
Abstract
Cancer is one of the killer diseases in humans and needs alternate curative measures despite recent improvement in modern treatment modalities. Oncolytic virotherapy seems to be a promising nonconventional way to treat cancers. Newcastle disease virus (NDV), a poultry virus, is nonpathogenic to human and domestic animals and has a long history of being used in oncotherapy research in several preclinical studies. The ability of NDV to successfully infect and destroy cancer cells is dependent on the strain and the pathotype of the virus. Adaptation of viruses to heterologous hosts without losing its replicative and oncolytic potential is prerequisite for use as cancer virotherapeutics. In the present study, velogenic NDV was adapted for replication in HeLa cells, and its cytotoxic potential was evaluated by observing morphological, biochemical, and nuclear landmarks of apoptosis. Our results indicated that the NDV-induced apoptosis in HeLa cells was dependent on upregulation of TNF-related apoptosis-inducing ligand (TRAIL) and caspases activation. Different determinants of apoptosis evaluated in the present study indicated that this strain could be a promising candidate for cancer therapy in future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Conventional chemo-/radiotherapy has many limitations and failed to cure many types of cancers in humans and animals [1, 2]; thus, there is an urgent need to develop an alternative approach for cancer therapy. Viruses are one of the nonconventional treatment modalities for cancers. The idea of using viruses for the treatment of cancer is not new. In the last two decades, advances in the field of molecular biology, tumor biology and host–pathogen interaction made possible the clinical evaluation of different viruses to combat cancer. Oncolytic virotherapy uses replication-competent viruses to selectively infect and kill cancer cells while leaving normal cells intact [3].
Newcastle disease virus (NDV), a poultry virus, is nonpathogenic to animals including humans. It belongs to the genus Avulavirus of the family Paramyxoviridae [4]. Newcastle disease viruses are grouped into three pathotypes, i.e., lentogenic, mesogenic, and velogenic strains depending on the severity of the disease they cause. The lentogenic strain causes mild or unapparent respiratory disease, whereas the mesogenic strain produces respiratory and nervous signs with moderate mortality. The velogenic strains are further classified as viscerotropic or neurotropic velogenic strain depending upon site of affection, causing severe intestinal lesions or neurological disease, respectively, resulting in mortality up to 100 % [5].
NDV has been studied extensively by many researchers for its inherent oncolytic potential [6, 7]. Different lentogenic and mesogenic strains of NDV like Cassel's 73-T, MTH 68/H, Ulster, Italien, Rokin, Lasota IV, PV 701 (MK107), and Malaysian isolates (AF2240, 01/C, Ijuk, S, F, and V4) have been studied (in vitro, in vivo, and in preclinical trials in cancer patients) as anticancer agents since 1950s [2, 8–16].
In clinical studies, the first lentogenic strain of NDV evaluated for cancer therapy was NDV-HUJ. It was derived from the parental NDV Hitchner B1 strain, which was attenuated (lentogenic) due to multiple passages in 10- to 11-day embryonated chicken eggs. Clinical evaluation of this lentogenic strain in patients with recurrent glioblastoma multiforme has been performed with encouraging results [7]. This strain induced cell death in C6 and RG2 rat malignant glioma cells, chemoresistant advanced melanoma cells and fibrosarcoma HT 1080 cells, but not in normal fibroblast MS5 cells [7]. However, NDV-HUJ did not show specificity for cancer cells when administered to human cancer patients systemically [1]. On the other hand, LaSota vaccine strain, another lentogenic NDV, has been shown to induce tumor regression in colorectal cancer patients [17]. Further, Italien, a lytic strain of NDV has also been shown to destroy several human tumor cells as well as cancer xenograft model in nude mice [6].
The three major mesogenic strains of NDV that have been evaluated in clinical studies are 73-T, MTH-68/H, and PV701 (MK109). 73 T, derived from MD 202, has been shown to induce apoptosis in mouse Ehrlich ascites carcinoma and also in many human tumor cells including HeLa cells [8]. This strain was used as viral oncolysate to augment antitumor immunologic responses against cancers. It had also been documented to cause regression in human tumor xenograft model. MTH-68/H, a derivative of Hertfordshire strain (Herts'33) isolated from England in 1933, has been known to cause regression of various human tumor cell lines and has also shown favorable results in patients with advanced tumor when administered by nasal inhalation [13, 18]. PV701, a replication-competent, naturally attenuated, nonrecombinant, oncolytic strain of NDV, caused regression of tumor xenografts after intravenous administration. However, desired responses were achieved only at higher dose levels [10]. Preclinical efficacy was shown in tumor patients with certain side effects like fluelike symptoms, tumor site-specific adverse events, and infusion reactions [15]. These side effects were minimized by repeated doses and potential tolerability of PV701 improved by slowing the i.v. infusion rate [19]. The Malaysian isolate of NDV, Af2240, has been shown to cause death of brain tumor cell line (glioblastoma multiforme) through apoptosis [16].
The oncolytic activity of NDV is either by a direct cytolytic effect from virus replication or by an indirect effect by syncytia formation and apoptosis depending on the tumor cells [20]. Immunomodulation also contribute to the in vivo oncolytic effect of NDV [6]. It kills tumor cells [21–23] and causes immune stimulation through induction of cytokines such as IFN-α, IFN-β, TNF-α, and IL-1 [18]. NDV exploits defects in interferon signaling pathway and mutated p53 expression in tumor cells to selectively kill the cells [24]; however, a newer mechanism has also been suggested [25]. Replication-competent NDV strains are able to replicate and produce viable progeny virus, which can infect and kill adjacent tumor cells. Virulent NDV pathotypes which usually do not affect animals including humans seem to be potential candidates for cancer therapy. As the virus-induced effects are cell line-dependent [26], it is important to study the outcome of infection in different cell lines to develop NDV-based viral therapeutics for the treatment of cancer. The prerequisite for the experimental investigation of viruses as an anticancer agent are their adaptation and active replication in the host system. In the present study, the NDV velogenic (virulent) strain has been adapted to replicate in the HeLa cells, and its cytotoxic potential and pathways of programmed cell death were evaluated by different apoptosis assays, which may be useful in developing viral therapeutics for cancer.
Materials and Methods
Virus
Chicken embryonic fibroblasts (CEFs) passaged velogenic NDV (UP1/93 [27] was used.
Cells
Established HeLa cell line procured from the National Centre for Cell Sciences (NCCS), Pune, India was maintained in DMEM supplemented with 10 % fetal bovine serum (FBS) and antibiotics (gentamycin 20 μg/ml).
Adaptation of NDV to HeLa Cells
HeLa cells were grown to 60–70 % confluency in a 25-cm2 tissue culture flask (Nunc, Denmark) infected with 1 ml of NDV parental stock (diluted 1:20 in DMEM). Cells along with NDV were incubated at 37 °C for 1 h with intermittent shaking. Thereafter, 6 ml DMEM containing 2 % FBS was added and it was again incubated at 37 °C under 5 % CO2. Mock control was prepared similarly without virus inoculum. A monolayer of infected and mock cells was observed under an inverted microscope at a 12-h interval for cytopathic effects (CPE) like cell shrinkage, cell rounding, detachment of cells from culture flask, etc. At 96 h of infection, the passage 1 (P. no. 1) virus was harvested by freezing (−20 °C) and thawing (25 °C) twice. Harvested virus (50 μl) was used to infect fresh 60–70 % confluent HeLa cells again as described earlier. Similarly, the virus was passaged 13 times, and CPE was observed carefully during all passages. HeLa-adapted virus (P. no. 5) was assayed for its growth kinetics and determinants of NDV-induced apoptosis.
Confirmation of NDV in HeLa Cells by Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
RT-PCR was carried out using total RNA extracted from NDV-infected cells using TRIzol reagent (Invitrogen, USA). Briefly, at 60 hpi (hours postinfection), culture supernatant was discarded and TRIzol (1 ml) reagent was added to the monolayer, and RNA was extracted as described by the manufacturer. The cDNA from total RNA was synthesized using random hexamer primers and MMLV-RT as described previously [27]. PCR was carried out using NDV HN gene-specific primers (forward 5′-gagaatgaggaaagagaagcaaagaac-3′ and reverse 5′-gccagcggggattcaagag-3′ with an expected product length of 253 bp. The PCR was carried out in a 25-μl reaction volume containing 1× Taq DNA polymerase buffer, 1.5 mM MgCl2, 200 μM of dNTPs mix, 10 pmol of each forward and reverse primers and 1.5 U of Taq DNA polymerase (Promega, USA). The PCR reaction was carried out in a thermal cycler with initial denaturation at 95 °C for 5 min followed by 35 cycles each of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s with a final extension at 72 °C for 5 min. PCR product was analyzed on 2 % agarose gel and visualized by ethidium bromide staining under gel documentation system (Alpha Innotech, Cell Biosciences).
Determination of NDV Growth Kinetics in HeLa Cells by Real-Time PCR
Relative quantification of virus load in HeLa cells was determined by real-time PCR using the aforementioned HN-specific primers. The reaction was carried out using a commercial HotStart-ITSYBR Green qPCR Master mix (USB, USA) as described by the manufacturer. GAPDH primers [28] were used for normalization. A melting curve analysis was performed to know the specificity of qPCR. For the test gene and endogenous control standards, tenfold serial dilutions were run in the study to estimate the efficiency of PCR, and the percentage efficiency ranged between 90 and 100 %. The relative expression of each sample was calculated using the 2−ΔΔCT method [29]. Results were analyzed and shown as fold change (log10 relative quantification) relative to the control group. An analysis of variance with Dunett's test was used to compare the different groups with the control (JMP 8.0). The data are mean triplicate of the three experiments. Differences were considered significant at P ≤ 0.01.
Hemagglutination (HA) and Hemagglutination Inhibition (HI)
HA and HI of HeLa-adapted NDV (P. no. 5) were performed using 1 % chicken RBC as described earlier [30]. The virus was confirmed by HI determination using NDV antiserum. For this, serial twofold dilutions of serum were made in 25 μl volume, and 4 HA unit of adapted NDV (P. no. 5) in 25 μl was added to each well. A plate was incubated at room temperature for 30 min and then 25 μl of 1 % (v/v) chicken RBCs were added to each well, mixed by gentle shaking, and incubated for 40 min at room temperature.
Apoptosis Studies I
A comparative analysis of parental NDV and NDV (P. no. 5) was performed along with mock- and staurosporin-treated HeLa cells.
-
1.
Cell viability assay: Viability of HeLa cells at 12, 24, 36, 48, 60, 72, 84, and 96 hpi was determined by trypan blue dye exclusion assay using an automated cell counter (Countess, Invitrogen, USA). Briefly, cell monolayer (60–70 % confluency) was infected with 0.01 and 0.1 multiplicity of infection (moi) of NDV in 50 μl of DMEM and incubated at 37 °C with intermittent shaking for adsorption. Cells were collected by mild trypsinization, washed twice with bovine serum albumin (PBS) containing 1 % FBS, and resuspended in 200 μl of PBS. Equal amount of trypan blue dye was mixed with the cell suspension, and total cell population having viable (unstained) and dead cells (dead) was counted. Data are reported as mean value ± SEM of quadruplicate observations. P values of <0.05 were considered statistically significant.
-
2.
Annexin V binding assay: Annexin V is a phospholipid-binding protein that has a high affinity for phosphatidyl serine (PS) [31]. PS translocation to the outer leaflet of plasma membrane was detected by annexin V staining using the Vybrant Apoptosis assay kit, (Invitrogen, USA) following the manufacturer's instruction. Cells were analyzed in a flow cytometry using FL1 (530 nm) bandpass filters of FACS Calibur (Becton Dickinson, USA), and data were analyzed by the CellQuest software (BD, USA).
-
3.
Propidium iodide (PI) staining: Hypodiploid population of cells was detected by a flow cytometer after PI staining [32]. Briefly, both floating and adherent cells (1 × 106) were harvested, washed once with PBS containing 1 % FBS, and fixed using 70 % ice chilled ethanol added dropwise with gentle mixing. Cells were stored at −20 °C overnight. After washing the cells twice with PBS, the cell pellet was resuspended in 1 ml hypotonic buffer (sodium citrate 0.1 %, Triton-X 0.1 %, PBS 100 ml, PI 50 μg/ml, RNase 20 μg/ml) and incubated for 15 min at 37 °C in water bath. Cells were immediately placed on ice. A total of 10,000 cells were acquired using FL2 filter of the flow cytometer and analyzed using the CellQuest software.
-
4.
Hoechst staining: For visualization of chromatin condensation in NDV-infected HeLa cells, Hoechst staining was performed as mentioned elsewhere [33]. Cells were visualized with a fluorescence microscope (Nikon, Japan) under 40× objective. Three independent observations were performed, and each time, four fields were observed for changes in chromatin morphology.
-
5.
Terminal deoxynucleotidyl transferase-mediated dUTP nick labeling (TUNEL) assay: TUNEL assay was performed with APO-BrdU™ tunnel assay kit (Invitrogen) following the protocol of the manufacturer. Briefly, HeLa cells (1 × 105) were seeded in a 6-well plate and incubated at 37 °C under 5 % CO2. NDV (0.01 moi)-infected HeLa cells were collected at 24, 48, and 72hpi by mild trypsinization, washed once with PBS containing 1 % FBS, and processed following the manufacturer's instructions. A total of 10,000 cells were examined using an FL2 channel of the flow cytometer and analyzed using the CellQuest software.
-
6.
DNA fragmentation assay: A DNA fragmentation assay was performed as mentioned elsewhere [34]. The DNA ladder was resolved on 2 % agarose gel electrophoresis and visualized with ethidium bromide staining under a UV illuminator.
Apoptosis Studies II
HeLa-adapted NDV (P. no. 5) was further studied for different determinants of apoptosis.
-
1.
Detection of TNF-related apoptosis-inducing ligand (TRAIL) expression by fluorescence microscopy: HeLa cells (1 × 104) were seeded in a 4-well plate and incubated at 37 °C under 5 % CO2. Cells grown to 60–70 % confluency were infected with NDV (0.1 moi). The cells (48 hpi) were fixed in 3 % PFA, and permeabilized by treating with 0.2 % Triton X-100 (Merck, Germany). The permeabilized cells were incubated with 2 % PBS. Cells were then incubated at room temperature with TRAIL-specific primary antibody (Cat. # AB16957, Millipore, USA) at 1:50 dilution for 1 h. Cells were washed twice with PBS and incubated with anti-species FITC-conjugated secondary antibody (Cat. # K0211703, goat polyclonal anti-rabbit IgG, FITC KOMABIOTECH, South Korea) at 1:100 dilution at room temperature. The stained cells were visualized under 40× objective in a fluorescence microscope.
-
2.
Detection of mitochondrial membrane potential (ΔΨm): Alteration in mitochondrial membrane potential caused by NDV (0.01 moi) infection to HeLa cells was detected using 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine (Mitoprobe™ JC-1 assay kit, Invitrogen, USA) by flow cytometry following the instructions of the manufacturer. Cells (1 × 106) were finally resuspended in 500 μl of PBS and analyzed in a flow cytometer with 488 nm excitation using FL1and FL2 filters. The chemical uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP) provided along with the kit (25 μm for 30 min) was used as positive control for the ∆Ψm decrease. The mean fluorescence intensity of 10,000 cells was captured for each sample and corrected for autofluorescence obtained from samples of unlabeled cells. Three independent experiments were performed.
-
3.
Flow cytometric estimation of caspase 8, caspase 9, and caspase 3: Quantitative measurement of caspase 8, caspase 9, and caspase 3 expressions in NDV (0.1 moi)-infected HeLa cells were done with anti-caspases antibodies using a flow cytometer. Fixed and permeabilized HeLa cells were incubated separately with primary antibodies (Cat. #04-573; anti-caspase 8 rabbit monoclonal, cat. #04-444; anti-caspase 9 rabbit monoclonal, and cat. #04-439; anti-caspase 3 rabbit monoclonal, Millipore) for 1 h at room temperature. After washing twice with PBS, cells were resuspended in 1 ml incubation buffer (0.5 % BSA in PBS) followed by incubation with FITC-conjugated secondary antibody (Cat. #K0211703, anti-rabbit IgG, FITC; Kom Biotech, South Korea) for 1 h at room temperature. Cells were washed twice with PBS and resuspended in 500 μl incubation buffer (0.5 % BSA in PBS). A total of 10,000 cells were examined using the FL1 channel of a flow cytometer. Caspase 8, caspase 9, and caspase 3-positive cells were analyzed using the CellQuest software.
Results
NDV was Adapted to Grow in HeLa Cells
NDV velogenic isolate was adapted to grow in HeLa cells. During three passages, no gross cytopathic changes were observed. In the fourth passage, some changes including cell rounding and vacuolation were observed after 24 h of infection. At passage 5, HeLa cells started showing cytopathic changes which included rounding of the cells, granulation and fusion of cells, and cell detachment from monolayer (Fig. 1). Further passages revealed similar cytopathic changes. Presence of NDV RNA in HeLa cells was observed by RT-PCR of total RNA harvested from NDV-infected HeLa cells after 48 hpi (Fig. 2).

Phase contrast microscopic view of NDV-induced cytopathic effects on HeLa cells, 48 hpi. a Healthy control, b parental NDV-infected HeLa cells, c HeLa-adapted NDV (P. no. 5)-infected cells. photographs are shown at 400× magnification

Reverse transcriptase-PCR amplification (253-bp gene fragment of HN gene) of viral RNA in HeLa cells (48 hpi). Lane 1 parental stock (P. no. 1), lane 2 P. no. 2, lane 3 P. no. 3, lane 4 P. no. 4, lane 5 P. no. 5, lane 6 positive control, lane 7 100-bp DNA ladder, lane 8 mock control. One representative gel is shown
NDV Growth Kinetics in HeLa Cells was Determined by Real-Time PCR
Real-time quantitation of HN mRNA level in parental NDV and HeLa-passaged NDV (P. no. 5)-infected HeLa cells were performed. NDV growth kinetics of HeLa-passaged NDV (P. no. 5) was significantly enhanced as compared to parental NDV (Fig. 3). The fold values of HN mRNA at 12 h after infection in parental NDV and HeLa-adapted NDV were 0.152 ± 0.117 and 3.380 ± 0.159 which were increased to 1.923 ± 0.176 and 5.445 ± 0.088, respectively, at 96 h after infection. These finding suggested that the infectivity of the virus increased as it adapted to grow in the HeLa cell microenvironment.

Determination of NDV growth kinetics in HeLa cells by real-time PCR: HeLa cells were mock-, NDV parental (0.1 moi)-, and NDV (P. no. 5) [0.1 moi]-infected. Total RNA was extracted from cells at 12, 24, 36, 48, 60, 72, 84, and 96 hpi and was used to quantitate viral RNA using HN gene-specific nested primers. GAPDH was used as internal control. The mRNA level for each gene is expressed as log10 relative quantification (RQ) unit, relative to the control group. Error bars indicate the calculated maximum (RQMax) and minimum (RQMin) expression levels and represent the standard error (SE) of the mean of the expression levels. The error bars are based on an RQMin/Max of the 95 % confidence level. Differences were considered significant at P ≤ 0.001 (two asterisks) and P ≤ 0.01 (asterisk). One representative result is shown
HA and HI of HeLa-Adapted NDV was Determined
The highest dilution of virus giving complete HA was 210. NDV (4HAU) was used for inhibition of hyperimmune sera against NDV. The highest dilution of serum causing complete inhibition was 212.
NDV Infection Leads to Loss of Cell Viability in HeLa Cells
A significant (P < 0.05) decrease in cell viability was observed in NDV (P. no. 5)-infected cells as compared to mock- and parental NDV-infected cells as judged by trypan blue dye exclusion test. Cell viability was negatively correlated with time and dose of NDV. Viability of HeLa cells at the time of NDV infection was 95.667 ± 0.333 %. After 96 hpi, viability percentages of NDV (P. no. 5)-infected cells were 40.667 ± 0.667 (0.01 moi) and 30.667 ± 1.2019 % (0.1 moi); however, viability of parental NDV and mock-infected cells were 69 ± 0.577 and 67.33 ± 0.667 %, respectively. The viability percentage of staurosporin (30 nM)-treated HeLa cell (96 hpi) was 49.667 ± 1.453 (Fig. 4).

Cell viability assay: HeLa cells were infected with NDV (P. no. 5) [0.01 and 0.1 moi] along with controls [parental NDV (0.1 moi), mock, and staurosporin (30 nM)]. Cell viability was determined by trypan blue dye exclusion assay using an automated cell counter at 12, 24, 36, 48, 60, 72, 84, and 96 hpi. Mean (four independent observations) values of viability percentages were plotted at different time intervals. Differences were considered significant at P < 0.05
Translocation of Phosphatidylserine (PS) was Observed in NDV-Infected HeLa Cells
Apoptosis can be assessed by evaluating annexin V binding to PS that externalized early in the process of apoptosis. A significant (P < 0.05) increase in percent annexin-positive cell population was observed in NDV (P. no. 5)-infected HeLa cells. The percent annexin V-positive cells at 48 hpi in mock-, parental NDV-, and NDV (P. no. 5)-infected and staurosporin-treated cells were 19.14, 39.64, 69.86, and 51.42 %, respectively (Fig. 5).

Annexin V binding assay: HeLa cells were infected with NDV (0.1 moi) and cells (48 hpi) were analyzed by flow cytometry. A total of 10,000 cells were acquired and data were analyzed by the CellQuest software. Histograms represent percent annexin V-positive cells: i mock infected, ii parental NDV infected, iii HeLa-adapted NDV (P. no. 5), and iv staurosporin (30 nM)-treated cells. Results from one out of three independent experiments are shown
NDV Causes Increase in Sub-G1 Population of Cells
Hypodiploid count of NDV-infected HeLa cells was estimated by flow cytometry. Our result indicated an increase in sub-G1 peak in NDV (0.1 moi)-infected cells at 48 hpi as compared with mock control. The percent PI-positive cells in mock control, parental NDV, and NDV (P. no. 5)-infected and staurosporin (30 nM)-treated cells were 30.72, 39.12, 64.79, and 66.78 %, respectively (Fig. 6).

Determination of sub-G1 peak by PI staining: HeLa cells were infected with NDV (0.1 moi) and cells (48 hpi) were processed for flow cytometry, and data were captured on an FL2 channel of a flow cytometer. A total of 10,000 cells were acquired and data were analyzed by the CellQuest software. Percentages of sub-G1 population of cells are shown in histograms: i mock, ii parental NDV, iii NDV (P. no. 5), and iv staurosporin (30 nM). Results from one out of three independent experiments are shown
Nuclear Condensation and Fragmentation was Observed in NDV-Infected HeLa Cells
Hoechst staining revealed condensed and fragmented nuclei in NDV (P. no. 5)-infected cells which was absent in mock-infected cells (Fig. 7a). Similarly, condensed and fragmented nuclei were observed in staurosporine (30 nM)-treated HeLa cells used as positive control for apoptosis. Furthermore, internucleosomal DNA fragmentation ability of NDV (0.01 moi) in HeLa cells was detected by TUNEL assay at 24, 48, and 72 hpi and by agarose gel electrophoresis of DNA on 2.0 % agarose gels. TUNEL assay (Fig. 7b) indicated a significant (P < 0.05) increase in DNA fragmentation of NDV (P. no. 5)-infected HeLa cells as compared with mock- and parental NDV-infected cells. DNA analysis displayed a characteristic ladder pattern of discontinuous DNA fragments (Fig. 7c), whereas no such fragmentation was noticed in mock control and NDV parental-infected cells.

Determination of changes in chromatin material caused by NDV infection in HeLa cells. a Hoechst staining: NDV-infected HeLa cells (48 hpi) were processed for Hoechst staining. i Mock control, ii cells infected with 0.1 moi of parental NDV, iii cells infected with 0.1 moi of NDV (P. no. 5), and iv cells treated with staurosporin (30 nM). b DNA fragmentation analysis by terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) assay: HeLa cells were infected with NDV (0.01 moi) at 24, 48, and 72 hpi, cells were processed for flow cytometry, and data were captured on an FL1 channel of a flow cytometer. A total of 10,000 cells were acquired, and data were analyzed by the CellQuest software. Percent TUNEL-positive cells (24, 48, and 72 hpi) are shown by bar diagrams, and data were analyzed by Tukey–Kramer HSD, SAS. Differences were considered significant at P <0.05. c Agarose gel electrophoresis of fragmented DNA in NDV (0.1 moi; 48 hpi)-infected HeLa cells: lane 1 NDV (P. no. 5), lane 2 staurosporin (30 nM), lane 3 parental NDV (0.1 moi), lane 4 mock control, lane 5 healthy cells, lane 6 100-bp DNA ladder. One representative gel is shown. a All photographs are shown at 400× magnification
NDV Infection Upregulated TRAIL Expression in HeLa Cells
A fluorescent microscopic examination of NDV-infected HeLa cells was performed to observe surface expression of the TRAIL. Results indicated that TRAIL acts as one of the effector molecules which were upregulated in NDV-infected HeLa cells (Fig. 8).

Immunofluorescent localization of TRAIL expression. HeLa cells were infected with NDV (0.1 moi) and cells (48 hpi) were processed for fluorescent microscopy: i mock control and ii NDV (P. no. 5)-infected cells. Representative photographs are shown at 400× magnification
Disruption of Mitochondrial Membrane Potential was Evident in NDV Infection
A significant (P < 0.05) increase in red to green shift were noticed in NDV-infected HeLa cells as compared with mock control. Mitochondrial depolarization was observed at 24 hpi in HeLa cells infected with 0.1 moi of NDV. This red to green shift in fluorescence increased at 48 hpi; however, it was decreased at 72 hpi. The result clearly indicated that mitochondrial depolarization occurred in NDV-infected HeLa cells (Fig. 9).

Flow cytometric analysis of alteration in mitochondrial membrane depolarization (ΔΨm) by JC-1 dye: HeLa cells were infected with NDV (0.01 moi) and cells (24, 48, and 72 hpi) were processed for flow cytometry. A total of 10,000 cells were acquired and percentages of red (detected by FL2) and green (detected by FL1) fluorescence-positive cells were determined by the CellQuest software. Percentages of green fluorescent-positive cells are shown. These dot plots are from one out of three independent experiments
Expression of Caspases was Observed in NDV-Infected HeLa Cells
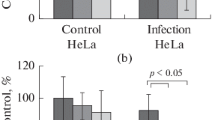
Caspase 8, caspase 9, and caspase 3 expressions were measured by flow cytometric analysis of NDV (0.1 moi)-infected HeLa cells at 48 hpi. The values of percent caspase-positive cells were 53.3867 ± 1.3153, 32.11 ± 3.5234, and 14.6367 ± 1.8566 in NDV-infected HeLa cells and 10.26 ± 0.3998, 4.3764 ± 0.1351, and 1.8033 ± 0.4893 in mock control for caspase 8, caspase 9, and caspase 3, respectively. As evident from Fig. 10, significant (P < 0.05) increase in caspases activity was observed in NDV-infected HeLa cells as compared to mock control suggesting that caspases play an important role in NDV-induced cell death.

A flow cytometric analysis of expression of caspase 8, caspase 9, and caspase 3: HeLa cells were infected with NDV (0.1 moi) and cells (48 hpi) were processed for flow cytometry. A total of 10,000 cells were acquired (FL1) for fluorescence-positive cells. Data were analyzed by the CellQuest software. Mean (three independent observations) values of percent caspase-positive cells in mock- and NDV (P. no. 5)-infected cells are shown by a bar diagram. Data are analyzed by a t test. Differences were considered significant at P ≤ 0.05
Discussion
NDV has potential to be used as therapeutics for cancers, which the virus achieves through apoptosis [35]. Several studies have been conducted worldwide to establish NDV strains as an effective virotherapeutics for cancer. In the present study, velogenic NDV was adapted to grow in HeLa cells (a human tumor cell line) with retained apoptotic potential. In the present study, HeLa cell-adapted NDV was used at P. no. 5 as cytopathic changes were stabilized at this passage onward. Cells undergoing apoptosis were identified by morphological changes which include typical apoptotic features such as cell shrinkage, rounding, detachment from the monolayer, and formation of giant cells (syncytium). Parental NDV (unadapted to HeLa cells)-infected HeLa cells appear normal in its gross morphology; however, rounding in around 5 % more cells was noticed as compared with mock control cells. The formation of large multinucleated cells (syncytia) observed in HeLa-adapted NDV (P. no. 5)-infected HeLa cells approves its oncolytic potential as discussed earlier [36]. More than nine passages (from passages 6 to 15) of NDV had been given in HeLa cells without alteration in its cytotoxicity potential. There are several other reports suggesting that NDV replicates in human cell lines and induces apoptosis [37–40]. Several previous workers including those in our laboratory have adapted NDV to Vero cell lines to study molecular mechanism of apoptosis and virus pathogenesis [41, 42].
NDV presence in HeLa cells were confirmed by RT-PCR by observing the HN gene fragment-specific PCR product on agarose gel electrophoresis. Viral RNA was detected in all five passages (P. nos. 1 to 5) studied. The growth kinetics of NDV in HeLa cells were assessed by real-time PCR. For this purpose, total RNA was isolated from the NDV-infected cells. Quantitative PCR analysis of parental NDV and HeLa-passaged NDV (P. no. 5) in HeLa cells suggested clearly that adapted virus has better growth kinetics as compared to parental NDV. However, a linear increase in NDV mRNA has not been observed. The most plausible explanation could be the differences in virus progeny release and cell growth kinetics. At some point of time (60 and 84 hpi in this case), there might be virus progeny released in culture supernatants which was not considered for quantification. Quantitative PCR methods could be a better representation of viral load and was helpful in estimating the virus growth kinetics of NDV in HeLa cells. Several other workers used real-time quantitation (relative and absolute) for determination of virus load in host tissues [43–45].
Multiplication of NDV in HeLa cells was also confirmed by HA and HI test. High titer of virus in HeLa cells indicated that NDV (P. no. 5) replicates well in the HeLa cells. Viruses cause stress to the cell metabolism and changes in cell membrane integrity. Trypan blue dye exclusion assay, which takes advantage of changes in membrane permeability, is used to assess the same, as viable cells do not uptake trypan blue and remains unstained [46]. The present study indicated that viability of HeLa cells infected with NDV (0.1 moi) was decreased significantly (P < 0.05) as compared to mock-infected cells at 96 hpi. Significant (P < 0.05) differences in viability percentage between parental NDV and NDV (P. no. 5) were observed. It seems that adaptation of virus to the cell microenvironment enhances cytotoxic potential of the virus. Further, increase in moi decreases the viability of HeLa cells. Trypan blue dye exclusion assay did not show significant (P < 0.05) differences in viability of parental NDV- and mock-infected cells.
There are several modalities of cell death in multicellular organisms which includes apoptosis, necrosis, autophagy, mitotic catastrophe, etc. Apoptosis is considered to be one of the safer modes of cell death causing very little bystander side effects [47]. It is characterized by distinct morphological, biochemical, and nuclear landmarks such as cytoplasmic shrinkage, loss of plasma membrane integrity, alteration in mitochondrial membrane potential, perinuclear redistribution of mitochondria, nuclear condensation, oligonucleosomal DNA fragmentation, and finally, separation of the nucleus into apoptotic bodies [48]. Another form of cell death, e.g., necrosis is often associated with many unwarranted side effects [49]. NDV-induced HeLa cell death is due to apoptosis and not to necrosis. To prove this hypothesis, annexin V binding assay, PI staining, Hoechst staining, nuclear DNA fragmentation, TRAIL expression, disruption of mitochondrial membrane potential, and expression of caspases in NDV-infected HeLa cells were determined.
A hallmark of apoptotic process is the loss of phospholipid asymmetry of the plasma membrane. Phosphatidyleserine (PS), which is confined to the inner leaflet of the plasma membrane in healthy cells, externalizes to the outer plasma membrane indicating that cells are programmed to die [50]. A significant (P < 0.05) increase in PS-positive cells in HeLa-adapted NDV (P. no. 5) indicated that cells entered into apoptosis. Further, NDV-infected HeLa cells were quantified for sub-G1 population as an apoptosis index. PI has ability to bind DNA and fluoresces under UV light. During apoptosis, cellular DNA gets fragmented which comes out from the apoptotic or necrotic cells, and fluorescence is reduced. More cell population in this region suggested an increase in apoptosis (reduced fluorescence) and vice versa. Our result clearly indicated that sub-G1 peaks were increased in NDV (P. no. 5)-infected HeLa cells as compared with parental NDV and mock control. Further, apoptosis inducing ability of parental NDV was significantly (P < 0.05) less as compared with NDV (P. no. 5).
Morphological changes in nuclear materials such as condensed and fragmented chromatin are associated with apoptotic cell death. Loss of chromosome integrity was considered to be an important determinant of mitotic death. During apoptosis, specific Ca-dependent endonucleases cleave genomic DNA and create fragments with double stranded breaks. In the present study, HeLa-passaged NDV (P. no. 5)-infected cells had shown condensed nuclei, which were absent in mock control and very few (three to four cells per field observed) in parental NDV-infected cells. Start time of DNA fragmentation, an indication of onset of apoptosis, can be assessed by TUNEL assay [51]. Significant differences in percent TUNEL-positive cells between mock and NDV infection were observed. In certain cases of apoptosis study, identification of TdT-labeled degrading DNA in the nucleus of cells is not considered sufficient to demonstrate that the cells are engaged in apoptosis [52, 53]. Therefore, DNA fragmentation ability of NDV in HeLa cells was further confirmed by agarose gel electrophoresis. TUNEL assays had also shown apoptotic DNA in parental NDV-infected HeLa cells, but the same could not be observed by gel electrophoresis indicating the need for more than one assay to demonstrate DNA fragmentation in apoptosis. DNA fragmentation is a late event during the process of apoptosis [54]. The event of nuclear fragmentation observed in NDV-infected HeLa cells is an important cellular event indicating the point of no return in apoptosis. Other workers had also suggested that oligonucleosomal laddering is an obvious indicator of apoptosis [55].
Different cytotoxicity assays indicated that HeLa-passaged NDV has more cytotoxic potential as compared with parental NDV. Apoptosis is a cellular event and associated with overexpression of several proteins and other biochemical changes like disruption in mitochondrial membrane potential [56]. Therefore, we further analyzed expression of key proteins and involvement of mitochondria in NDV (P. no. 5)-infected HeLa cells. Our results indicated that TRAIL expression is upregulated in NDV-infected HeLa cells. TRAIL, a ligand belonging to the TNF family, is known to induce cell death in various cell lines [57]. TRAIL-mediated signaling is involved in the killing of tumor cells by NDV-stimulated macrophages [58].
The key proteins that modulate the apoptotic response are represented by a family of cysteine-requiring aspartate protease called caspase. Caspases participated in intracellular signaling cascade that leads to apoptosis. Major caspase-dependent pathways are divided mainly into extrinsic and intrinsic pathways. Extrinsic pathways are mediated by death receptor-mediated signaling leading to activation of caspase 8, which is one of the initiator caspases known to be involved in the extrinsic pathway of apoptosis. Upregulation of TRAIL expression and caspase 8 in NDV-infected HeLa cells suggested that the extrinsic pathway of apoptosis play a role in NDV-induced apoptosis in HeLa cells. The intrinsic pathway involved breakdown of the mitochondrial membrane potential and release of proapoptotic effector proteins mainly cytochrome c. The collapse of electrochemical gradient across the mitochondrial membrane is one of the early events during cellular apoptosis [59]. Disruption of mitochondrial membrane potential and significant (P < 0.05) increase in caspase 9 (an initiator caspase of mitochondria-mediated pathway of apoptosis) expression in NDV-infected HeLa cells indicated the involvement of the intrinsic pathways of apoptosis. Active forms of these initiator caspases activate caspase 3 that cleaves a multitude of intracellular substrates.
The results of the present study correlate well with previous work in NDV [40]. Furthermore, this study confirmed that the velogenic NDV caused apoptosis (oncolysis) in HeLa cells and can be a potential strain for virotherapy of cancer because apoptosis (type I cell death) is considered the safest mode of cell death which causes minimum or no harm to adjacent tissues in multicellular organization.
References
Dey, M., Ulasov, I. V., & Lesniak, M. S. (2010). Cancer Letters, 289, 1–10.
Lazar, I., Yaacov, B., Shiloach, T., Eliahoo, E., Kadouri, L., Lotem, M., et al. (2010). Journal of Virology, 84(1), 639–646.
Zemp, F. J., Corredor, J. C., Lun, X., Muruve, D. A., & Forsyth, P. A. (2010). Cytokine Growth F. R, 21, 103–117.
Mayo, M. A. (2002). Archives of Virology, 147(8), 1655–1656.
Alexander, D. J. (1997). In diseases of poultry, 10th ed. (B. W. Calnek (ed.),), Iowa State University Press, Iowa, pp. 541-569.
Schirrmacher, V., Griesbach, A., & Ahlert, T. (2001). International Journal of Oncology, 18, 945–952.
Freeman, A. I., Zakay-Rones, Z., Gomori, J. M., Linetsky, E., Rasooly, L., Greenbaum, E., et al. (2006). Molecular Therapy, 13, 221–228.
Cassel, W. A., & Garret, R. E. (1965). Cancer, 18, 863–868.
Cassel, W. A., & Murray, D. R. (1992). Medicine Oncology Tumor Pharmacy, 9(4), 169–171.
Pecora, A. L., Rizvi, N., Cohen, G. I., Meropol, N. J., Sterman, D., Marshall, J. L., et al. (2002). Journal of Clinical Oncology, 20, 2251–2266.
Steiner, H. H., Bonsanto, M. M., Beckhove, P., Brysch, M., Geletneky, K., Ahmadi, R., et al. (2004). Journal of Clinical Oncology, 22(21), 4272–4281.
Csatary, L. K., Moss, R. W., Beuth, J., Torocsik, B., Szeberenyi, J., & Bakacs, T. (1999). Anticancer Research, 19, 635–638.
Csatary, L. K., Gosztonyi, G., Szeberenyi, J., Fabian, Z., Liszka, V., Bodey, B., et al. (2004). Journal of Neuro-Oncology, 67, 83–93.
Lorence, R. M., Pecora, A. L., Major, P. P., Hotte, S. J., Laurie, S. A., Roberts, M. S., et al. (2003). Current Opinion in Molecular Therapeutics, 5, 618–624.
Lorence, R. M., Roberts, M. S., O'Neil, J. D., Groene, W. S., Miller, J. A., Mueller, S. N., et al. (2007). Current Cancer Drug Targets, 7, 157–167.
Ali, R., Alabsi, A. M., Ali, A. M., Ainideris, Omar, A. R., & Khatijah, Y. (2011). International Journal of Cancer Research, 7(1), 25–35.
Liang, W., Wang, H., Sun, T. M., Yao, W. Q., Chen, L. L., Jin, Y., et al. (2003). World Journal of Gastroenterology, 9(3), 495–498.
Csatary, L. K., Eckhardt, S., Bukosza, I., Czegledi, F., Fenyvesi, C., Gergely, P., et al. (1993). Cancer Detection and Prevention, 17, 619–627.
Hotte, J. S., Lorence, R. M., Hirte, H. W., Polawski, S. R., Bamat, M. K., O'Neil, J. D., et al. (2007). Clinical Cancer Research, 13(3), 977–985.
Omar, A. R., Ali, A. I. A. M., Othman, F., Yusoff, K., Abdullah, J. M., Wali, H. S. M., et al. (2003). Malaysian Journal of Medical Sciences, 10(1), 4–12.
Reichard, K. W., Lorence, R. M., Cascino, C. J., Peeples, M. E., Walter, R. J., Fernando, M. B., et al. (1992). Journal of Surgical Research, 52, 448–453.
Bar-Eli, N., Giloh, H., Schlesinger, M., & Zakay-Rones, Z. (1996). Journal of Cancer Research and Clinical Oncology, 122, 409–415.
Yaacov, B., Eliahoo, E., Lazar, I., Ben-Shlomo, M., Greenbaum, I., Panet, A., et al. (2008). Cancer Gene Therapy, 15, 795–807.
Krishnamurthy, S., Takimoto, T., Scroggs, R. A., & Portner, A. (2006). Journal of Virology, 80, 5145–5155.
Mansour, M., Palese, P., & Zamarin, D. (2011). Journal of Virology, 84(12), 6015–6023.
Nykky, J., Tuusa, J. E., Kirjavainen, S., Vuento, M., & Gilbert, L. (2010). International Journal of Nanomedicine, 5, 417–428.
Nanthakumar, T., Kataria, R. S., Tiwari, A. K., Butchiah, G., & Kataria, J. M. (2000). Veterinary Research Communications, 24, 275–286.
Kalali, B. N., Kollisch, G., Mages, J., Muller, T., Bauer, T., Wagner, H., et al. (2008). Journal of Immunology, 181, 2694–2704.
Schmittgen, T. D., & Livak, K. J. (2008). Nature Protocols, 3(6), 1101–1108.
Available from www.oie.int/fileadmin/Home/eng/2.03.14, Accessed June 20, 2011.
Kenis, H., Genterbern, H. V., Rinia, H. A., Narula, T., & Hafstra, L. (2004). Journal of Biological Chemistry, 279, 52623–52629.
Hughes, D. and Mehmet H. (2003). Cell proliferation and apoptosis, 1st edn., (pp.341-342), BIOS Scientific, Oxford.
Blau, H. M., Chiu, C. P., & Webster, C. (1983). Cell, 32(4), 1171–1180.
Herrmann, M., Lorenz, H. M., Voll, R., Grunke, M., Woith, W., & Kalde, J. R. (1994). Nucleic Acids Research, 22(24), 5506–5507.
Ravindra, P. V., Tiwari, A. K., Ratta, B., Chaturvedi, U., Palia, S. K., & Chauhan, R. S. (2009). Virus Research, 141, 13–20.
Vigil, A., Park, M. S., Martinez, O., Chua, M. A., Xiao, S., Cros, J. F., et al. (2007). Cancer Research, 67(17), 8285–8292.
Ahlert, T., & Schirrmacher, V. (1990). Cancer Research, 50(18), 5962–5968.
Zorn, U., Dallman, I., Grosse, J., Kirchner, H., Poliwoda, H., & Atzpodien, J. (1994). Cancer Biotherapy, 9, 225–235.
Washburn, B., & Schirrmacher, V. (2002). International Journal of Oncology, 21, 85–93.
Fabian, Z., Csatary, C. M., Szeberenyi, J., & Csatary, L. K. (2007). Journal of Virology, 81, 2817–2830.
Ahamed, T., Hossain, K. M., Billah, M. M., Islam, K. M. D., Ahasan, M. M., & Islam, M. E. (2004). International Journal of Poultry Science, 3(2), 153–156.
Ravindra, P. V., Ratta, B., Chaturvedi, U., Palia, S. K., Subudhi, P. K., & Tiwari, A. K. (2008). Journal of Hellenic Veterinarian Medical Society, 59(4), 341–345.
Mendy, M. E., Kaye, S., Sande, M. V., Rayco-Solon, P., Waight, P. A., Shipton, D., et al. (2006). Virology Journal, 3, 23.
Lay, M. L. J., Lucas, R. M., Ratnamohan, M., Taylor, J., & Ponsonby, A. L. (2010). Virology Journal, 7, 252.
Li, G., Li, W., Guoc, F., Xuc, S., Zhaod, N., Chena, S., et al. (2010). Journal of Virological Methods, 165, 9–14.
Katsates, V., Petsa, A., Felesakis, A., Paparidis, Z., Nikolaidou, E., Gargani, S., et al. (2009). Laboratory Medicine, 40(9), 557–560.
Fink, S. L., & Cookson, B. T. (2005). Infection and Immunity, 73(4), 1907–1916.
Kroemer, G., Zamzami, N., & Susin, S. A. (1997). Immunology Today, 18, 44–51.
Krysko, D. V., Berghe, V., D'Herde, K., & Vandenabeele, P. (2008). Methods, 44(3), 205–221.
Vermes, I., Haanen, C., Steffens-Nakken, H., & Reutelingperger, C. (1995). Journal of Immunological Methods, 184, 39–51.
Mo, H., & Elson, C. E. (1999). Journal of Nutrition, 129, 804–813.
Kraupp, B. G., Ruttkay-Nedecky, B., Koudelka, H., Bukowska, K., Bursch, W., & Schulte-Hermann, R. (1995). Hepatology, 21(5), 1465–1468.
Heatwole V. M. (1999) In: Methods in molecular biology, vol. 115 III (Javois, L. C. ed.) Humana Press Inc. Totowa, NJ, pp. 141-148.
Kerr, J. F. R., Wyllie, A. H., & Currie, A. R. (1972). British Journal of Cancer, 26, 239–257.
Couderc, T., Guivel-Benhassine, F., Calaora, V., Gosselin, A. S., & Blondel, B. (2002). Journal of General Virology, 83, 1925–1930.
Zamzami, N., Marchetti, P., Castedo, M., Zanin, C., Vayssiere, J. L., Petit, P. X., et al. (1995). The Journal of Experimental Medicine, 181, 1661–1672.
Muhlenbeck, F., Haas, E., Schwenzer, R., Schubert, G., Grell, M., Smith, C., et al. (1998). Journal of Biological Chemistry, 273(49), 33091–33098.
Washburn, W., Weigand, M. A., Grosse-Wilde, A., Janke, M., Stahl, H., Riser, E., et al. (2003). Journal of Immunology, 170, 1814–1821.
Litz, J., Carlson, P., Warshamana-Greene, S. G., Grant, S., & Krystal, G. W. (2003). Clinical Cancer Research, 9, 4586–4594.
Acknowledgments
We are thankful to the director, Indian Veterinary Research Institute, Izatnagar for providing necessary facilities and the Department of Science and Technology (DST), Government of India for funding (Grant no. SR/SO/AS-0062/2007).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kumar, R., Tiwari, A.K., Chaturvedi, U. et al. Velogenic Newcastle Disease Virus as an Oncolytic Virotherapeutics: In Vitro Characterization. Appl Biochem Biotechnol 167, 2005–2022 (2012). https://doi.org/10.1007/s12010-012-9700-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-012-9700-1