
Abstract
Hepatitis B is a major public health problem worldwide, which may lead to chronic liver diseases such as cirrhosis and hepatocellular carcinoma. The hepatitis B core antigen (HBcAg) is one of the major viral proteins, which forms the inner core of hepatitis B virus (HBV) particles. In this study, filamentous bacteriophage M13 was genetically modified to display the polypeptides of HBcAg in order to develop an alternative carrier system. HBcAg gene was inserted into the minor coat protein (pIII) gene of M13, and HBcAg was expressed on the phage surface as a whole protein. Antigenicity and immunogenicity of HBcAg were tested by immunizing BALB/c mice three times with HBcAg-displaying recombinant phages. After successful immunization, one of the mice with high antibody titer to HBcAg was selected for fusion, and four monoclonal antibodies specific for HBcAg were developed. This result showed that HBcAg-displaying recombinant bacteriophages are immunogenic and can potentially be used for the development of monoclonal antibodies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatitis B virus (HBV) infection, a major cause of human liver diseases, can lead to chronic liver diseases such as cirrhosis and hepatocellular carcinoma; it results in 620,000 annual deaths worldwide [1]. According to the World Health Organization (WHO) 2008 data, about 2 billion people worldwide have been infected with the virus and about 350 million live with chronic infection. Even though HBV infections can cause severe health problems, they can be prevented by vaccination. Early diagnosis of HBV infection is also important for determining the extent of infection and for the prognosis of HBV-infected patients.
HBV belongs to the Hepadnaviridae family of viruses and consists of a nucleocapsid containing the viral genomic DNA and viral DNA polymerase encapsulated in an outer lipoprotein envelope [2]. HBV is a partially double-stranded DNA virus with a genome of 3.2 kb. The genome has four open-reading frames, which encode seven different proteins, i.e., hepatitis B surface antigens (HBsAg; S, preS1, and preS2), DNA polymerase, X protein, hepatitis B core antigen (HBcAg), and hepatitis B e antigen (HBeAg). HBcAg is one of the major viral proteins, which forms the inner core of HBV particles. It is a C gene-encoded cytoplasmic protein composed of 183–185 amino acids depending on serosubtypes [3–5]. One of the specific serological markers of HBV infection is the presence of anti-HBcAg antibodies in sera. Therefore, anti-HBcAg antibodies are used for initial diagnosis, patient monitoring, differentiation between acute and chronic infection (determined on the basis of immunoglobulin M [IgM] and IgG titers, respectively), and epidemiological investigations [5, 6]. Moreover, anti-HBcAg antibody detection is important in the identification of HBV-infected individuals who are negative for HBsAg but positive for anti-HBcAg antibody, in an effort to prevent the contamination of blood transfusion products with HBV [7]. To date, several monoclonal antibodies against HBcAg have been successfully developed [8–10] by immunizing mice with the antigen.
Phage display is a powerful molecular technique by which foreign proteins are expressed at the surface of phage particles [11]. DNA sequences of interest are inserted into a location in the genome of a filamentous bacteriophage such that the encoded proteins are expressed or “displayed” on the surface of the filamentous phage as fusion products with pIII or pVIII phage coat protein [11, 12]. Phage display technology is widely used for the production of peptides, including recombinant antibodies such as Fabs or single-chain variable fragments, for diagnostic or therapeutic purposes [13, 14]. Because of their immunogenic activity, phages are also used as potent carriers for immunization [15]. In a previous study, the cyclic disulfide peptide corresponding to the variable region 2 of the outer membrane protein PorA from Neisseria meningitidis strain B385 was displayed on a filamentous phage and showed immunogenic activity [16]. Whole phage particles displaying antigenic proteins have been used as vaccine vehicles in animal models [17, 18]. Repeat regions of the circumsporozoite protein gene of Plasmodium falciparum were cloned into the pIII protein gene of a filamentous phage, and the genetically engineered filamentous phage displaying the recombinant protein was both antigenic and immunogenic in rabbits [19].
In the present study, we describe a new approach for the production of monoclonal antibodies against whole HBcAg by using HBcAg-displaying phages as a source of immunogen. Immunization assays similar to that performed in this study have been described previously [20, 21], where peptides were fused with the phage coat protein pVIII and the recombinant phages were used for immunization of mice to develop a vaccine candidate. In this study, whole HBcAg was expressed on the surface of a filamentous phage as a fusion product with the minor phage coat protein pIII, and mice were immunized with the recombinant phages to generate HBcAg-specific monoclonal antibodies. Conventional hybridoma technology was used to generate the monoclonal antibodies against HBcAg. The method described in this paper is suitable for the production of large proteins such as HBcAg by phage display technology, and the recombinant phage particles can be used for further immunization with the aim of producing monoclonal antibodies to different epitopes.
Materials and Methods
Bacterial Strain
Escherichia coli strain TG1 {supE, hsdΔ5, thiΔ(lac-proAB), F’[traD36 proAB + lacIq lacZΔM15]; Amersham Pharmacia Biotech, Buckinghamshire, England} was used as the bacterial host for recombinant pCANTAB5E phagemid vector (Amersham Pharmacia Biotech) and wild-type pCANTAB5E phagemid vector.
Construction of HBcAg-Displaying M13 Phages
HBcAg gene was amplified by polymerase chain reaction (PCR) with the primers CForward (5′-ACTCGCGGCCCAGCCGGCCTTGGACATTGACCCTTATAA-3′) and CReverse (5′-CATTCTGCGGCCGCACATTGGGAAGCTGGAGATTG-3′). The underlined sequences in CForward and CReverse indicate the restriction sites for SfiI and NotI, respectively. PCR was performed in a reaction mixture (50 μl) containing 10× reaction buffer (200 mM Tris-HCl [pH 8.8], 100 mM KCl, 100 mM (NH4)2SO4, 20 mM MgSO4, 1% [v/v] Triton X-100, 1 mg/ml nuclease-free bovine serum albumin [BSA]) with 2.5 mM MgCl2, 10 mM of each deoxynucleoside triphosphate (dNTP; 1 μl), 20 pmol each of the forward and reverse primers, plasmid pCR2.1/HBc (kindly provided by Dr. M. Yapar, Department of Virology, Gulhane Military Medical Academy, Ankara, Turkey; 100 ng/μl; 2 μl), and Taq DNA polymerase (1 U/μl; 1 μl; Roche Molecular Biochemicals, Mannheim, Germany) in thermal cycler (T3000 Thermocycler; Biometra, Goettingen, Germany). PCR was completed at 30 cycles (at 94 °C for 1 min, at 55 °C for 2 min, at 72 °C for 2 min), followed by a final elongation step at 72 °C for 10 min. The amplified fragments were digested with SfiI and NotI restriction enzymes and purified with High Pure PCR Purification Kit (Roche Molecular Biochemicals). The purified DNA fragments were then ligated to the SfiI/NotI-digested pCANTAB5E phagemid vector in a ligation reaction mixture (30 μl) containing 10× ligase buffer (400 mM Tris-HCl, 100 mM MgCl2, 100 mM dithiothreitol [DTT], 5 mM ATP) and T4 DNA ligase (2.5 U; Fermentas, Helsingborg, Sweden), at 16 °C overnight. The ligation reaction product was transformed into E. coli TG1 cells by calcium chloride transformation [22]. Clones were controlled by colony PCR for the insertion of HBcAg gene into the vector. PCR-positive transformant clones were then subjected to plasmid isolation with High Pure Plasmid Isolation Kit (Roche Molecular Biochemicals). DNA sequencing of the plasmids was performed using GenomeLab DTCS Quick Start Kit (Beckman Coulter, Brea, USA) with reverse primer 458 (5′-TTTTGTCGTCTTTCCAGACGTT-3′) and forward primer 459 (5′-TATGACCATGATTACGCCAAG-3′), and the sequences were read using the CEQ 8800 genetic analysis system (Beckman Coulter).
Generation of Phage-Displayed HBcAg
To amplify wild-type phages and phage-displayed HBcAg, a single transformant E. coli TG1 clone containing the correct HBcAg gene insert or the wild-type phagemid, was selected and grown in 2× tryptone-yeast (TY) medium containing 100 μg/ml ampicillin and 2% glucose at 37 °C until optical density at 600 nm (OD600) reached 0.5. Thereafter, M13KO7 helper phages (1 × 1010 plaque-forming units [pfu]/ml; New England BioLabs, Beverly, MA) were added to the culture. After incubation at 37 °C for 45 min without agitation and at 37 °C for 45 min with agitation, the cells were centrifuged at 3,000 × g for 10 min. They were resuspended in the same volume of 2× TY medium containing 100 μg/ml ampicillin and 50 μg/ml kanamycin. The culture was incubated at 30 °C overnight with agitation. The next day, the cell culture was centrifuged at 3,000 × g, and supernatants containing the recombinant or wild-type infective phages were precipitated at 4 °C by adding one fourth of the supernatant volume of 1 M polyethylene glycol 6000 (PEG 6000; 2.5 M NaCl). After 2 h of incubation, the phages were centrifuged at 12,000 rpm for 40 min. Pellets containing the phage particles were resuspended in 1 ml of phosphate-buffered saline (PBS). The purified phage particles were then filter sterilized using Millex-HV (Millipore, Billerica, MA) and used for immunization and phage enzyme-linked immunosorbent assay (ELISA).
Phage Titration
Infectious titer of phages was determined by plaque counting assay. A single colony of E. coli TG1 was inoculated into 5 ml of 2× TY medium and grown at 37 °C to mid-log phase (OD600, 0.5–0.8). Subsequently, 100 μl of the culture was infected with a serial dilution of phages at 37 °C for 30 min. Infected bacteria were spread on 2× TY agar plates (with 100 μg/ml of ampicillin) and incubated overnight at 37 °C. The number of plaques formed were counted and translated to pfu per unit volume.
Phage ELISA
Phage ELISA was used to analyze the specific binding of anti-HBcAg polyclonal antibodies to phage-displayed HBcAg. Two methods were used to detect the HBcAg-displaying phages. In the first method, ELISA plates were coated with HBcAg-displaying phages (1 × 1010 pfu/well) at 4 °C overnight. The next day, the wells were washed six times with PBS containing 0.1% Tween 20 (TPBS) and blocked with a blocking solution (1% BSA in TPBS). Thereafter, rabbit anti-HBcAg polyclonal antibodies (1/200) were applied to the wells at room temperature for 1 h. The wells were rewashed with TPBS and anti-rabbit polyclonal antibodies conjugated with alkaline phosphatase (1/1,000) were added. After 1 h of incubation, the wells were washed six times with TPBS. Subsequently, para-nitrophenylphosphate (pNPP) substrate solution (1 mg/ml of pNPP in substrate buffer; 0.1 M glycine, 1 mM ZnCl2, 1 mM MgCl2, pH 10.4) was added to each well and incubated for 1 h. The reaction was terminated with 1 M NaOH and absorbance was read at 405 nm.
In the second method, ELISA plates were coated with anti-HBcAg polyclonal antibodies (1/200) at 4 °C overnight. The next day, the wells were washed six times with TPBS and blocked with the blocking solution. Thereafter, HBcAg-displaying phages (1 × 1010 pfu/well) were added to each well and incubated for 1 h. The wells were washed six times with TPBS, and horseradish peroxidase (HRP)/anti-M13 conjugate (1/1,000; Amersham Pharmacia Biotech, Buckinghamshire, England) was added to each well. Absorbance was read at 405 nm after 1 h of incubation with 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) substrate solution (Amersham Pharmacia Biotech).
Immunization
Eight-week-old BALB/c mice were used for immunization. Four different mice groups were administered different injections. The first group received 1 × 1011 pfu of HBcAg-carrying recombinant phage (recPhage) prepared in incomplete Freund’s adjuvant (IFA; Sigma, USA). The second group received 1 × 1011 pfu of recPhage only; the third group received 1 × 1011 pfu of wild-type phage (M13); and the last group received 5 μg of recombinant HBcAg (Fitzgerald, MA, USA) prepared in IFA. All immunizations were performed three times intraperitoneally at 3-week intervals.
Fusion
The mouse with the highest antibody titer to HBcAg was used for fusion. Lymphocytes were isolated from the spleen and lymph nodes and fused with mouse myeloma cells (F0; American Type Culture Collection [ATCC] CRL 1646) in the ratio of 5:1 in the presence of PEG 4000 (Merck, Darmstadt, Germany). Fusion was performed according to the original fusion protocol [23]. Hybrid cells were selected in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with hypoxanthine–aminopterin–thymidine (HAT) medium. After selecting the positive clones, monoclonal antibody-producing hybrid cells were applied to limiting dilution method.
Detection of Antibody Activity by ELISA
Microtiter ELISA plates were coated with HBcAg (400 ng/ml) prepared in PBS (10 mM, pH 7.2) and incubated overnight at 4 °C. The plates were washed with washing buffer (0.5% TPBS) and then saturated with 100 μl of blocking solution (1% milk powder) at 37 °C for 1 h. After washing, 100 μl of hybrid cell supernatant or 1/100 diluted mouse serum was added and incubated at 37 °C for an additional hour. After washing again, 100 μl of 1/1,000-diluted alkaline phosphatase conjugates of goat anti-mouse IgM, IgG, and polyvalent antibodies (Sigma) were added and incubated at 37 °C for 1 h. After a careful rewash, pNPP at 1 mg/ml in substrate buffer (0.1 M glycine, 1 mM ZnCl2, 1 mM MgCl2, pH 10.4) was added, and absorbance at 405 nm was determined.
Results
Construction of HBcAg-Displaying M13 Phages
With the aim of cloning HBcAg gene into pCANTAB5E phagemid vector, the gene coding for HBcAg was amplified by PCR, using pCR2.1/HBc plasmid as the template. The PCR amplification products were checked by agarose gel electrophoresis, and a 580-bp amplification product was monitored. The amplified band was extracted from the agarose gel, digested with SfiI and NotI restriction enzymes, and inserted into the pCANTAB5E phagemid vector. The recombinant vector was then transferred into E. coli TG1 cells by calcium chloride transformation. HBcAg gene insert sequence was amplified by colony PCR and sequenced using CEQ 8800. The transformant E. coli TG1 clone confirmed for the presence of HBcAg gene insert was infected with M13KO7 helper phages, and infective phages displaying HBcAg on their surface as HBcAg-pIII fusion protein were produced.
Phage ELISA
The binding property of phages displaying HBcAg to anti-HBcAg polyclonal antibodies was tested with phage ELISA. For this purpose, two different approaches were used. All the assays were performed in triplicate. For both strategies, an interaction between the anti-HBcAg polyclonal antibodies and phage-displayed HBcAg was detected (A405 for the first and second ELISA: 3.69 ± 0.02 and 1.97 ± 0.28, respectively, for HBcAg-displaying phages and 0.33 ± 0.057 and 0.12 ± 0.01, respectively, for wild-type phages; Fig. 1). These results showed that rabbit anti-HBcAg polyclonal antibodies recognized the HBcAg displayed on the phage surface as HBcAg-pIII fusion protein.

Optical density at 405 nm (OD 405 ) in the first- and second-phage enzyme-linked immunosorbent assay (ELISA). HBcAg Hepatitis B core antigen
Immunization
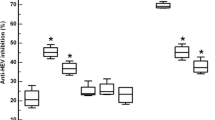
Immunogenicity of the HBcAg-displaying recombinant phage was analyzed using BALB/c mice. Mice groups were immunized with recPhage (1 × 1011 pfu) prepared in IFA, recPhage only (1 × 1011 pfu), wild-type phage (1011 pfu), and commercial HBcAg prepared in IFA. Mice were bled through the tail vein and sera were tested by ELISA for antibody reactivity. Figure 2 shows the antibody response induced by different carrier systems after three subsequent immunizations.

Antibody responses to HBcAg in mice after three subsequent immunizations. recPhage Recombinant phage, IFA incomplete Freund’s adjuvant, control nonimmunized mouse sera
Overall, both recPhages and recPhages prepared in IFA induced a strong immune response. No significant difference was detected in the immune response between the phages injected alone or with IFA, showing that the phages themselves were antigenic and generated the immune response. Moreover, no significant difference was observed in the immune response between the mice having recombinant phages or HBcAg (5 μg), indicating that 1 × 1011 pfu of phages displaying HBcAg are as good as pure HBcAg prepared in IFA. These results showed that immunization of mice with HBcAg-displaying phages generated antibody response and that these mice could be used for the production of monoclonal antibodies. However, the mice injected with wild-type phages also showed an immune response against HBcAg. This might be because of nonspecific antibody interactions due to the use of M13 phage, since M13 phages are large molecules and act as nonspecific polyclonal activators. In another study conducted in the laboratory, the mice sera from wild-type phage immunized groups were also tested for cross-reactivity with other antigens such as HBsAg, HBeAg, BSA, transferrin, and casein. As tested by ELISA, nonspecific binding to all of these antigens, as much as HBcAg, was obtained. Our results suggest that M13 phage acts as a nonspecific polyclonal activator and induces vigorous humoral response (data not shown).
Fusion and ELISA
After booster immunization, the mouse with the highest antibody response against HBcAg was selected for fusion. Lymphocytes from the spleen and lymph nodes were isolated and fused with mouse myeloma cells. As shown in Table 1, 5 × 108 lymphocytes were fused with 1 × 108 mouse myeloma cells. From this fusion, 783 hybridoma colonies were obtained, and 52 of them showed antibody activity against HBcAg. Four clones (5E5, 6D6, 7 F2, and 8 F7) were characterized as HBcAg-specific clones; of these, two clones (5E5 and 7 F2) showed better reactivity to HBcAg, while no clone showed any cross-reactions with HBeAg, HBsAg, human serum, wild-type phage M13, BSA, and powdered milk (Fig. 3). Subisotyping of monoclonal antibodies classified them as IgM.

Cross-reactivity of four monoclonal antibodies (5E5, 6D6, 7 F2, and 8 F7) against HBcAg, hepatitis B e antigen (HBeAg), hepatitis B surface antigen (HBsAg), human serum, wild-type phage M13, bovine serum albumin (BSA), and milk powder
Discussion
Phage display of protein and peptide libraries offers a powerful technology for the selection and isolation of ligands and receptors [11]. A filamentous phage has a number of characteristics that make it an excellent model system for studying antibody responses [15–18].
Filamentous bacteriophages are highly immunogenic particles, while phages elicit T cell responses [24]; their immunogenicity is also enhanced by virion-associated lipopolysaccharide [25, 26]. Van Houten and colleagues [27] demonstrated that antibody response was better focused against a synthetic peptide when the peptide was conjugated to a phage than when it was conjugated to the classical carrier ovalbumin.
Several studies have reported the cloning and expression of HBcAg in E. coli and the binding of the antigen has been analyzed with serological diagnostic assays [5, 6], but this is the first report on the display of HBcAg on phage surface. Phages have been used to generate immune responses to several antigens and mostly pVIII coat protein has been used to display antigens on phage surface [15, 28, 29]; this is because pVIII has 2,700 copies by which the amount of the displayed antigen is increased. However, the sequences fused to pVIII protein must be short (about 6–8 amino acids) owing to the closeness of the abundant proteins on phage surface [15, 30]. In this study, we preferred to use pIII protein, which has three to five copies on the phage surface, because HBcAg is a relatively large molecule to be properly displayed on pVIII. We selected only three to five copies of HBcAg displayed on the phage because of the closeness of each expressed protein. To control the presence of phage-displayed HBcAg, we used two different ELISA methods (Fig. 1) and the results showed that only HBcAg displaying phages were binding to the rabbit anti-HBcAg polyclonal antibodies but not to the wild-type phages.
Phages have been used as antigen carriers for vaccine development. Several studies on HIV-1 [31], hepatitis C virus (HCV) [32], Alzheimer’s disease [33], and cancer [20, 34] demonstrated the potential of using phage displayed antigens for vaccine development in mice and rabbits. Also, multiple epitope vaccine against Taenia solium pig cysticercosis was the first-phage display-based vaccine developed for large animal such as a pig [35]. The immunization methodology used in these works are differing either by the injection locations (intraperitonal [15, 31, 32], intranasal [33], or subcutaneous [34] or the amount of phages particles injected (1011 pfu [29, 33], 1012 pfu [34], 6 × 1012 pfu [31], 2.5 × 1013 pfu [15, 32]). All the different methods were successful for the activation of the immune response. This variability might be dependent on the characteristic of the antigen displayed on the phages. We have tested different amount of recombinant phages (1010 to 1012 pfu), and we have seen that 1012 recombinant phages displaying HBcAg were lethal for BALB/c mice. Furthermore, we have seen that optimum immune response was obtained with 1011 recombinant phages injections (data not shown).
The immunogenicity and antigenicity of HBcAg displayed on phage surface were shown by hybridoma technology. As shown in Fig. 2, pIII protein copy number was enough to generate immunogenic response against recombinant HBcAg without IFA. Immunogenicity of recombinant phage without IFA was also high, similar to pure HBcAg. The mice injected with wild-type phages showed an immune response against HBcAg; this is because the phages are large molecules and may stimulate polyclonal antibody response.
Antigenic parts of HBsAg, another HBV antigen, have been expressed on phage surface [29, 36]. To the best of our knowledge, this is the first study explaining the display of full length of an HBV antigen on phage surface. We expressed HBcAg on phage surface by cloning HBcAg gene into the minor coat protein gene of the phage, and the recombinant phage was shown to be highly immunogenic in mice, with the antibody response as good as that of commercial HBcAg. This result showed that M13 filamentous phage was a good carrier for recombinant HBcAg and induced generation of monoclonal antibodies specific for HBcAg.
Taken together, our results indicate that M13 phage is an efficient carrier for the cloning of full-length HBcAg, which was displayed efficiently on the phage and exhibited immunogenic activity. Most importantly, M13 phage would be an alternative choice to reduce the cost and time of the production and purification of HBV antigen for future applications.
Conclusions
Filamentous phages can display proteins and peptides on their surface, and they can also be carriers for immunogenic particles. To date, in several studies, a short length of a hepatitis B antigen has been expressed on phage surface. This is the first study reporting that full-length viral proteins are displayed on M13 phage surface by fusion with the minor coat protein of the phage, and the recombinant phages can further be used for the development of monoclonal antibodies to the antigen displayed on the phage surface without using additional adjuvants.
References
Zanetti, A. R., Van Damme, P., & Shouval, D. (2008). The global impact of vaccination against hepatitis B: a historical overview. Vaccine, 26, 6266–6273.
Shepard, C. W., Simard, E. P., Finelli, L., Fiore, A. E., & Bell, B. P. (2006). Hepatitis B virus infection: epidemiology and vaccination. Epidemiological Reviews, 28, 112–125.
Schödel, F., Peterson, D., Zheng, J., Jones, J. E., Hughes, J. L., & Milich, D. R. (1993). Structure of hepatitis B virus core and e-antigen. A single precore amino acid prevents nucleocapsid assembly. Journal of Biological Chemistry, 268, 1332–1337.
Malik, A. H., & Lee, W. M. (2000). Chronic hepatitis B virus infection: treatment strategies for the next millennium. Annals of Internal Medicines, 132, 723–731.
Watelet, B., Quibriac, M., Rolland, D., Gervasi, G., Gauthier, M., Jolivet, M., et al. (2002). Characterization and diagnostic potential of hepatitis B virus nucleocapsid expressed in E. coli and P. pastoris. Journal of Virological Methods, 99, 99–114.
Li, Z. X., Hong, G. Q., Hu, B., Liang, M. J., Xu, J., & Li, L. (2007). Suitability of yeast- and Escherichia coli-expressed hepatitis B virus core antigen derivatives for detection of anti-HBc antibodies in human sera. Protein Expression and Purification, 56, 293–300.
Katchaki, J. N., Siem, T. H., Brouwer, R., Brandt, K. H., & van der Waart, M. (1980). Detection and significance of anti-HBc in the blood bank: preliminary results of a controlled prospective study. Journal of Virological Methods, 2, 119–125.
Hlozánek, I., Korec, E., Dostálová, V., Stará, J., König, J., Bichko, V. V., et al. (1987). Monoclonal antibodies against genetically manipulated hepatitis B core antigen. Folia Biologica (Praha), 33, 295–300.
Ueno, Y., Kobayashi, K., Suzuki, H., Yamamoto, T., & Toyota, T. (1990). Production of monoclonal antibodies against recombinant HBcAg. Tohoku Journal of Experimental Medicine, 161, 253–255.
Tedder, R. S., Guarascio, P., Yao, J. L., Lord, R. B., & Eddleston, A. L. (1983). Production of monoclonal antibodies to hepatitis B surface and core antigens, and use in the detection of viral antigens in liver biopsies. Journal of Hygeine (London), 90, 135–142.
Smith, G. P. (1985). Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science, 228, 1315–1317.
Vidová, B., Godány, A., & Šturdík, E. (2008). Phage display — a tool for detection and prevention against pathogens. Nova Biotechnologica, 8, 23–33.
Paschke, M. (2006). Phage display systems and their applications. Applied Microbiology Biotechnology, 70, 2–11.
Erdag, B., Balcioglu, K. B., Kumbasar, A., Celikbicak, O., Zeder-Lutz, G., Altschuh, D., et al. (2007). Novel short peptides isolated from phage display library inhibit vascular endothelial growth factor activity. Molecular Biotechnology, 35, 51–63.
Meola, A., Delmastro, P., Monaci, P., Luzzago, A., Nicosia, A., Felici, F., et al. (1995). Derivation of vaccines from mimotopes. Immunologic properties of human hepatitis B virus surface antigen mimotopes displayed on filamentous phage. Journal of Immunology, 154, 3162–3172.
Menendez, T., De Haz, I., Delgado, M., Garay, H., Martin, A., & Vispo, N. S. (2001). Immunisation with phage-displayed variable region 2 from meningococcal PorA outer membrane protein induces bactericidal antibodies against Neisseria meningitidis. Immunological Letters, 78, 143–148.
Wang, L. F., & Yu, M. (2004). Epitope identification and discovery using phage display libraries: applications in vaccine development and diagnostics. Current Drug Targets, 5, 1–15.
Irving, M. B., Pan, O., & Scott, J. K. (2001). Random-peptide libraries and antigen-fragment libraries for epitope mapping and the development of vaccines and diagnostics. Current Opinion in Chemical Biology, 5, 314–324.
De la Cruz, V. F., Lal, A. A., & McCutchan, T. F. (1988). Immunogenicity and epitope mapping of foreign sequences via genetically engineered filamentous phage. Journal of Biological Chemistry, 263, 4318–4322.
Fang, J., Wang, G., Yang, Q., Song, J., Wang, Y., & Wang, L. (2005). The potential of phage display virions expressing malignant tumor specific antigen MAGE-A1 epitope in murine model. Vaccine, 23, 4860–4866.
Solomon, B. (2007). Active immunization against Alzheimer’s beta-amyloid peptide using phage display technology. Vaccine, 25, 3053–3056.
Sambrook, J., Fritsch, E. F., & Maniatis, T. (1989). Molecular cloning: a laboratory manual (2nd ed., Vol. 1, pp. 1.82–1.84). Plainview, New York: Cold Spring Harbor Laboratory Press.
Galfre, G., & Milstein, C. (1981). Preparation of monoclonal antibodies: strategies and procedures. Methods in Enzymology, 73, 3–46.
Willis, A. E., Perham, R. N., & Wraith, D. (1993). Immunological properties of foreign peptides in multiple display on a filamentous bacteriophage. Gene, 128, 79–83.
Grabowska, A. M., Jennings, R., Laing, P., Darsley, M., Jameson, C. L., Swift, L., et al. (2000). Immunisation with phage displaying peptides representing single epitopes of the glycoprotein G can give rise to partial protective immunity to HSV-2. Virology, 269, 47–53.
Eriksson, F., Tsagozis, P., Lundberg, K., Parsa, R., Mangsbo, S. M., Persson, M. A., et al. (2009). Tumor-specific bacteriophages induce tumor destruction through activation of tumor-associated macrophages. Journal of Immunology, 182, 3105–3111.
Van Houten, N. E., Zwick, M. B., Menendez, A., & Scott, J. K. (2006). Filamentous phage as an immunogenic carrier to elicit focused antibody responses against a synthetic peptide. Vaccine, 24, 4188–4200.
Minenkova, O. O., Ilyichev, A. A., Kishchenko, G. P., & Petrenko, V. A. (1993). Design of specific immunogens using filamentous phage as the carrier. Gene, 128, 85–88.
Tan, G. H., Yusoff, K., Seow, H. F., & Tan, W. S. (2005). Antigenicity and immunogenicity of the immunodominant region of hepatitis B surface antigen displayed on bacteriophage T7. Journal of Medical Virology, 77, 475–480.
Wan, Y., Wu, Y., Bian, J., Wang, X. Z., Zhou, W., Jia, Z. C., et al. (2001). Induction of hepatitis B virus-specific cytotoxic T lymphocytes response in vivo by filamentous phage display vaccine. Vaccine, 19, 2918–2923.
Scala, G., Chen, X., Liu, W., Telles, J. N., Cohen, O. J., Vaccarezza, M., et al. (1999). Selection of HIV-specific immunogenic epitopes by screening random peptide libraries with HIV-1-positive sera. Journal of Immunology, 162, 6155–6161.
Puntoriero, G., Meola, A., Lahm, A., Zucchelli, S., Ercole, B. B., Tafi, R., et al. (1998). Towards a solution for hepatitis C virus hypervariability: mimotopes of the hypervariable region 1 can induce antibodies cross-reacting with a large number of viral variants. EMBO Journal, 17, 3521–33.
Frenkel, D., Katz, O., & Beka Solomon, B. (2000). Immunization against Alzheimer’s b-amyloid plaques via EFRH phage administration. Proceedings of the National Academy of Sciences of the United States of America, 97, 11455–11459.
Wu, Y., Wan, Y., Bian, J., Zhao, J., Jia, Z., Zhou, L., et al. (2002). Phage display particles expressing tumor-specific antigens induce preventive and therapeutic anti-tumor immunity in murine p815 model. International Journal of Cancer, 98, 748–53.
Manoutcharian, K., Diaz-Orea, A., Gevorkian, G., Fragoso, G., Acero, G., Gonzalez, E., et al. (2004). Recombinant bacteriophage-based multiepitope vaccine against Taenia solium pig cysticercosis. Veterinary Immunology and Immunopathology, 99, 11–24.
Kok, W. L., Yusoff, K., Nathan, S., & Tan, W. S. (2002). Cloning, expression and display of the PreS domain of hepatitis B virus on filamentous bacteriophage M13. Journal of Biochemistry, Molecular Biology and Biophysics, 6, 55–58.
Acknowledgments
This work was supported by a grant from Tubitak (TUBITAK KAMAG-1007, Project no. 105G056). The authors wish to thank Aydin Bahar, Harun Kocaaga, and Sakir Sekmen for their excellent technical assistance and Dr. Esma Yolcu and Dr. Digdem Aktoprakligil Aksu for their comments on this paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bahadir, A.O., Balcioglu, B.K., Uzyol, K.S. et al. Phage Displayed HBV Core Antigen with Immunogenic Activity. Appl Biochem Biotechnol 165, 1437–1447 (2011). https://doi.org/10.1007/s12010-011-9365-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-011-9365-1