
Abstract
Metschnikowia reukaufii W6b isolated from marine environment was found to produce a cell-bound acid protease. The full-length cDNA (cDNASAP6 gene) of the acid protease (SAP6) from the marine-derived yeast M. reukaufii W6b was cloned. The insert was 1,755-bp long and contained an open reading frame of 1,527-bp encoding 508 amino acids. The deduced amino acid sequence included a signal peptide of 16 amino acids. The consensus motifs contained a VLLDTGSSDLRM active site and an ALLDSGTTITQF active site. The protein sequence deduced from the cDNASAP6 gene exhibited 12.9% overall identity with Cwp1 of Saccharomyces cerevisiae and a hydropathy profile characteristic of glycosylphosphatidylinositol cell-wall proteins. The cDNASAP6 gene without 48 bp encoding the signal peptide sequence was subcloned into an expression plasmid pET-24a (+) and fused with a 6-His Tag and transformed into Escherichia coli BL21 (DE3) for recombinant expression of the protease. The expressed fusion protein was found to have a unique band with molecular mass of about 54 kDa. The crude acid protease of the culture of the marine yeast strain W6b and the crude recombinant acid protease had milk clotting activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acid proteases, commonly known as aspartic proteases including pepsin, chymosin, rennin, cathepsin D, and related microbial enzymes such as endothiapepsin and penicillopepsin, with a pH optimum in the acidic range (pH 3–4) have been reported in a variety of microorganisms as intracellular and extracellular enzymes [1]. They have two reactive aspartyl residues (Asp32 and Asp215, according to pepsin numbering) in the active site within the characteristic sequences (hydrophobic generally Phe) Asp32-Thr-Gly-Ser in the N-terminal domain, and a corresponding (hydrophobic)-Asp215-Thr-Gly-Ser/Thr in the C-terminal domain [2]. These proteases are recognized by their specific inhibition by pepstatin.
The enzymes from fungi have been studied the most extensively, and several of them have been purified and cloned [1]. For example, acid protease from Rhodotorula glutinis K-24 [3] has been purified and characterized in some detail. Saccharomyces carlsbergensis has been reported to secrete four different proteases, each of which had a double pH optimum: one acidic between pH 2 and 4 and the other near neutrality between pH 6 and 8 [4]. Abdehl et al. [5] reported that Yarrowia lipolytica 37–1 when grown with casein at pH 3.2 produced extracellular acid protease(s) active at pH 3.2. Y. lipolytica [6] and Candida olea [7] are also reported to secrete aspartic protease. All of these yeasts secrete acidic proteases ranging in molecular mass from 30 to 45 kDa and with a pH optimum varying from 2.5 to 3.9. A psychrotrophic, dimorphic yeast Candida humicola, isolated from Antarctic soil, secretes an acidic protease with a molecular mass of 36 kDa [8]. However, its pH optimum (pH 1.0) seems to be much lower than those of the other yeasts.
Among the acid proteases, some are of particular interest for their successful commercial applications, e.g. as a rennet substitute in the cheese industry, or as a catalyst in brewing industry [1]. Acid protease also plays an important role in fermentation industry because it hydrolyzes protein in the fermentation mash to liberate amino acids or peptides under the acidic condition [9]. This may imply that acid protease can play an important role in degradation of proteinous materials in acid environment. For example, it may be used to remove proteins in shrimp shell to obtain chitin and chitosan. However, many acid proteases are also involved in infection of plant pathogen and human pathogen. For example, Candida albicans, the common human fungal pathogen, possesses at least eight genes encoding enzymes of this type [10].
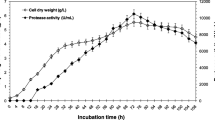
In recent years, marine yeasts and their bioactive substances have received increasing attention [11–15]. In this study, we found that Metschnikowia reukaufii W6b, isolated from sea sediment of South China Sea could produce cell-bound acid protease (58.8 U/ml) with an optimum pH 3.5. To extend knowledge of the acid protease and its encoding gene from marine yeasts, a study of protease produced by M. reukaufii and its encoding gene was performed in this study.
Materials and Methods
Strains, Plasmids, and Media
The marine-derived yeast M. reukaufii W6b was isolated from sediment at South China Sea and maintained in this laboratory. Escherichia coli DH5α was used to amplify the plasmids carrying the cloned gene and E. coli BL21 (DE3) was used as host strains for expression. Plasmids pMD19T (given ampicillin resistance) and pET-24a (+) (given kanamycin resistance) were used as the cloning and expression vectors, respectively. Yeast strain M. reukaufii W6b was grown at 25 °C in YPD medium which contained 10.0 g/l yeast extract, 20.0 g/l polypeptone, and 20.0 g/l glucose. The E. coli strains were grown at 37 °C in LB medium in the presence of 100 μg/ml of ampicillin or 30 μg/ml of kanamycin.
DNA Sequence and Computer Analysis
BLAST and ORF Finder programs at the National Center for Biotechnology Information (NCBI) were used for the nucleotide sequence analysis, deduction of the amino acid sequence and database searches. Multiple sequence alignments of DNA and amino acid were carried out using the programs of DNAMAN 6.0 (http://www.lynnon.com) and Clustal X 1.8 [16].
PCR-Based Cloning of Partial Acid Protease Gene
PCR was used for the partial acid protease DNA amplification. Genomic DNA was prepared as described by Chi et al. [17]. The conserved motifs were usually used to design the degenerate primers to clone these homologs. In this case, amino acid sequences of extracellular aspartic proteases from different species of eukaryotic microorganisms were downloaded from GenBank (http://www.ncbi.nlm.nih.gov/) and aligned. The degenerate forward primers and reverse primers used in this study were shown in Table 1. PCR was performed using a program of 94 °C for 5 min, 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min, followed by extension for 10 min at 72 °C. The PCR was run for 30 cycles and the PCR cycler was GeneAmp PCR System 2400 (PerkinElmer, Waltham, MA, USA). The PCR products were separated by agarose gel electrophoresis and recovered using UNIQ-column DNA gel recovery kits (BIOASIA, Shanghai). The recovered PCR products were ligated into pMD19-T and transformed into the competent cells of E. coli DH5α. The transformants were selected on LB plates with ampicillin. The plasmids in the transformant cells were extracted using the methods as described by Sambrook et al. [18]. The cloned DNA fragments inserted on the vector were sequenced by Shanghai Sangon Company. The amino acid sequences of the cloned DNA fragments were deduced and protein sequences were aligned using the programs of DNAMAN 6.0 (http://www.lynnon.com) and Clustal X 1.8 [16].
Total RNA Extraction and First-strand cDNA Synthesis
Total RNA was extracted from marine yeast strain M. reukaufii W6b using Trizol® reagent (RNA Extraction Kit, TAKARA, Japan) according to the manufacturer’s protocol. The concentration and quality of the total RNA were estimated by measuring the absorbance ratio of 260/280 nm and agarose-gel electrophoresis, respectively. Approximately, 5 μg of isolated RNA was reversely transcribed into the first-strand cDNA using a SMART™ RACE cDNA Amplification Kit (Clontech Laboratories, USA).
5′-RACE PCR and 3′-RACE PCR
The full-length cDNA was obtained using SMART RACE PCR. Four gene specific primers were designed according to the DNA fragment sequence obtained from the PCR using the degenerate primers (Table 1). The first round of 3′-RACE PCR reaction was carried out according to the SMART RACE Amplification kit User Manual with a total volume of 50.0 μl PCR mixture, including 2.5 μl of 3′-RACE-Ready cDNA (used as template), 5.0 μl of 10× BD Advantage 2 PCR buffer, 1.0 μl of 10 mM dNTP mix, 5.0 μl of 10 × UPM, 1.0 μl of GSP2, 34.5 μl of sterile deionized water, and 1.0 µl of 50× BD Advantage 2 Polymerase Mix. The products of the first round 3′-RACE PCR reaction were diluted 50 times and utilized as the templates in the 3′ nested PCR reaction. The contents of the PCR mixture used in this reaction were similar to those in the first round 3′ PCR, except using the diluted templates and the nested primer 3RNGSP instead of the RACE-ready first-strand cDNA and the first primer 3RGSP, respectively. In the 5′-RACE PCR, the 10× universal primer A mix was used as forward primer in both the first round and nested round reactions, while the primers 5RGSP and 5RNGSP (Table 1) were utilized as reverse primers in the first and nested round PCR reactions, respectively. Amplification reactions of the 3′- and 5′-RACE were performed (1) at 94 °C for 5 min followed by 5 cycles at 94 °C for 1 min and at 72 °C for 2 min; (2) then, this was followed by 6 cycles at 94 °C for 1 min, at 72 °C for 1 min, (1 °C of the annealing temperature was decreased per cycle), and at 72 °C for 2 min; (3) then, it was followed by 30 cycles at 94 °C for 1 min, at 65 °C for 1 min, at 72 °C for 2 min; (4) at last followed by an extension at 72 °C for 10 min. The 5′- and the 3′-RACE products of the cDNA were then sequenced and analyzed.
Cloning of Full-Length cDNA and Genomic DNA
The forward primer FLup was designed based on the sequence of the 5′-RACE product, whereas the reverse primer FLdown was designed based on the sequence of the 3′-RACE product (Table 1). This pair of primers was used to clone the full-length cDNA and the genomic DNA encoding acid protease from M. reufaukii W6b. The PCR reaction was performed in a total volume of 50.0 μl PCR mixture containing 5.0 μl of 10× ExTaq buffer, 4.0 μl of 2.5 mM dNTPs, 1.0 μl of 50 μM each primer (FLup and FLdown), 2.0 μl of (10.0 ng/ml) template DNA, 36.5 μl of sterile deionized water and 0.5 μl of ExTaq DNA polymerase. The conditions for the PCR amplification were as follows: initial denaturation at 94 °C for 5 min, denaturation at 94 °C for 1 min, annealing temperature at 56 °C for 1 min, extension at 72 °C for 2 min, and final extension at 72 °C for 10 min. PCR was run for 30 cycles. The PCR products were cloned into pMD-19T vector and sequenced. The full-length acid protease gene cloned from cDNA was named cDNASAP6 (Accession number: EU186020) while the full-length acid protease gene cloned from genomic DNA was designated as SAP6.
Plasmid Construction and Expression of the Recombinant SAP6
To construct a recombinant expression vector, the primers for amplification of the gene encoding the acid protease were designed according to the sequence of the gene cDNASAP6. The forward primer was BamH I pu: GGATCC GAA AAA GGC CAC TTA AAG TTA G (contained a BamH I cleavage site, italicized) and the reverse primer was Hind III pd: AAGCTT GAA GTA AGC AAA GAA AGC TGC G (contained a Hind III cleavage site, italicized). The PCR products were separated by agarose gel electrophoresis and ligated into plasmid pMD19-T simple. The recombinant vector was transformed into E. coli DH5α. The recombinant vectors carrying the PCR products were then extracted from the E. coli transformants and purified. The purified recombinant vectors carrying the PCR products were digested with BamH I and Hind III, and ligated into pET-24a (+) digested with the same enzymes, and transformed into E. coli DH5α. The resulting plasmid was confirmed by sequencing and named pET-24a(+)-SAP6-6His. It was transformed into E. coli BL21 (DE3) and the transformants obtained were selected on the LB plate containing 30 μg/ml of kanamycin.
To induce expression of the recombinant protein, the transformants were inoculated in LB broth supplemented with 30 μg/ml of kanamycin and cultured with shaking at 37 °C overnight. Twenty microliters of the seed culture were transferred into fresh LB broth supplemented with 30 μg/ml of kanamycin and cultured until OD600nm reached 1.0. Then isopropyl-thiogalactopyranoside (IPTG) was added to the E. coli cultures (final concentration of 1.0 mM) and the cultures were incubated at 20 °C overnight to induce expression of the target proteins. The cells were harvested and washed by centrifugation at 8,000×g and 4 °C for 5 min with 20 mM citrate buffer (pH 6.0). The washed cells were added to 5 × sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer and boiled for 5 min. SDS-PAGE and Western blotting combined with immunostaining were applied for confirmation of the expressed products in the cells as described below.
Preparation of the Crude Recombinant Aspartic Protease
In order to improve expression of the soluble recombinant protein, after IPTG (final concentration 1.0 mM) was added for induction, the cultures were further cultured at 20 °C overnight, according to the pET System Manual (10th edition, www.novagen.com). Then, the cells were washed and resuspended in 5.0 ml of 20 mM citrate buffer (pH 3.4) followed by repeated treatments of 10 s by ultrasonication in Ultrasonic Homogenier (400W model, ampl, Sonics&Material Ins. USA) at maximum output in the ice [19]. After disruption by ultrasonication and removal of cell debris by centrifugation at 14,006×g and 4 °C for 20 min, the supernatant (cell-free extract) obtained was used as the crude recombinant acid protease. SDS-PAGE and Western blotting were then applied for detecting the expressed protein. The supernatant (cell-free extract) prepared from the transformants with the plasmid pET-24a (+) was used as the control.
SDS-PAGE Analysis and Western Blotting
The presence of the recombinant acid protease in the E. coli cells was confirmed in non-continuous denaturing SDS-PAGE [20] with a two-dimensional electrophoresis system (Amersham Biosciences, Sweden) and stained by Coomassie Brilliant Blue R-250 [21]. The molecular mass standards for SDS-PAGE comprised β-galactosidase (116 kDa), bovine serum albumin (66.2 kDa), ovalbumin (45 kDa), lactate dehydrogenase (35 kDa), restriction endonuclease Bsp981 (25 kDa), and β-lactoglobulin (18.4 kDa).
To confirm the recombinant protease expression as a His-tagged fusion protein, Western blot analysis was carried out using monoclonal mouse-anti-His-Tag antibody (TianGen, China) as the primary antibody and goat anti-mouse IgG antibody conjugated with horseradish peroxidase (HRP) (TianGen, China) as the secondary antibody. After SDS-PAGE, proteins were transferred to PVDF membrane for 2 h at 11 mA. The membrane was blocked with 5% skimmed milk in PBST (phosphate-buffered saline (PBS) buffer containing 0.05% Tween-20) for 2 h at 37 °C and then was incubated with the mouse-anti-His-Tag antibody at a dilution of 1:1,500 for 2 h at 37 °C and washed three times for 15 min in PBST. Subsequently, the membrane was incubated with the goat anti-mouse IgG antibody conjugated with horseradish peroxidase (TianGen, China) at a dilution of 1:200 for 2 h at 37 °C. After washing in PBST four times and PBS (without Tween-20) once, the membrane was applied to 3, 3-diaminobenzideine (DAB) reagents (TianGen, China) according to manufacturer’s instruction.
Protease Assay
The crude recombinant acid protease activity and crude acid protease activity in the culture of the marine yeast strain M. reukaufii W6b were determined according to the methods described by Larson and Whitaker [22]. The substrate contained 1.0% bovine hemoglobin (Sigma, USA) dissolved in citrate buffer (20 mM, pH 3.4). Enzyme (0.4 ml) and substrate (0.4 ml) were combined and incubated for 1 h at 40 °C. The reaction was stopped by addition of 0.4 ml of 15% trichloroacetic acid, the precipitate was removed by centrifugation and the absorbance of the supernatant was recorded at 280 nm. One acid protease unit was defined as the amount of protease causing an increase in absorbance of 0.1 at 280 nm after 1 h. Assays were done in duplicate.
The assay of the recombinant acid protease activity and crude acid protease activity in the culture of the marine yeast strain M. reukaufii W6b was also performed with a skimmed-milk agar (pH 6.0) plate using 6-mm-diameter sterile Oxford-cups which were put on the assay plates. Two hundred microliters of the crude enzyme of the marine-derived yeast M. reukaufii W6b and the recombinant acid protease were added to each cup and incubated at 30 °C for 24 h, respectively and the clear zone formed was observed and photographed.
Skimmed Milk Clotting Test
In order to obtain the evidence that the crude enzyme of the marine-derived yeast M. reukaufii W6b and the recombinant enzyme from E. coli transformants carrying pET-24a(+)-SAP6-6His were the acid protease, the milk clotting ability was assayed by the method of Berridge [23]. Ten grams of skimmed milk powder was suspended by stirring with glass rod in 100 ml of 0.05 M CaCl2. The pH of milk substrate was adjusted to 5.8 with 0.1 N NaOH or HCl. Marine-derived yeast M. reukaufii W6b or the recombinant acid protease from the E. coli transformants, 0.5 ml, was added to 4.0 ml of pre-incubated skimmed milk for complete coagulation at 50 °C, respectively. Water, 0.5 ml, and 0.5 ml of the supernatant obtained from E. coli BL21/pET-24a (+) were used as negative controls, respectively.
Results
Screening of the Marine Yeast Strains with Acid Protease Activity
After over 400 yeast strains isolated from seawater, sediments, the guts of marine fish, and marine algae were tested [24], only one strain (W6b) among them was found to be able to produce acid protease (Table 2) when they were grown in the medium with 20.0 g/l glucose. This yeast strain was isolated from sediment of South China Sea. The crude acid protease of the strain W6b had the optimal temperature of 40 °C and the optimal pH of 3.5 (Table 2). It also can be seen from Table 2 that most of the acid protease produced by strain W6b was cell-bound enzyme. The results in Table 2 also show that the activity of the crude acid protease produced by the yeast strain W6b was sensitive to pepstatin A, suggesting that the acid protease belonged to aspartic protease. After identification using the routine and molecular methods (accession number of 26S rDNA was EU439452), it was found to be closely related to M. reukaufii [25].
Cloning of the Full-Length cDNA-Encoding Acid Protease
Because most of the acid protease produced by the marine-derived yeast was cell-bound enzyme (Table 2), the gene (cDNASAP6) encoding the enzyme was amplified from cDNA of the yeast cells (Fig. 1).To clone the full-length cDNA encoding the acid protease in M. reukaufii W6b, we first tried to obtain a partial sequence of the genomic DNA of the acid protease. The conserved motifs were used to design the degenerate primers to clone the partial gene (Table 1). In this case, amino acid sequences of extracellular aspartic proteases from different species of eukaryotic microorganisms were downloaded from GenBank (http://www.ncbi.nlm.nih.gov) and aligned. The PCR-generated fragments were sequenced. Analysis of the sequence by BLAST program indicated that the fragment (617 bp) of the putative aspartic protease gene was isolated as the amino acid sequences deduced from the fragment contained the consensus motifs VLLDTGSSDLRM and ALLDSGTTITQF of aspartic proteases (Fig. 1). Then, the new primers were designed according to the sequence of the fragment (617 bp) of the putative aspartic protease gene and used for amplification of 3′-RACE cDNA and 5′-RACE cDNA of the aspartic protease gene from the first-strand cDNA of M. reukaufii M6b by SMART RACE PCR as described in Materials and methods. Finally, the 5′- and 3′-RACE PCR products were obtained and sequenced. By aligning the nucleotide sequence of the two fragments, the 3′ and 5′ full-length cDNA (1,035 bp and 515 bp, respectively) were obtained. Then, the full-length cDNA of the aspartic protease was amplified from the 5′ first-strand cDNA and the 3′ first-strand cDNA with the primers FLup and FLdown (Table 1), which were located at the 5′- and 3′-ends of the full-length cDNA sequence. A 1,755 bp cDNA fragment was amplified and named cDNASAP6 (accession number EU186020) (Fig. 1). To confirm the identity of the full-length cDNA of the aspartic protease, efforts were made to clone the genomic DNA of the aspartic protease gene. The genomic DNA (SAP6 gene) of the protease was amplified from genomic DNA of M. reufaukii W6b with the primers FLup and FLdown (Table 1). The obtained genomic DNA fragment was also 1,755 bp long and the DNA sequence was the same as that of the cDNA fragment (Fig. 1). This means that there are no introns in the SAP6 gene.

The cDNA sequence and deduced amino acid sequence of the M. reufaukii M6b aspartic protease. The nucleotide and/or amino acid sequence is numbered on the left. Translation initiation codon ATG was positioned at nt + 1.The putative signal peptide is underlined and the cleavage site is indicated by a vertical arrow. The translation initial and stop codons are designated with a frame and an asterisk (*) respectively. The sequences of consensus motifs flanking these conserved active sites are shaded. The amino acid sequence for N-linked glycosylation site was boxed
Analysis of the Acid Protease Protein Deduced from cDNASAP6
Signal peptide analysis of the protein deduced from cDNASAP6 gene at http://cbs.dtu.dk/services/SignalP/ showed that the N-terminal signal peptide had 16 amino acids and the peptide bond between 16th and 17th amino acid would be cleaved by signal peptidase (Fig. 1). N-glycosylation sites of the protein were also analyzed at http://cbs.dtu.dk/services/NetNGlyc and the results indicate that there were six potential N-linked glycosylation sites of the protein, among them, -N-G-T- at 155 amino acid and -N-S-T- at 182 amino acid were the most possible N-glycosylation sites (Fig. 1). The alignment of conserved domain database (CDD) of the deduced acid protease at NCBI (http://www.ncbi.nlm.nih.gov/Structure) reveals a typical conserved region (from 55 to 390 amino acids) which was the characteristics of eukaryotic aspartyl protease (Fig. 2). Analysis of the deduced aspartic protein at http://motif.genome.jp/ showed that the protein had the conserved eukaryotic and viral aspartyl proteases active site “VLLDTGSSDLRM” from 67 to 88 amino acids and “ALLDSGTTITQF” from 278 to 289 amino acids, respectively (Fig. 1). Therefore the results demonstrate that the acid protease obtained in this study belonged to one member of aspartic proteases in the eukaryotic aspartyl protease (EC 3. 4. 23) family.

Conserved domains of the SAP6 gene obtained in this study. Fifty-five to three hundred ninety amino acids deduced from the SAP6 gene were the same conserved domain as eukaryotic aspartyl protease
Expression of the Recombinant SAP6 in E. coli
The cDNASAP6 gene amplified with the primers BamH I pu and Hind III pd (Table 1) was cloned into the expression vector pET-24a (+) and the recombinant plasmid containing the cDNASAP6 gene was transformed into E. coli BL21 (DE3). BL21 (DE3)/pET-24a (+)-SAP6-6His obtained was induced by IPTG. SDS-PAGE shows that cell-free extracts from the induced cells of E. coli BL21 (DE3) harboring pET-24a (+)SAP6-6His exhibited one unique band with a molecular mass of about 54 kDa (lanes 1 and 2 in Fig. 3a), which was the similar size range (53.5 kDa) as estimated from the deduced amino acid sequence of the cDNASAP6 gene. The results of Western blotting (Fig. 3b) confirm that this unique band was indeed the His-tagged fusion protein of the recombinant aspartic protease. The results demonstrate that the cDNASAP6 gene obtained from the marine yeast strain M. reukaufii W6b could be expressed in E. coli cells and the recombinant SAP6 produced by E. coli cells became the mature form with acid protease activity.

Expression of the aspartic protease of M. reufaukii W6b in E. coli. a The mature peptide of the aspartic protease was expressed with pET-24a (+) as a fusion protein in BL21 cells at 20 °C for overnight. Lane 1 cell free extract obtained from E. coli BL21/pET-24a (+)SAP6-6His; lane 2 cell debris obtained from E. coli BL21/pET-24a (+)SAP6-6His; lane M protein markers. b Western blotting analysis of the expressed fusion protein SAP6-6His. M protein molecular markers (116, 66.2, 45, 35, 25, and 18.4 kDa from top to bottom, respectively)
After disruption by ultrasonication and removal of cell debris of the induced cells of BL21(DE3)/pET-24a (+)-SAP6-6His, the crude recombinant protease activity in the supernatant was determined against the transformants with the plasmid pET-24a (+) as the control. The results in Table 3 show that specific protease activity of the induced cells of BL21 (DE3)/pET-24a (+)SAP6-6His was 36.1 ± 0.84 U/mg, while no aspartic protease activity was detected in BL21 (DE3)/pET-24a(+). Figure 4 shows that clear zones around the Oxford-cup containing the crude recombinant aspartic protease and crude acid protease from the culture of the marine yeast strain M. reukaufii W6b were formed. These results also indicate that the aspartic protease cDNA gene cloned from M. reufaukii W6b has been expressed in E. coli cells successfully and the recombinant SAP6 became the mature form with acid protease activity.

The clear zones formed on the skimmed-milk double plates. a Front; b back
The milk clotting activity is the common characteristics of all the aspartic protease [26]. Therefore, the milk clotting activity of the crude acid protease produced by the marine yeast strain M. reukaufii M6b and the recombinant SAP6 from the positive E. coli transformants was determined. It can be clearly seen from the results in Fig. 5 that there was the skimmed milk coagulability of the crude acid protease from the culture of the yeast strain M. reukaufii W6b (C in Fig. 5) and the recombinant SAP6 (D in Fig. 5), respectively, while no such coagulability was observed either on the addition of water (A in Fig. 5) or the cell-free extract obtained from E. coli harboring only the expression vector pET-24a (+) (B in Fig. 5). The results in Fig. 5 demonstrate that the cDNASAP6 gene amplified from the marine yeast strain M. reufaukii W6b indeed encoded the aspartic protease.

Milk clotting test of the recombinant SAP6 from the positive E. coli transformants and the crude SAP6 from the marine yeast strain M. reukaufii W6b. a Water + milk, b milk + cell-free extract obtained from E.coliBL21/pET-24a (+). c Milk + culture of the marine yeast M. reukaufii W6b. d Milk + cell-free extract obtained from E. coli BL21/pET-24a (+)SAP6
Discussion
In this study, it was found that M. reukaufii M6b could produce acid protease. Although it has been reported that Y. lipolytica [27], C. clea [7], Rhodotorula glutinis [3], S. carlsbergensis [4], Candida humicola [8], Candida albicans [10] and Saccharomycopsis fibuligera [5] can produce acid protease, this is the first report that M. reukaufii isolated from marine environment could produce acid protease. Gonzalez-Lopez et al. [27] found that the type of protease synthesized in the yeast Y. lipolytica is strictly dictated by ambient pH. At acidic pH, induction of the AXP1 gene leads to secretion of an acid protease (Axp), whereas at neutral pH, an alkaline protease (Aep) is produced as the XPR2 gene becomes induced. Therefore, it is unusual that M. reukaufii W6b isolated from marine environment could produce acid protease as pH of sea sediment is around 8.0. However, it is completely unknown if M. reukaufii W6b can yield such protease in natural sea sediment.
The gene cDNASAP6 encoding the acid protease had 1,527 bp long and did not contain any intron. The ORF encoded 508 amino acid residues with an estimated molecular mass of 53.5 kDa and the pI of the deduced protein was 4.2. It has been reported that a secreted aspartic proteinase from Glomerella cingulata (GcSAP) has a molecular mass of 36.0 kDa as estimated by SDS-PAGE [28]. The complete nucleotide sequence of a 2,753-bp-long DNA fragment from S. fibuligera is essential for acid protease production in Saccharomyces cerevisiae [29]. The fragment contained an open reading frame of 1,170 bp that encoded a 390-amino acid polypeptide [30]. The purified protease from the psychrotrophic yeast C. humicola had a molecular mass of 36 kDa [8]. Despite the prevalent cold temperature in Artarctica, the extracellular protease of the yeast had optimal activity at 37 °C [8]. Protease I from Y. lipolytica had a molecular weight near 28 kDa, an isoelectric point of pH 4.9 and a pH optimum of 3.5. Protease II from Y. lipolytica had a molecular weight near 32 kDa and a pH optimum of 4.2. Protease III from Y. lipolytica had a molecular weight near 36 kDa, an isoelectric point of 3.8, and a pH optimum of 3.1 [28]. However, the acid protease-encoding gene (PepA) of Aspergillus oryzae encodes 404 amino acid residues and contains three putative introns ranging in length from 50 to 61 nucleotides [31]. This may imply that the molecular mass of the acid protease produced by the marine yeast M. reukaufii M6b were much higher than that of the acid proteases produced by other yeasts.
Based on the alignment and comparison of the protein (SAP6) sequence deduced from cDNASAP6 gene with sequences in the protein databases using BLAST program, it was found that the SAP6 shared identity with aspartic protease from other fungi, A. oryzae (BAA02994, 18.11%), Neolitsea fischeri (EAW20369, 14.26%), S. cerevisiae (P32329, 20.52%), Cryphonectria parasitica (5ER1E, 18.53%), Candida parapsilosis (B47701, 28.6%), and Y. lipolytica (CAA65778, 19.63%), respectively. The topology of the phylogram confirms that the amino acid sequence deduced from the cDNASAP6 gene obtained in this study also had high match with that of S. cerevisiae GPI-anchored aspartic protease 1, S. cerevisiae GPI-anchored aspartyl protease and S. cerevisiae GPI-anchored aspartic protease 2 (Fig. 6). This suggest that the SAP6 was GPI-anchored aspartic protease in the marine yeast M. reukaufii M6b. It has been reported that such protease was covalently linked to cell membrane by GPI-anchor [32]. The results in Table 2 indeed show that most of the acid protease activity in M. reukaufii M6b was cell bound. The Kyte and Doolittle hydropathy profiles of the Cwp1 from S. cerevisiae and the SAP6 from M. reukaufii M6b were both characteristic of glycosylphosphatidylinositol cell-wall proteins (GPI-CWPs) (Fig. 7). Like the gene encoding the Cwp1 in S. cerevisiae, C-terminal anchor domain of the gene encoding GPI-CWP in M. reukaufii M6b can be used to construct surface display vector in yeast cells [32, 33]. Therefore, the cDNASAP6 gene obtained in this study may have many applications in biotechnology.

Phylogenetic tree of amino acid sequences of aspartic proteases. The phylogenetic tree of the deduced amino acid sequence of the SAP6 was constructed by using PHYLIP 3.56

Kyte and Doolittle hydropathy profile of Cwp1 (a) from S. cerevisiae and SAP6 (b) from M. reukaufii W6b
The cDNASAP6 gene obtained from the marine yeast strain M. reukaufii W6b could be expressed in E. coli cells and the crude recombinant enzyme had acid protease activity and the milk clotting activity, demonstrating the recombinant SAP6 produced by E. coli cells became the mature form. The recombinant acid protease had a molecular mass of about 54 kDa (lanes 1 and 2 in Fig. 3a), which was the similar size range (53.5 kDa) as estimated from the deduced amino acid sequence of the cDNASAP6 gene. The gene encoding the acid protease from Scytalidium lignicolum was expressed in S. cerevisiae AH22 and the recombinant acid protease was secreted in mature form [34]. The recombinant acid protease had a pH optimum of 2.3 for activity and was stable between pH 2 and 5, at 50 °C for 15 min at pH 4.0 and insensitive to DAN as well as pepstatin. The DNA coding for a secretable acid protease gene of Saccharomycopsis fibuligera was isolated from a genomic DNA library of the organism. S. cerevisiae cells transformed with a plasmid carrying the cloned gene secreted acid protease having the same enzymatic properties as those of the S. fibuligera protease [35].
References
Kocabiyik, S., & Ozel, H. (2007). Bioresource Technology, 98, 112–117. doi:10.1016/j.biortech.2005.11.016.
De Viragh, P. A., Sanglard, D., Togni, G., & Falcetto, R. (1993). Journal of General Microbiology, 139, 335–342.
Kamada, M., Oda, K., & Murao, S. (1972). Agricultural and Biological Chemistry, 36, 1095–1101.
Maddox, I. S., & Hough, J. S. (1970). The Biochemical Journal, 117, 843–852.
Abdehl, A. T. H., Kennedy, E. H., & Ahearn, D. G. (1977). Journal of Bacteriology, 130, 1125–1129.
Tobe, S., Takami, T., Ikeda, S., & Mitsuzi, K. (1976). Agricultural and Biological Chemistry, 40, 1087–1092.
Nelson, G., & Young, T. W. (1987). Journal of General Microbiology, 133, 1461–1469.
Ray, M. K., Uma devi, K., Umar, G. S., & Shivaji, S. (1992). Applied and Environmental Microbiology, 58, 1918–1923.
Kitano, H., Kataoka, K., Furukawa, K., & Hara, S. (2002). Journal of Bioscience and Bioengineering, 93, 563–567.
Hube, B., Monod, M., Schofield, D. A., Brown, A. J. P., & Gow, N. A. R. (1994). Molecular Microbiology, 14, 87–99. doi:10.1111/j.1365-2958.1994.tb01269.x.
Chi, Z. M., Liu, Z. Q., Gong, F., & Li, H. F. (2006). Journal of Ocean University of China, 3, 243–247.
Ferri, S., Miura, S., Sakaguchi, A., Ishimura, F., Tsugawa, W., & Sode, K. (2004). Marine Biotechnology (New York, N.Y.), 6, 625–632. doi:10.1007/s10126-004-0001-8.
Gao, L. M., Chi, Z. M., Sheng, J., & Ni, X. M. (2007). Applied Microbiology and Biotechnology, 77, 825–832. doi:10.1007/s00253-007-1210-7.
Ni, X. M., Chi, Z. M., Ma, C. L., & Madzak, C. (2008). Marine Biotechnology (New York, N.Y.), 10, 319–327. doi:10.1007/s10126-007-9067-4.
Rhishipal, R., & Philip, R. (1998). Bioresource Technology, 65, 255–266. doi:10.1016/S0960-8524(97)00179-X.
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., & Higgins, D. G. (1997). Nucleic Acids Research, 24, 4876–4882. doi:10.1093/nar/25.24.4876.
Chi, Z. M., Ma, C. L., Wang, P., & Li, H. F. (2007). Bioresource Technology, 98, 534–538. doi:10.1016/j.biortech.2006.02.006.
Sambrook, J., Fritsch, E. F., & Maniatis, T. (1989). Molecular cloning: A laboratory manual (2nd ed., pp. 367–370). Beijing: Cold Spring Harbor Laboratory, (Chinese translating ed.).
Chi, Z. M., He, S., & Yao, S. M. (2005). Enzyme and Microbial Technology, 37, 395–401. doi:10.1016/j.enzmictec.2005.01.040.
Laemmli, U. K. (1970). Nature, 227, 680–685. doi:10.1038/227680a0.
George, V., & Diwan, A. M. (1983). Analytical Biochemistry, 132, 481–483. doi:10.1016/0003-2697(83)90037-4.
Larson, M. K., & Whltaker, J. R. (1970). Journal of Dairy Science, 3, 253–261.
Berridge, N. J. (1952). Analyst (London), 77, 57–62. doi:10.1039/an952770057b.
Wang, L., Chi, Z. M., Wang, X. H., Liu, Z. Q., & Li, J. (2007). Annals of Microbiology, 57, 495–501.
Kurtzmam, C. P., & Fell, J. W. (1998). The yeast: A taxonomic study (2nd ed., pp. 1–100). The Netherlands: Elsevier.
Kumar, S., Sharma, N. S., Saharan, M. R., & Singh, R. (2005). Process Biochemistry, 40, 1701–1705. doi:10.1016/j.procbio.2004.06.047.
Gonzalez-Lopez, C. I., Szabo, R., Blanchin-Roland, S., & Gaillardin, C. (2002). Genetics, 160, 417–427.
Clark, S. J., Templeton, M. D., & Patrick, A. (1997). Microbiology, 143, 1395–1403.
Yamada, T., & Orydziak, T. D. M. (1983). Journal of Bacteriology, 154, 23–31.
Hirata, D., Fukui, S., & Yamashita, I. (1988). Agricultural and Biological Chemistry, 52, 2647–2649.
Gomi, K., Arikawa, K., Kamiya, N., Kitamoto, K., & Kumagai, C. (1993). Bioscience Biotechnology and Biochemistry, 57, 1095–1100.
Ueda, M., & Tanaka, A. (2000). Biotechnology Advances, 18, 121–140. doi:10.1016/S0734-9750(00)00031-8.
Yue, L. X., Chi, Z. M., Wang, L., Liu, J., Madzak, C., Li, J., & Wang, X. H. (2008). Journal of Microbiological Methods, 72, 116–123. doi:10.1016/j.mimet.2007.11.011.
Shimuta, K., Oda-Ueda, N., Washio, M., Oyama, H., Oda, K., & Tsura, D. (2000). Bioscience, Biotechnology, and Biochemistry, 64, 1542–1546. doi:10.1271/bbb.64.1542.
Yamashita, I., Hirata, D., Machida, M., & Fukui, S. (1986). Agricultural and Biological Chemistry, 50, 109–113.
Acknowledgement
This work was supported by National Infrastructure of Natural Resources for Science and Technology Program of China (No. 2005DKA21209).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, J., Chi, Z., Liu, Z. et al. Cloning and Characterization of a Novel Aspartic Protease Gene from Marine-Derived Metschnikowia reukaufii and its Expression in E. coli . Appl Biochem Biotechnol 159, 119–132 (2009). https://doi.org/10.1007/s12010-008-8400-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-008-8400-3