
Abstract
Development of endoproteases, programmed to promote degradation of peptides or proteins responsible for pathogenic states, represents an attractive therapeutic strategy, since such biocatalytic agents could be directed against a potentially unlimited repertoire of extracellular proteinaceous targets. Difficulties associated with engineering enzymes with tailor-made substrate specificities have, however, hindered the discovery of proteases possessing both the efficiency and selectivity to act as therapeutics. Here, we disclose a genetic system, designed to report on site-specific proteolysis through the survival of a bacterial host, and the implementation of this method in the directed evolution of proteases with a non-native substrate preference. The high sensitivity potential of this system was established by monitoring the activity of the Tobacco Etch Virus protease (TEV-Pr) against co-expressed substrates of various recognition level and corroborated by both intracellular and cell-free assays. The genetic selection system was then used in an iterative mode with a library of TEV-Pr mutants to direct the emergence of proteases favoring a nominally poor substrate of the stringently selective protease. The retrieval of mutant enzymes displaying enhanced proteolytic properties against the non-native sequence combined with reduced recognition of the cognate hexapeptide substrate demonstrates the potential of this system for evolving proteases with improved or completely unprecedented properties.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recent advances in biotechnology have highlighted biomacromolecules as potent and selective therapeutic agents, commonly characterized by superior effectiveness and a reduced propensity for harmful side effects. Enzyme-based therapeutic strategies, in general, hold the promise of unrivalled efficacy due to the potential for both low drug-to-target ratios and exquisite specificity, with the ability to address disease targets that are inaccessible to small molecule agents [1]. Endoproteases that could be programmed to promote the degradation of peptides and proteins responsible for pathogenic states are particularly attractive as therapeutics, since such agents can be used to degrade a potentially unlimited repertoire of proteinaceous targets [2]. Several examples of proteases used as oncolytics [3], anti-inflammatory agents [4], antibacterials [5], thrombolytics [6], pro-apoptotics [7], and replacements for metabolic deficiencies, such as celiac sprue [8], have recently appeared in biomedical literature, substantiating the therapeutic promise of proteolytic enzymes as a unique class of drugs. Developing therapeutic proteases with novel substrate specificities, aimed at proteins and peptides involved in disease states, may open unprecedented opportunities in both efficiency and selectivity of pharmaceutical interventions.
Endowing proteases with tailor-made specificities is one of the principal challenges of this approach [9]. The current level of understanding of the relationship between protein structure and function has limited the extent to which desired properties can be engineered rationally [10]. Recent advances in recombinant DNA technology have, however, provided access to vast collections of sequence permutations, with an improved potential for yielding rare properties [11, 12]. Despite these advances, the major obstacle that remains is the shortage of functionally flexible, high-throughput assays that are capable of rapidly sifting through a vast pool of variants, while simultaneously examining them directly for a desired property [13].
Difficulties associated with purification and in vitro characterization of proteases suggest that a cell-based genetic assay could be highly advantageous for evaluating numerous candidates in a search for a desired function. Importantly, a cellular system enables the straightforward implementation of a selection format, whereby the host vitality is employed as the reporter of a specific function, allowing concurrent sorting of large libraries. Unlike affinity-based display methods [14], such assays could also monitor directly the level of a desired function, which should be advantageous in reliably identifying the optimal individuals from large pools of inferior variants [15]. Moreover, in an intracellular format, positive candidates are required to function within the context of the entire host proteome, requiring them to have an enhanced level of selectivity for their target. The selection step can be repeated as many times as necessary, using the inputs derived from previous rounds, while applying a higher level of stress challenge to each consecutive iteration until a desired level of activity is achieved. The stepwise protocol constitutes a process, known as “directed evolution,” which allows a requisite property to emerge in the course of the iterative search, even if it is only weakly present at the outset [16].
To implement these features for the discovery of proteases with de novo substrate specificities, a previously disclosed genetic system [17, 18] was converted to report on site-specific proteolysis. We disclose herein the implementation of this system for the detection of proteolytic activity in live cells and its utilization for the evolution of proteases with novel substrate specificities.
Materials and Methods
Materials
Unless specified otherwise, all chemicals were purchased from VWR Scientific or Sigma Aldrich Chemical Company. Restriction and DNA-modifying enzymes were purchased from Promega, New England Biolabs, and Fermentas. DNA polymerases were from GenScript Corporation. Oligonucleotides were obtained from Sigma Genosys. Fluorogenic peptides (>97% purity) were synthesized by Chem-Impex International, Inc. Plasmid isolation, PCR purification, and gel extraction kits were purchased from Qiagen.
Culture Media
Escherichia coli cultures were maintained in Luria–Bertani (LB) broth, unless specified otherwise. In liquid media, antibiotics were used at the following concentrations: ampicillin (Amp), 100 μg/ml; chloramphenicol (Cm), 30 μg/ml; kanamycin (Kan), 50 μg/ml; and tetracycline (Tet), 20 μg/ml. For chromosomally integrated markers, the concentration of antibiotics was reduced by half. Minimal Media A (MMA) [19], supplemented with 2% glycerol, 1 mM MgSO4, 10 μg/ml thiamine, 10 mM 3-amino-1,2,4-triazole (3AT), and 75 μg/ml Kan, was used in conditional growth experiments and is referred to as selective medium. Isopropyl-β-d-1-thiogalactopyranoside (IPTG) and l-arabinose (Ara) were added to media preparations, when appropriate, to induce protein expression.
Recombinant DNA Techniques
DNA manipulations were carried out with DH5α-E (Invitrogen) and DH5αpir [20] cells. Plasmids were transformed into E. coli by heat-shock or electroporation. All DNA sequencing was performed at Purdue Genomics Core Facility. Plasmid and strain construction details can be found in the Supporting Information.
Intracellular Proteolysis Assay
The single chain repressor (SCR) genes were subcloned into pET28a(+) vector (Novagen), and the expression strain BL21(DE3) was sequentially transformed with repressor- and protease-expressing plasmids to provide the requisite combinations. The SCR substrates and proteases were co-expressed under constant induction (100 μM IPTG) of pET plasmids expressing SCRs and variable induction (0, 13, 130, and 1,300 μM Ara) of protease plasmids, while maintaining them with Kan (50 μg/ml) and Cm (30 μg/ml). After 4 h of induction at 30 °C, the cultures were centrifuged at 4,000 rpm for 15 min at 4 °C. The cells were resuspended in 50 mM Tris–Cl pH 8.0 buffer and lysed with BugBuster® reagent (Novagen). The crude lysate pellet was resuspended in SDS Laemmli loading buffer and analyzed by SDS-PAGE gel (15%).
Preparation of the Naive Protease Library
The initial protease library for the directed evolution was prepared by performing error-prone PCR (epPCR) amplification of the coding region in the pAR-wtTEV plasmid, utilizing methods adapted from published protocols [21, 22]. Primers 5′-GAAGAACATGGGAAGCGGTCATCATCATCATCATCATCATGGAG-3′ and 5′-GTTGTTTCTAGATTAATTCATGAGTTGAGTCGCTTCC-3′ were used to generate DNA fragments coding for polyhistidine-tagged Tobacco Etch Virus protease (TEV-Pr) variants. The buffered mixtures, containing the primers (1 mM each), plasmid template (0.2 pmol), and Taq DNA polymerase (5 U), were supplemented with a biased mixture of dNTPs (0.2 mM dATP/dGTP, 1 mM dCTP/dTTP, and 0.25 mM MnCl2). The epPCR amplification was performed using the following thermocycling conditions: 94 °C 2 min, [94 °C 30 s, 59 °C 30 s, 72 °C 1 min] × 25 cycles, 72 °C 10 min. Restriction digest of the amplified product was performed with NcoI and XbaI, followed by purification through gel extraction. Ligation of the endonuclease-modified library into the corresponding linearized pARCBD-p plasmid and subsequent electroporation of the circularized plasmids into DH5α-E cells provided the naive library (2.5 × 108 cfu) for subsequent selection and evolution experiments.
Directed Evolution
The naive plasmid library was electroporated into the OC108 cells, and an aliquot of the transformants was plated onto rich media agar supplemented with Cm to establish transformation efficiencies of 107–108 cfu per electroporation. Transformed cells were centrifuged at 4,000 rpm and resuspended in 1× MMA buffer containing 10% glycerol. The resulting suspensions were plated on selective MMA media containing 13 μM Ara and variable amounts of IPTG (5.6–18 μM). The plates were incubated at 37 °C for 3–5 days until individual colonies emerged among the background growth. The fastest growing colonies from each induction level were streaked onto rich media for further analysis, while the remaining plate growth was harvested en masse and processed using a miniprep kit (Qiagen), with the isolated plasmids serving as templates for subsequent rounds of epPCR. Selection stringency of the subsequent evolution rounds was gradually enhanced by increasing the concentration of IPTG up to 32 μM.
Results and Discussion
Construction of the System for Detecting Site-Specific Proteolysis
Genetic Reporter Design and Development
Several genetic systems for monitoring endoproteolytic activity in live cells—yeast and bacteria—have been disclosed in the past, principally for exploring substrate scopes of native proteases [23–26], inhibitor searches [27–31], and in vivo solubility and stability optimizations [32, 33]. Most of these multi-plasmid systems have only been implemented as screens, limiting their utility in high-throughput assays. The need for a rapid and reliable retrieval of rare properties from large pools of variants prompts the development of a robust selection-based system that is protected against genetic instability induced by an applied stress and, as a result, yields a minimal number of false positives [28, 30]. Using bacterial hosts in genetic assays presents several important advantages over eukaryotic systems, such as higher transformation efficiencies, faster growth rate, and ease of manipulation, which collectively impart significantly higher processing capacity [27, 28]. Finally, we were interested in developing a method that could even detect and amplify relatively weak activities, likely to be found during the early stages of directed evolution [25]. Guided by these criteria, an existing genetic selection system, utilized previously in systematic searches for inhibitors of protein–protein interactions [17, 18], was adopted as the starting point.
The E. coli strain selected as a host for the reporter system, SNS126 [18], is a derivative of BW27786 [34] that lacks the imidazole glycerol phosphate dehydratase (IGPD) gene, involved in the biosynthesis of histidine. This auxotrophy is complemented by a chromosomally integrated tricistronic reporter cassette comprised of the HIS3, kanR, and lacZ genes, which code for yeast’s IGPD, aminoglycoside 3′-phosphotransferase responsible for resistance to Kan, and β-galactosidase, respectively. The reporter operon is placed under the control of the promoter and the corresponding operator from bacteriophage 434 lysis–lysogeny regulatory circuit [29]. Furthermore, the reporter activity can be chemically tuned by adjusting the concentrations of Kan and 3AT, a competitive inhibitor of yeast’s IGPD [28]. The expression level of β-galactosidase can be used for quantitative reporting on the operator status with chromogenic substrates [19].
The extent of reporter expression in this system is conditional, relying on the functional status of a transcription factor capable of regulating the bacteriophage-derived promoter. This regulator was engineered to be a SCR composed of two covalently linked DNA-binding domains (DBDs) of repressor protein cI from bacteriophage 434 [31]. Insertion of a proteolytically sensitive sequence into the flexible hinge region of an SCR renders the transcriptional inhibitor sensitive to site-specific proteolysis. The DBD segments of the resulting construct are expected to withstand proteolysis, due to their globular nature, while presenting the solvent-exposed linker region, the “cleavage cassette,” for site-specific processing by a cognate protease [26].
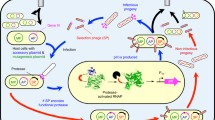
The SCR-expressing vector was generated as a monocistronic derivative of a previously disclosed pTHCP14 plasmid [18] under tac promoter (PTAC) control (see Supporting Information for details). The resulting construct, named pOC1, codes for two DBDs separated by a multiple cloning site (MCS) for in-frame and directional insertion of substrate fragments (Fig. 1a). Upon co-expression of a repressor–protease pair in the reporter strain, the SCR that is not recognized as a substrate by the protease should act as a powerful constitutive repressor of the reporter cassette, resulting in the death of the host cell on selective media (Fig. 1b). A combination of an active protease and a cognate SCR, on the other hand, is expected to allow the rescue of the host from the applied stress via unimpeded reporter expression (Fig. 1c). In addition, the host’s viability is expected to report on the substrate selectivity of an expressed protease, as variants with broad substrate scope are expected to exhibit enhanced toxicity [25].

Schematic representation of the genetic system for intracellular detection and evolution of site-specific proteases. a A plasmid for expressing SCR substrates is engineered by inserting a multiple cloning site between two DBD genes placed under the PTAC promoter control. The resulting construct can be used for directional and in-frame subcloning of pre-annealed oligonucleotides coding for proteolysis substrates (cleavage cassettes). b A defective protease (bladeless scissors) does not recognize a co-expressed SCR as a substrate, leading to the lack of expression of the tricistronic reporter system (HIS3–kan R–lacZ), compromising both the host’s survival on selective media and β-galactosidase activity on indicating media. c An active protease (intact scissors) cleaves a cognate SCR, resulting in the host’s survival and an enhanced level of β-galactosidase activity
Construction of Strains Sensitive to TEV-Pr
Of the known site-specific proteases, those of viral origin have been the most extensively studied and present convenient architectural models for understanding structure–activity relationships involved in site-specific proteolysis [35]. We have focused our initial efforts on TEV-Pr, a stringently specific representative of picornaviral processing 3C proteases, which displays structural similarity to serine proteases, but utilizes a thiol of cysteine as the nucleophilic center of the catalytic triad [36]. TEV-Pr cleaves its cognate sequence ENLYFQX (P6–P1′, where X is S or G) in a variety of structural contexts [37] and has never been observed to target proteins at unintended sites [38]. Substrate recognition segments of TEV-Pr span three relatively unstructured loops, which are adequately insulated from the bulk of the enzyme [39]. The catalytic triad residues—His46, Asp81, and Cys151— are positioned deep at the base of the substrate recognition loops. Taken together, these architectural features are expected to permit significant sequence variation in the substrate-binding regions, without compromising the activity of protease variants.
The MCS linker within pOC1 was used to insert three pairs of pre-annealed oligonucleotides, coding for the native substrate of TEV-Pr (G-TTENLYFQS-G), a weakly recognized P1′-aspartate mutant (G-TTENLYFQD-G) [40], and a scrambled control (G-STYFELNQT-G). The glycines flanking each sequence were incorporated to reduce steric influences from adjacent DBD residues on linker proteolysis. The resulting plasmids, coding for the wild-type (wtSCR), weak (wkSCR), and scrambled (scrSCR) substrates were named pOC2, pOC4, and pOC21, respectively. The SCRs expressed from these plasmids contain 101 N-terminal residues of DBD434* and 101 N-terminal residues of DBD434 separated by the 11-residue “cleavage cassette” linkers. Next, to ensure the stable expression of both proteases and substrates under conditions of selection stress, the SCR expression operons and non-repressing (nil) control were integrated into the chromosomes of the reporter strain, using a conditional-replication, integration, and modular plasmid system [41]. The resulting nil, wtSCR, scrSCR, and wkSCR integrants were named OC83, OC85, OC86, and OC108, respectively.
Performance of the Genetic Reporter System
The four SCR-integrated strains were evaluated for the extent of reporter transcription. A gene-reporter assay, based on the hydrolysis of o-nitrophenyl-β-d-galactoside (ONPG) by expressed β-galactosidase [19], was used to provide a quantitative measure of the repression power exhibited by the SCRs (Fig. S1A). The optimal repression could be achieved in the SCR-expressing OC85, OC86, and OC108 strains under relatively mild induction (10 μM) of the repressor genes with IPTG. A growth study on kanamycin-supplemented medium confirmed the cooperative power of the SCR architecture in controlling transcription (Fig. S1B, C). Importantly, these results indicate that the extent of transcriptional repression is relatively uniform for SCRs of different composition, ensuring a broad and consistent dynamic range in the growth rates of strains expressing active SCR–protease pairs.
Next, a protease-encoding plasmid compatible with co-expression of SCRs was generated. The TEV-Pr gene was amplified from the pRK793 vector [38] expressing autoinactivation-resistant mutant S219V. A restriction-modified PCR product was subcloned into the pARCBD-p vector [42], controlled by the l-arabinose (Ara)-sensitive PBAD promoter, to provide pAR-wtTEV. To access an inactive mutant control, the nucleophilic cysteine of the catalytic triad (C151) was replaced with alanine via site-directed mutagenesis, providing the pAR-mtTEV plasmid. SCR-expressing strains OC85, OC86, and OC108, as well as unrepressed control OC83, were transformed with pAR-wtTEV. The strain integrated with wtSCR (OC85) was also modified with pAR-mtTEV to generate a control for protease specificity. The transformants were then subjected to an analysis of growth rates under a variety of inducing and selecting conditions. The growth trends were ascertained by inoculating cell suspensions as microdroplets (ca. 2 μl) of variable concentration (10–106 colony-forming units, cfu, per aliquot) in arrays for side-by-side comparison. The strains displayed the following phenotypes: (1) indistinguishable growth rates under non-selective inducing conditions for all strains, (2) repressed growth for the SCR-expressing integrants in the presence of Kan and 3AT (Fig. 2a), and (3) partial rescue of growth for the strains expressing wtSCR/wtTEV and wkSCR/wtTEV combinations in the presence of Ara, with a significantly larger growth rate for the former (Fig. 2b). The strains were further tested for the amounts of β-galactosidase expressed under the control of the SCR-sensitive operator (Fig. 2c). The observed levels of ONPG hydrolysis replicated the growth trend reported for the wtSCR/wtTEV strain along with the small enhancement observed for the wkSCR/wtTEV strain when compared to the scrSCR/wtTEV strain, establishing the important link between host survival and conditional transcription of the reporter genes.

Detection of intracellular TEV-Pr-mediated proteolysis in the reporter strains expressing no SCR (1), wtSCR (2), scrSCR (3), and wkSCR (4). a Phenotypes observed for serially diluted inoculations (10–106 cfu per 2 μl drop) on selective media inducing the expression of the SCRs (IPTG 10 μM); b inoculations of the same four strains onto the selective media inducing expression of both SCRs (10 μM IPTG) and TEV-Pr (13 μM Ara); and c ONPG assay data
To confirm that the distinct growth rates of the test strains were indeed the result of site-specific proteolysis, the substrate and non-substrate SCRs were overexpressed in the presence of either wild-type or mutant enzymes using a two-plasmid format. Initially, the co-expression experiment was performed with constant SCR induction (100 μM IPTG) and variable induction of proteases (0, 13, 130, and 1,300 μM Ara). SDS-PAGE was used to visualize the extent of proteolytic processing of the substrate SCRs. As shown in Fig. 3a, the site-specific cleavage of wtSCR by TEV-Pr could be readily detected at 0.13 and 1.3 mM Ara, while the scrambled control was inert to all expression levels of the wild-type protease, as expected. Other substrate–enzyme combinations, grown at the optimized induction conditions (100 μM IPTG and 1.3 mM Ara), are demonstrated in Fig. 3b. While the strains expressing either the mutant protease or the scrambled substrate retained intact SCRs at all expression levels, the gel bands corresponding to both wtSCR and wkSCR were depleted upon induction of TEV-Pr. The reduced intensity of the wkSCR band indicates extensive processing of even a relatively poor substrate by the wild-type enzyme under the conditions of co-expression. Significantly, the observed selective processing of the cognate SCR substrates by the active protease links the intracellular, site-specific proteolysis to the survival propensities of the corresponding strains on selective media.

SDS-PAGE analysis of enzyme–substrate co-expression experiments. a Intracellular activity of TEV-Pr expressed from pAR-wtTEV under variable induction conditions (0, 13, 130, and 1,300 μM Ara) and detected by the processing of the scrambled (lanes 2, 3, 4, and 5, respectively) and native (lanes 7, 8, 9, and 10, respectively) SCRs (100 μM IPTG induction). b Processing of the SCR substrates (native, weak, and scrambled) with active and inactive variants of TEV-Pr: molecular weight ladder (lane 1), background protein expression in pET28-scrSCR/pAR-wtTEV strain (lane 2), and whole cell protein content upon induction with IPTG (100 μM) and Ara (1.3 mM) of pET28-wtSCR/pAR-mtTEV, pET28-scrSCR/pAR-mtTEV, pET28-wtSCR/pAR-wtTEV, pET28-wkSCR/pAR-wtTEV, and pET28-scrSCR/pAR-wtTEV strains (lanes 3, 4, 5, 6, and 7, respectively)
Directed Evolution
To demonstrate the potential of the assembled selection system for identifying proteolytic variants with improved properties, an iterative directed evolution sequence was initiated. The target of this search became a protease variant with superior activity against the P1′-modified substrate TTENLYFQD, which is only weakly recognized by the wild-type enzyme [40]. The naive library of TEV-Pr mutants was generated via PCR-based random mutagenesis [21] of the wild-type gene, insertion of the restriction-modified DNA duplexes into the pARCBD-p expression plasmid [42], and transformation of DH5α-E with the corresponding ligation mixture. The resulting pool of successful transformants (2.5 × 108) provided the naive library of mutant plasmids, an aliquot of which was subsequently electroporated into the strain integrated with wkSCR (OC108). The transformed cells were plated at a density of 1 × 106 cfu on mildly selective media and allowed to develop their phenotypes at 37 °C for 3–5 days. The surviving colonies were pooled and lysed to yield a focused collection of plasmids rendering their hosts with a growth advantage. A representative fraction of the first-round selectants was used as a collection of PCR templates for the second round of diversification. Altogether, three rounds of directed evolution were carried out, while increasing the stringency of selection at each stage, so that only a nominal number (ca. 100) of colonies with superior survival advantage could be visually identified before pooling the surviving colonies together.
After each round of selection, the best candidates (ca. 60), based on colony size and plating concentrations, were isolated and re-analyzed in a separate screen to yield mutants providing the wkSCR strain with optimal growth rates. The top performers from the first, second, and third rounds of directed evolution, as judged by comparative screens and ONPG hydrolysis assays [43], were designated as M1, M2, and M3, respectively. Sequencing of the plasmids coding for M1, M2, and M3 proteases revealed a likely hierarchical genetic relationship of the selected individuals, with mutations accumulating progressively with each round of evolution (Table 1). Specifically, all mutants contained four identical mutations: M2, an apparent progeny of M1, accumulated five additional amino acid changes; and M3 appears to be derived from M2 by a single mutagenesis event (see Supporting Information for additional analysis).
For a side-by-side analysis of both activities and selectivities of the evolved proteases, the plasmids encoding the selected mutants were isolated and transformed back into the selection strain expressing wkSCR, as well as the strain expressing wtSCR. The micro-droplet inoculation arrays confirmed that wkSCR strain transformed with mutant proteases M1, M2, and M3 exhibited readily detectable enhancements in growth compared to the pre-evolution performance of TEV-Pr (Fig. 4a), which was also reflected in the results of the corresponding ONPG assay (Fig. 4b). In addition to the relatively modest gains in activity toward the wkSCR substrate, the evolved mutants have also demonstrated notable decreases in the recognition of the wtSCR substrate, particularly following the first round of evolution, as demonstrated by both the growth analysis (Fig. 4c) and ONPG assay (Fig. 4d).

Analysis of intracellular proteolysis using droplet inoculation (a, c) and ONPG (b, d) assays. The reporter strains regulated by wkSCR (a, b) and wtSCR (c, d) were induced to co-express TEV-Pr (row 1), evolved protease M1, M2, and M3 (rows 2–4, respectively), and C151A TEV-Pr (row 5). Assays were performed under constant induction of SCRs (10 μM IPTG) and proteases (13 μM Ara)
Direct confirmation of the role played by site-specific proteolysis in the survival advantage of the selected mutants was established using the substrate–enzyme co-expression assay. The SDS-PAGE analysis of cells overexpressing both TEV-Pr variants and SCR substrates confirmed the proteolytic processing of the wkSCR by all of the selected proteases, as well as the significant activity loss of M2 and M3 toward wtSCR (Fig. S3). Additional cell-free characterization of the mutant proteases using fluorogenic versions of the native, weak, and scrambled substrates was performed to obtain quantifiable confirmation of the gradual increase in the selectivity of evolved mutants toward the weak substrate (see Supporting Information for additional details).
The need for a sensitive, rapid, and high-throughput analysis of mutant enzymes isolated in the process of the directed evolution experiment, combined with limited and unpredictable solubilities of enzymes derived from TEV-Pr [32], restricted the utility of a monophasic implementation of the kinetic analysis. Several recent reports have, however, highlighted the unique utility of heterogeneous formats for activity assays, which rely on solid-phase-supported enzymes [44–46]. We envisioned that the cobalt-based Talon® metal-affinity resin could be used for immobilization, purification, and on-bead refolding of the hexahistidine-tagged proteases. The wild-type and mutant (C151A TEV-Pr, M1, M2, and M3) proteases were, therefore, overexpressed in inclusion bodies, and the urea-solubilized denaturated enzymes were selectively captured by the resin. Washing and brief incubation of the beads in the refolding buffer (50 mM Tris–Cl, 10% glycerol, pH 8.0) and their subsequent resuspension in the protease activity buffer (50 mM Tris–Cl, 10% glycerol, 1 mM EDTA, 5 mM DTT, pH 8.0) restored the proteolytic activity of the immobilized biocatalysts, as confirmed by the observed steady-state rates obtained with SDS-PAGE-normalized amounts of supported TEV-Pr (Fig. S5).
For direct comparison of the observed activities, equal amounts of resin-bound enzymes were combined with fluorogenic versions of the substrates (see Supporting Information for details) in the wells of a 96-well plate and allowed to incubate at 25 °C for up to 16 h in a bioplate reader for real-time fluorescence measurements. The immobilized TEV-Pr displayed approximately 39-fold faster processing of the glycine substrate over the aspartate variant, and no fluorescence increase was noted for the scrambled control, as expected (Fig. S5A). The immobilized mutants were analyzed in a similar fashion using normalized amounts for side-by-side comparisons of substrate selectivities. The fluorescence time course profile displayed by the mutants (Fig. 5) confirmed the gradual loss of activity toward the native substrate (Fig. 5a), concomitant with the enhanced processing of the weak one (Fig. 5b). On the basis of pseudo first-order rate constants (Fig. S6 and Table S2), the most advanced selectant, M3, was found to exhibit a 92-fold decrease in activity against the P1′-glycine peptide and 3.4-fold improvement in the recognition of the P1′-aspartate fluorogen. These preliminary observations are consistent with the results of the cellular assays and confirm the unique potential of the combination of genetic selection and directed evolution for generating enzymes displaying novel substrate specificities.

Fluorescence characterization of immobilized proteases against a P1′-glycine peptide (H-(2-ABz)-ENLYFQG-(3-NT)-D-OH) and b P1′-aspartate peptide (H-(2-ABz)-ENLYFQD-(3-NT)-D-OH), at a concentration of 40 and 80 μM fluorogenic substrate, respectively. a Comparison of proteolytic activity against P1′-glycine substrate for TEV-Pr (1), M1 (2), M2 (3), and M3 proteases (4). b Comparison of TEV-Pr, M1, M2, and M3 proteases against P1′-aspartate substrate
Conclusion
Proteolytic enzymes, programmed to degrade specific peptides and proteins responsible for pathogenic states, are attractive therapeutic agents, since they could be aimed against a potentially unlimited repertoire of extracellular targets. The challenges inherent in the discovery of proteases with desired substrate specificities demand a robust, sensitive, and flexible assay that both reports with high sensitivity on a desired function and is amenable to high-throughput processing. Herein, we have described a novel genetic assay, capable of reporting on intracellular proteolytic activity via a powerful selection format, whereby the cleavage of a specific peptide bond is linked to the host’s survival. Moreover, the use of tandem reporters allowed the enzymatic efficiency to be amplified in the form of growth rates, establishing both a high dynamic range and a tunable stringency control for the discovery of unique activities. The potential of the genetic reporter system to yield enzymes with novel properties was demonstrated by a directed evolution sequence that allowed a gradual shift in the substrate recognition by the evolved proteases from the native substrate of the parent enzyme to a nominally weak one. Although full kinetic characterization of the evolved proteases would be required to report definitively on the source of the observed effects, the preliminary results demonstrate the utility of this genetic system for identifying proteolytic activities with improved or entirely novel properties. We plan to exploit this selection strategy in the discovery of proteases with tailor-made substrate specificities, which may open new avenues in both medicine and biotechnology.
References
Vellard, M. (2003). The enzyme as drug: Application of enzymes as pharmaceuticals. Current Opinion in Biotechnology, 14, 444–450.
Christie, R. B. (1980). The medical uses of proteolytic enzymes. In A. Wiseman (Ed.), Topics in enzyme and fermentation biotechnology (pp. 25–113). Chichester: Ellis Horwood.
Chobotova, K., Vernallis, A. B., & Majid, F. A. A. (2010). Bromelain’s activity and potential as an anti-cancer agent: Current evidence and perspectives. Cancer Letters, 290, 148–156.
Zhou, Y.-X., Karlee, S., Taguchi, H., Planque, S., Nishiyama, Y., & Paul, S. (2002). Prospects for immunotherapeutic proteolytic antibodies. Journal of Immunological Methods, 269, 257–268.
Kumar, J. K. (2008). Lysostaphin: An antistaphylococcal agent. Applied Microbiology and Biotechnology, 80, 555–561.
Uesugi, Y., Usuki, H., Iwabuchi, M., & Hatanaka, T. (2011). Highly potent fibrinolytic serine protease from Streptomyces. Enzyme and Microbial Technology, 48, 7–12.
Kurschus, F. C., & Jenne, D. E. (2010). Delivery and therapeutic potential of human granzyme B. Immunology Reviews, 235, 159–171.
Stenman, S. M., Venalainen, J. I., Lindfors, K., Auriola, S., Mauriala, T., Kaukovirta-Norja, A., Jantunen, A., Laurila, K., Qiao, S. W., Sollid, L. M., Mannisto, P. T., Kaukinen, K., & Maki, M. (2009). Enzymatic detoxification of gluten by germinating wheat proteases: Implications for new treatment of celiac disease. Annals of Medicine, 41, 390–400.
Pogson, M., Georgiou, G., & Iverson, B. L. (2009). Engineering next generation proteases. Current Opinion in Biotechnology, 20, 390–397.
Yuen, C. M., & Liu, D. R. (2007). Dissecting protein structure and function using directed evolution. Nature Methods, 4, 995–997.
Labrou, N. E. (2010). Random mutagenesis methods for in vitro directed enzyme evolution. Current Protein & Peptide, 11, 91–100.
Wang, T. W., Zhu, H., Ma, X. Y., Zhang, T., Ma, Y. S., & Wei, D. Z. (2006). Mutant library construction in directed molecular evolution: Casting a wider net. Molecular Biotechnology, 34, 55–68.
Jackel, C., & Hilvert, D. (2010). Biocatalysts by evolution. Current Opinion in Biotechnology, 21, 753–759.
Fernandez-Gacio, A., Uguen, M., & Fastrez, J. (2003). Phage display as a tool for the directed evolution of enzymes. Trends in Biotechnology, 21, 408–414.
Boersma, Y. L., Droge, M. J., & Quax, W. J. (2007). Selection strategies for improved biocatalysts. FEBS Journal, 274, 2181–2195.
Powell, K. A., Ramer, S. W., del Cardayre, S. B., Stemmer, W. P. C., Tobin, M. B., Longchamp, P. F., & Huisman, G. W. (2001). Directed evolution and biocatalysis. Angewandte Chemie International Edition, 40, 3948–3959.
Datta, S., Bucks, M. E., Koley, D., Lim, P. X., & Savinov, S. N. (2009). Functional profiling of p53-binding sites in Hdm2 and Hdmx using a genetic selection system. Bioorganic & Medicinal Chemistry, 18, 6099–6108.
Horswill, A. R., Savinov, S. N., & Benkovic, S. J. (2004). A systematic method for identifying small molecule modulators of protein–protein interactions. Proceedings of the National Academy of Sciences of the United States of America, 101, 15591–15596.
Miller, J. (1972). Experiments in molecular genetics. Cold Spring Harbor Laboratory: Cold Spring Harbor.
Platt, R., Drescher, C., Park, S. K., & Phillips, G. J. (2000). Genetic system for reversible integration of DNA constructs and lacZ gene fusions into the Escherichia coli chromosome. Plasmid, 43, 12–23.
Brakmann, S., & Lindemann, B. F. (2004). Generation of mutant libraries using random mutagenesis. In S. Brakmann & A. Schwienhorst (Eds.), Evolutionary methods in biotechnology: Clever tricks for directed evolution (pp. 5–11). Weinheim: Wiley-VCH Verlag Gmbh & Co. KgaA.
Cadwell, R. C., & Joyce, G. F. (1994). Mutagenic PCR. PCR Methods Applied, 3, S136–S140.
Cottier, V., Barberis, A., & Lüthi, U. (2006). Novel yeast cell-based assay to screen for inhibitors of human cytomegalovirus protease in a high-throughput format. Antimicrobial Agents and Chemotherapy, 50, 565–571.
Kim, S. Y., Park, K. W., Lee, Y. J., Back, S. H., Goo, J. H., Park, O. K., Jang, S. K., & Park, W. J. (2000). In vivo determination of substrate specificity of hepatitis C virus NS3 protease: Genetic assay for site-specific proteolysis. Analytical Biochemistry, 284, 42–48.
O’Loughlin, T. L., Greene, D. N., & Matsumura, I. (2006). Diversification and specialization of HIV protease function during in vitro evolution. Molecular and Biological Evolution, 23, 764–772.
Sices, H. J., & Kristie, T. M. (1998). A genetic screen for the isolation and characterization of site-specific proteases. Proceedings of the National Academy of Sciences of the United States of America, 95, 2828–2833.
Hu, J. C., Kornacker, M. G., & Hochschild, A. (2000). Escherichia coli one- and two-hybrid systems for the analysis and identification of protein–protein interactions. Methods, 20, 80–94.
Joung, J. K., Ramm, E. I., & Pabo, C. O. (2000). A bacterial two-hybrid selection system for studying protein–DNA and protein–protein interactions. Proceedings of the National Academy of Sciences of the United States of America, 97, 7382–7387.
Lallo, G. D., Castagnoli, L., Ghelardini, P., & Paolozzi, L. (2001). A two-hybrid system based on chimeric operator recognition for studying protein homo/heterodimerization in Escherichia coli. Microbiology, 147, 1651–1656.
Park, S.-H., & Raines, R. T. (2000). Genetic selection for dissociative inhibitors of designated protein–protein interactions. Nature Biotechnology, 18, 847–851.
Simoncsits, A., Chen, J., Percipalle, P., Wang, S., Törö, I., & Pongor, S. (1997). Single-chain repressors containing engineered DNA-binding domains of the phage 434 repressor recognize symmetric or asymmetric DNA operators. Journal of Molecular Biology, 267, 118–131.
van den Berg, S., Lofdahl, P. A., Hard, T., & Berglund, H. (2006). Improved solubility of TEV protease by directed evolution. Journal of Biotechnology, 121, 291–298.
Kostallas, G., & Samuelson, P. (2010). Novel fluorescence-assisted whole-cell assay for engineering and characterization of proteases and their substrates. Applied and Environmental Microbiology, 76, 7500–7508.
Khlebnikov, A., Datsenko, K. A., Skaug, T., Wanner, B. L., & Keasling, J. D. (2001). Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiology, 147, 3241–3247.
Tong, L. (2002). Viral proteases. Chemical Reviews, 102, 4609–4626.
Seipelt, J., Guarne, A., Bergmann, E., James, M., Sommergruber, W., Fita, I., & Skern, T. (1999). The structures of picornaviral proteinases. Virus Research, 62, 159–168.
Mondigler, M., & Ehrmann, M. (1996). Site-specific proteolysis of the Escherichia coli SecA protein in vivo. Journal of Bacteriology, 178, 2986–2988.
Kapust, R. B., Tözsér, J., Fox, J. D., Anderson, D. E., Cherry, S., Copeland, T. D., & Waugh, D. S. (2001). Tobacco etch virus protease: Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Engineering, 14, 993–1000.
Phan, J., Zdanov, A., Evdokimov, A. G., Tropea, J. E., Peters, H. K., III, Kapust, R. B., Li, M., Wlodawer, A., & Waugh, D. S. (2002). Structural basis for the substrate specificity of tobacco etch virus protease. Journal of Biological Chemistry, 277, 50564–50572.
Kapust, R. B., Tözsér, J., Copeland, T. D., & Waugh, D. S. (2002). The P1′ specificity of tobacco etch virus protease. Biochemical and Biophysical Research Communication, 294, 949–955.
Haldimann, A., & Wanner, B. L. (2001). Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure–function studies of bacteria. Journal of Bacteriology, 183, 6384–6393.
Scott, C. P., Abel-Santos, E., Wall, M., Wahnon, D. C., & Benkovic, S. J. (1999). Production of cyclic peptides and proteins in vivo. Proceedings of the National Academy of Sciences of the United States of America, 96, 13638–13643.
Verhoeven, K.D. (2011) Genetic system for detecting, monitoring, and evolving site-specific proteases: Development of therapeutic proteases targeting amyloid-beta peptide, Ph.D. Thesis, Purdue University, West Lafayette.
Arenkov, P., Kukhtin, A., Gemmell, A., Voloshchuk, S., Chupeeva, V., & Mirzabekov, A. (2000). Protein microchips: Use for immunoassay and enzymatic reactions. Analytical Biochemistry, 278, 123–131.
Puhl, A. C., Giacomini, C., Irazoqui, G., Batista-Viera, F., Villarino, A., & Terenzi, H. (2009). Covalent immobilization of tobacco-etch-virus NIa protease: A useful tool for cleavage of the histidine tag of recombinant proteins. Biotechnology and Applied Biochemistry, 53, 165–174.
Xie, W. L., & Ma, N. (2010). Enzymatic transesterification of soybean oil by using immobilized lipase on magnetic nano-particles. Biomass and Bioenergy, 34, 890–896.
Acknowledgments
The authors would like to thank B. L. Wanner (Purdue University, West Lafayette, Indiana) for donation of strains and plasmids. This study was sponsored by the National Institutes of Health (grant number: 1R21AG031437-01).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM. 1
(DOC 1239 kb)
Rights and permissions
About this article
Cite this article
Verhoeven, K.D., Altstadt, O.C. & Savinov, S.N. Intracellular Detection and Evolution of Site-Specific Proteases Using a Genetic Selection System. Appl Biochem Biotechnol 166, 1340–1354 (2012). https://doi.org/10.1007/s12010-011-9522-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-011-9522-6