRNAs and their targets. FEBS Lett, 580(15), 3753–3762.
Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A., & Eddy, S. R. (2003). Rfam: an RNA family database. Nucleic Acids Res, 31(1), 439–441.
Tamura, K., Dudley, J., Nei, M., & Kumar, S. (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol, 24(8), 1596–1599.
Tamura, K., Nei, M., & Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA, 101(30), 11030–11035.
Chen, S., Zhang, A., Blyn, L. B., & Storz, G. (2004). MicC, a second small-RNA regulator of Omp protein expression in Escherichia coli. J Bacteriol, 186(20), 6689–6697.
Esau, C., Kang, X. L., Peralta, E., Hanson, E., Marcusson, E. G., Ravichandran, L. V., et al. (2004). MicroRNA-143 regulates adipocyte differentiation. J Biol Chem, 279(50), 52361–52365.
Zhao, F., Xuan, Z., Liu, L., & Zhang, M. Q. (2005). TRED: a transcriptional regulatory element database and a platform for in silico gene regulation studies. Nucleic Acids Res, 33, D103–D107.
Poy, M. N., Eliasson, L., Krutzfeldt, J., Kuwajima, S., Ma, X., Macdonald, P. E., et al. (2004). A pancreatic islet-specific microRNA regulates insulin secretion. Nature, 432(7014), 226–230.
Lecellier, C., Dunoyer, P., Arar, K., Lehmann-Che, J., Eyquem, S., Himber, C., et al. (2005). A cellular microRNA mediates antiviral defense in human cells. Science, 308(5721), 557–560.
Martello, G., Rosato, A., Ferrari, F., Manfrin, A., Cordenonsi, M., Dupont, S., et al. (2010). A microRNA targeting dicer for metastasis control. Cell, 141(7), 1195–1207.
Chatterjee, R., & Chaudhuri, K. (2006). An approach for the identification of microRNA with an application to Anopheles gambiae. Acta Biochim Pol, 53(2), 303–309.
Hackl, M., Jadhav, V., Jakobi, T., Rupp, O., Brinkrolf, K., Goesmann, A., et al. (2012). Computational identification of microRNA gene loci and precursor microRNA sequences in CHO cell lines. J Biotechnol, 158(3), 151–155.
Mead, E. A., & Tu, Z. (2008). Cloning, characterization, and expression of microRNAs from the Asian malaria mosquito, Anopheles stephensi. BMC Genomics, 9, 244. doi:10.1186/1471-2164-9-244.
Hussain, M., Frentiu, F. D., Moreira, L. A., O’Neill, S. L., & Asgari, S. (2011). Wolbachia uses host microRNAs to manipulate host gene expression and facilitate colonization of the dengue vector Aedes aegypti. Proc Natl Acad Sci USA, 108(22), 9250–9255.
Winter, F., Edaye, S., Huttenhofer, A., & Brunel, C. (2007). Anopheles gambiae miRNAs as actors of defense reaction against Plasmodium invasion. Nucleic Acids Res, 35(20), 6953–6962.
Skalsky, R., Vanlandingham, D. L., Scholle, F., Higgs, S., & Cullen, B. R. (2010). Identification of microRNAs expressed in two mosquito vectors, Aedes albopictus and Culex quinquefasciatus. BMC Genomics, 11, 119. doi:10.1186/1471-2164-11-119.
Acknowledgments
All the authors are thankful to the Pondicherry Centre for Biological centre (PCBS) for providing the necessary facility to carry out the work. Financial support as startup loan from State Bank of India (RASMECC), Pondicherry, India to establish the institute is also gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Thirugnanasambantham, K., Hairul-Islam, V.I., Saravanan, S. et al. Computational Approach for Identification of Anopheles gambiae miRNA Involved in Modulation of Host Immune Response. Appl Biochem Biotechnol 170, 281–291 (2013). https://doi.org/10.1007/s12010-013-0183-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-013-0183-5