
Abstract
Purification of β-1,3-1,4-glucanase from the cell-free culture fluid of Bacillus subtilis GN156 by affinity chromatography of epoxy-activated sepharose 6B and ultrafiltration technique resulted in homogeneous J1 and partially purified pJ2 enzymes. The molecular weight and pI of J1 were 25 kDa and 3.5, respectively, while those for J2 were 90 kDa and 3.6, respectively. Both β-1,3-1,4-glucanase J1 and pJ2 had optimum pH values of 6–6.5 and an optimum temperature of 60°C. Both enzymes were not inhibited by Li2+ but were inhibited significantly by Ca2+, Cu2+, Mn2+ and Zn2+. However, J1 was slightly inhibited by Fe2+, while pJ2 was inhibited by Mg2+ as well. They were highly specific to only barley β-glucan. K m and V max values of J1 were 1.53 mg/ml and 8,511 μU/ml.min, respectively, while those for pJ2 were 4.36 mg/ml and 7,397 μU/ml.min, respectively. Degradation of barley β-1, 3-1,4-glucan resulted in four different oligosaccharides with 1,3 linkages triose, tetrose, pentose and a high molecular weight (HMW) with 1,3 linkage estimated from their mobilities. The quantitative degradation by the crude enzyme after of incubation yielded in descending order: triose, pentose and tetrose, while that of J1 in descending order yielded: pentose, triose and tetrose. The pJ2 showed low activity yielding a degradation pattern in descending order: pentose, triose, tetraose and a HMW polysaccharide.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
β-1,3-1,4-Glucan is a unique component of the cell walls in grasses [1–3], endosperm of cereal and brewer’s spent grain [4, 5]. To make use of these materials as carbohydrate sources for production of silage and valuable products such as lactic acid [6], in addition to enhanced digestibility in animal feed, the techniques of enzymatic treatment were applied [6, 7]. β-1,3-1,4-Glucanase or 1,3-1,4-β-d-glucan-4-glucano hydrolase [EC 3.2.1.73] is one of the key hydrolytic enzymes required. It is an endo-glycosidase which hydrolyses 1,4-β-d-glycosidic linkages in β-d-glucans containing mixed 1,3 and 1,4 linkages, but does not hydrolyse β-d-glucans containing only 1,3 or 1,4 linkages [8]. It is produced by various sources of organisms such as Bacillus brevis [9], Bacillus licheniformis [10], Bacillus subtilis [11–13], Bacteroides succinogenes [14], Clostridium thermocellum [15], Orpinomyces sp. PC-2 [16], Pseudomonas spp. [17], Ruminococcus flavefaciens [18], Streptomyces bovis JB1 [19], barley [20], wheat [21] and rice [22]. The important enzymes obtained for industrial application are usually from bacteria, especially hyperproducing selected strains of B. subtilis and Bacillus amyloliquefaciens [7]. B. subtilis GN156 was isolated from silage. It is able to produce various hydrolytic enzymes of β-1,3-1,4-glucanase, CM-cellulase, dextrinase, cellobiase, xylanase, polygalacturonase, polymethylgalacturonase and grass-degrading enzymes. Among these hydrolytic enzymes, β-1,3-1,4-glucanase did exhibit the highest activity. Its activities appeared at a wide range of pH (5.5–9.0) and temperature (40–60°C) [23]. These properties would be most appropriate for silage production which requires the active enzyme only at the beginning of hydrolysis where the pH is above 5.0. To understand more regrading grass degradation by this particular enzyme, purification and characterization of β-1,3-1,4-glucanases were undertaken in this study.
Materials and Methods
Microorganism
B. subtilis GN156 was used as an enzyme producer and was obtained from the culture collection at the Department of Biotechnology, Faculty of Agro-Industry, Kasetsart University, Thailand.
Enzyme Production
A colony of B. subtilis GN156 was grown in 5 ml of Nutrient Broth (NB) under aerobic condition with rotary agitation at 150 rpm for 18–20 h at 37°C. A 1.0% (v/v) of inoculum was transferred into 100 ml of NB, which contained 1% (w/v) carboxymethylcellulose (CMC), in a 250 ml flask. After 24 h of incubation with rotary agitation (150 rpm) at 37°C, the culture was centrifuged at 4°C for 15 min at 11,000×g, and the supernatant was stored at −20°C for further study.
Enzyme Purification
Purification of β-1,3-1,4-glucanase using affinity chromatography was performed by coupling Epoxy-activated Sepharose 6B (Bio-Rad, USA) with barley β-glucan according to instructions of the manufacturer (Bio-Rad, USA) and equilibrated with 5 mM citrate buffer pH 3.0 with a flow rate of 4 ml/min. Approximately 50 ml of cell-free culture supernatant (CFS) was dialyzed against 5 mM citrate pH 3.0 overnight and further loaded to β-glucan-coupled Epoxy-activated Sepharose (2.5 × 10 cm). The unbound proteins were removed with two bed volumes of 5 mM citrate buffer pH 3.0. Fractions were collected and eluted with a linear gradient of 0–0.25 M sodium chloride in the same buffer. Active fractions were pooled and dialyzed against 5 mM citrate buffer pH 6.0. Then, the dialysis solution was subjected to ultrafiltration using a membrane with a molecular weight cutoff of 50 kDa. Both filtrate and retentate were analysed for β-1,3-1,4-glucanase activity.
Determination of Enzyme Activity
β-1,3-1,4-Glucanase activity was determined by a modification of method of Okeke and Obi [24] using reaction mixtures containing 0.1 ml of sample and 0.1 ml of 1% (w/v) barley β-glucan (Sigma) in 50 mM citrate phosphate buffer (pH 5.5) at 50°C for 20 min. The amount of reducing sugar released was determined by the dinitrosalicylic acid (DNS) method [25]. One unit of enzyme was defined as the amount of enzyme that released 1 μmol of glucose per min.
The activities of carboxymethylcellulase (CMCase), xylanase, laminarinase and dextrinase were determined by the same method as above when CMC, xylan, laminarin and dextrin were used as substrates. One unit of CMCase, laminarinase and dextrinase was defined as β-1,3-1,4-glucanase activity while that for xylanase was as the amount of enzyme that released 1 μmol of xylose per min.
Protein Analysis
Protein concentrations were determined by absorbance measurements at 280 nm and the method of Lowry et al. [26]. Bovine serum albumin was used as a standard.
Molecular Weight Determination
Molecular weight of the purified enzyme was determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) according to Laemmli [27] using 10% acrylamide in the separating gel and stained with a Silver Stain Plus kit (Bio-Rad, USA). The pre-stained marker kit (catalog number 161-0318 Bio-Rad, USA) was used as a marker in this study.
Analysis of Native-Polyacrylamide Gel Electrophoresis (Native-PAGE)
The native purified enzyme was visualized using 7.5% acrylamide in a separating gel and stain with a Silver Stain Plus kit (Bio-Rad, USA). A HMW native protein marker kit (catalog number 17-0445-01 Amersham Bioscience, Sweden) was used as a marker in this study.
Determination of Isoelectric Point
Isoelectricfocusing was performed with an Ampholine™ PAGplate unit at pH 3.5–9.5 (Amersham Biosciences, Sweden) according to instructions of the manufacturer. The broad range pI calibration kits (catalog number 17-0471-01 Amersham Bioscience, Sweden) were used as markers.
Determination of Optimum pH and pH Stability
The optimum pH of enzyme activity was determined with a modification of the reaction mixture containing 100 μl of the enzyme solution and 100 μl of 1% (w/v) β-glucan in various pH buffers of 50 mM citrate pH 3.0–6.0, 50 mM phosphate buffer pH 6.0–8.0 and 50 mM glycine–NaOH pH 9.0–10.0. The reactions were incubated at 50°C for 20 min. For determination of pH stability, the enzyme reaction at each pH was incubated at 4°C for 2 h. Then, the remaining activities were assayed at the optimum pH and temperature of 50°C.
Determination of Optimum Temperature and Temperature Stability
For optimum temperature determination, enzyme reactions were performed at optimum pH in the same manner as pH determination. The optimum temperature of enzyme activity was determined by performing assays at various temperatures (70–20°C) for 20 min. To analyse the stability of enzyme, the reaction mixtures were incubated at the temperatures mentioned above for 30 min. The residual activities were then assayed at optimum pH and temperature.
Determination of K m and V max
The reactions were performed with various concentrations of substrate from 0.1 to 1% (w/v) under the optimum conditions and assayed for activity every 5 min for 0–30 min. The K m and V max were calculated from Lineweaver–Burke plots.
Determination of Enzyme Degradation Products by Thin-Layer Chromatography
β-1,3-1,4-Glucanase activity of 0.06 units/ml from the crude enzyme and purified enzyme were performed for the reaction mixtures of 0.6 ml of enzyme sample and 0.6 ml of 1% β-glucan and carried out at optimum pH and temperature for 24 h. The reactions after 6, 12 and 24 h of incubation were stopped by placing the tubes in boiling water for 10 min and oligosaccharides determined by thin-layer chromatography (TLC). The TLC was determined by a modified method of Akiyama et al. [22] using glucose and cellulooligosaccharides C2–C5 (Sigma) as standards. Approximately 2 μl of the degradation products and markers containing 2 μg/μl reducing sugar was applied on Kieselgel 60 (Merck) and developed for 90 min in a solvent mixture of butanol/isopropanol/ethanol/deionized water in the ratio of 2:3:3:2, respectively. The brown spots of sugars were developed by dipping the plates into 0.2% (w/v) orcinol in 10% (v/v) sulfuric acid in ethanol and later placing them at 100°C for 15 min. The relative percent intensity of each spot was further analysed by the GeneTools program version 3.06.04 (Syngene, USA) and was based on the spot of highest intensity defined as 100%.
The major degradation derived from enzymatic digestion of β-glucan (DPG) was defined as ratio of the intensity of each polysaccharide product and total products as shown in the equation below. ITP and ITT were the intensity of product and total degraded products, respectively.
Results
Purification of β-1,3-1,4-Glucanase
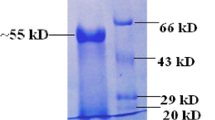
The crude enzyme from B. subtilis GN156 was loaded onto an Epoxy-activated Sepharose 6B column. Two peaks of enzyme activity were obtained using a linear elution gradient of 0–0.25 M sodium chloride derived from fractions 7–10 and 18–28 named AJ1 and AJ2, respectively (Fig. 1). Both AJ1 and AJ2 were ultrafiltrated with a filter membrane having a molecular weight cutoff of 50 kDa. The activity of AJ1 was detected from only the filtrate and named J1, while the activity of AJ2 was found only in the retentate and named pJ2. The β-1,3-1,4-glucanase J1 had a specific activity of 82.45 U/mg with a 0.65% recovery, while the specific activity of pJ2 was 555.11 U/mg with a 2.96% recovery, respectively. The results are summarized in Table 1. SDS-PAGE analysis of J1 yielded only band of approximately 25 kDa (Fig. 2), while four protein bands of approximately 40, 32, 25 and 18 kDa were resolved from pJ2 (Fig. 3). Further study on native PAGE of pJ2 was undertaken. A band of HMW and two of low molecular weight (LMW) are evident, shown in Fig. 4. The LMW band of pJ2 was in the same position as the protein J1 determined with GeneTools version 3.06.04 analysis (Syngene, USA). Thus, it could be concluded that pJ2 was a mixture of protein, some having the same size as J1 and HMW named J2. J2 was composed of three protein subunits of 40, 32 and 18 kDa. The sum of these three subunits was approximately 90 kDa.

Affinity chromatography of β-1,3-1,4-glucanases on Epoxy-activated Sepharose 6B column (2.5 × 10 cm column, equilibrated with 5 mM citrate phosphate buffer pH 5.5, with flow rate 4 ml/min, eluted with a linear gradient of 0–0.25 M sodium chloride in the same buffer)

SDS-PAGE of the purified J1 β-1,3-1,4-glucanase. M Prestained marker; B sample buffer; J1 purified J1

SDS-PAGE of β-1,3-1,4-glucanase preparation samples from each purification step. M Prestained marker; C crude enzyme; pJ2 retentrate J2 from UF 50 kDa

Native-PAGE of purified β-1,3-1,4-glucanase. J1 Purified J1; pJ2 partial purified J2; HMW high molecular weight band; LMW low molecular weight band; NM native marker
Further confirmation of J1 and pJ2 was determined by isoelectricfocusing. J1 and pJ2 exhibit pI values of 3.5 and 3.5 as well as 3.6, respectively. It was clearly shown that pJ2 was a mixture of at least two proteins of J1 and J2. However, the question still arose as to whether the activity in J2 was from only J1 appearing in both fractions or from synergism of J1 and J2.
Physical Characterization of β-1,3-1,4-Glucanase
The optimum pH values of J1 and pJ2 were pH 6.0 and 6.5, respectively (Fig. 5). Both J1 and pJ2 were stable at a pH range of 6.0–9.0 (Fig. 5) with 50% of the activity remaining at pH 9.0.

The pH effects on purified β-1,3-1,4-glucanase J1 (a) and pJ2 (b) from B. subtilis GN156 were carried out at 50°C for 20 min. The pH stability of the enzymes were determined at various pH values at 4°C for 2 h. Remaining activity was assayed at pH 6, 50°C for 20 min
The optimum temperature of J1 and pJ2 was 60°C for both (Fig. 6). J1 was stable at 20–50°C for 30 min with about 50% of activity remaining at 50°C. In contrast, pJ2 was less thermally stable; the remaining activity decreased to 20% at 20–40°C and was completely destroyed at 50–70°C (Fig. 6).

The effects of temperature on purified β-1,3-1,4-glucanase J1 (a) and pJ2 (b) from B. subtilis GN156 were carried out in citrate buffer pH 6. Temperature stability of the enzymes were treated at various temperatures for 30 min. The remaining activity was assayed at 50°C for 20 min
Effect of Divalent Cations
The effect of 10 mM Ca2+, Cu2+, Fe2+, Li+, Mg2+, Mn2+ and Zn2+ on enzyme activity of the β-1,3-1,4-glucanase J1 and pJ2 were studied as shown in Table 2. Neither enzyme was inhibited by Li +, but both were inhibited by Mn2+. Both were partially inhibited by Ca2+, Cu2+ and Zn2+ ions. J1 was slightly inhibited by Fe2+ but was not significantly inhibited by Mg2+. In contrast, pJ2 acted in the opposite way by being slightly inhibited by Mg2+ but was not significantly by Fe2+.
Substrate Specificity
The activities of J1 and pJ2 on 1% (w/v) barley-β-glucan, CMC, xylan, laminarin and dextrin were determined under optimum condition. J1 and pJ2 were active only on barley β-glucan. No activity was found against CMC, xylan, laminarin and dextrin. This indicated that β-1,3-1,4-glucanases from the GN156 strain could degrade only β-1,4 linkages of β-1,3-1,4-glucan. It is different from the β-1,3-1,4-glucanase from Streptococcus bovis and B. brevis which hydrolyse β-1,3 linkages of β-1,3-glucan in laminarin and β-1,3-1,4-glucan in lichenan [9, 19].
Kinetics Of J1 And pJ2
From Lineweaver–Burk plots, the K m V max and k cat of J1 were 1.53 mg/ml, 8,511 μU/ml.min and 0.074 sec−1, respectively, while these values for pJ2 were 4.36 mg/ml, 7,397 μU/ml.min and 0.091 sec−1, respectively. J1 had a 2.84-fold higher binding capacity than pJ2. However, the reaction rate and catalytic activity of both were nearly identical.
β-1,3-1,4-Glucan Degradation of Crude Enzyme, J1 and pJ2
Beta-glucan degradation by the crude enzyme preparation (J1 plus pJ2) was followed for 24 h. The mobilities of four different degradation products observed by TLC (Fig. 7) appeared at the intermediate position of the standard oligosaccharides (cellobiose, cellotriose, cellotetrose and cellopentose). Akita et al. [28] found that laminaribiose and laminaritriose (standard sugar for β-1,3 linkages) migrated ahead of standard sugars for β-1,4 linkages of cellobiose and cellotetrose, respectively. In addition, Johansson et al. [29] proposed that the action of β-1,3-1,4-glucanase cleaves β-1,4 linkages adjacent to a 1,3-linkage; the oligosaccharides thus obtained are the 1,4-linked building blocks of β-glucan with a 1,3-linked end group. Therefore, it was possible that the degraded products obtained in this study contained a β-1,3 linkage at the end providing different mobility compared to the standard β-1,4 linkage compound used here. It also appeared that the products obtained from each reaction had stopped at a specific position.

Thin-layer chromatography of degradation products of barley β-glucan by the β-1,3-1,4-glucanases from B. subtilis GN156 at various times of 0, 1, 6,12 and 24 h. M Standard oligosaccharide; C degradation products from crude enzyme; J1 degradation products from J1; pJ2 degradation products from pJ2; Cr crude enzyme
Four different oligosaccharides of triose (A), tetrose (B), pentose (C) and the oligosaccharides, which were larger than cellopentose (D), were further quantitatively analysed for the relative intensity of the TLC spots as shown in Fig. 8. Treatment with the crude enzyme preparation for 6 h yielded cellotriose as the major product. However, the amount of A, B and C products significantly increased from 12–24 h incubation (Fig. 8). The amount of A increased to 33 and 100% when the reaction time reached 12 and 24 h, respectively. Meanwhile, the higher-molecular-weight products B, C and D were detected at relative levels of 16.5, 43 and 20% at 12 h and 59, 86 and 0% at 24 h. It was clearly shown that degradation of barley β-glucan by the crude enzyme preparation yielded cellotriose in greatest quantity followed by cellopentose and cellotetrose in descending order as shown by DPG values in Table 3, while the HMW product D was not detected after 24 h.

Oligosaccharide products from barley β-glucan hydrolysis by β-1,3-1,4 glucanases from B. subtilis GN156. A Trioses, B tetraose, C penta, D oligosaccharides, which molecules were larger than cellopentaose
The degradation reaction product by J1 treatment was quite similar to those of the crude enzyme. After 24 h, the relative amounts of both tetrose and pentose produced were similar to the crude enzyme treatment (Fig. 8). However, the amount of triose was lower by about 60%. Therefore, treatment with J1 alone would delay degradation activity compared to the crude enzyme.
The degradation products with pJ2 which contained J1 and J2 were notably different from J1. After 24 h of incubation, of highest quantity was pentose (51%). The amount of degraded products was clearly lower than obtained with both crude enzyme and J1 alone (Fig. 8). The large oligosaccharides (D) were present in greater quantity (16%, 12 h; 21%, 24 h) than those from the crude enzyme and J1 treatment for 16%, respectively. It seems that the combination of J1 and J2 resulted in a lower degradation rate.
From the data in Table 3, it can be concluded that J1 and pJ2 exhibited different activities against barley β-glucan with respect to each oligosaccharide product. It could be proposed that the crude enzyme, J1 and pJ2 preferred different pattern of C3–C5–C4, C5–C3–C4 and C5–C3–C4–Coligomer in decreasing affinity, respectively.
Discussion
β-1,3-1,4-Glucanases from B. subtilis GN156 were effectively purified by two steps involving affinity chromatography and ultrafiltration. Using SDS-PAGE, native-PAGE and isoelectricfocusing, it was concluded that strain GN156 had two enzyme components of J1 and J2. Since both J1 and J2 occurred together in the same fraction (pJ2) and displayed β-1,3-1,4-glucanase activity, it was possible that the activity might come from only J1. However, when biochemical properties of metal ion effects and kinetic parameters of J1 and pJ2 were taken into account, both were clearly different in the effect of Fe2+ and Mg2+ as well as K m and V max values. Therefore, it was clearly shown that J2 also possessed β-1,3-1,4-glucanase activity as well. Hence, we concluded that strain GN156 had two enzyme components of J1 and J2 showing glucanase activity.
Recently, both unique and two enzyme components were found from various organisms. Their molecular weights were in the range of 25–35 kDa [9, 13, 15, 19, 20, 22]. The GN156 had two β-1,3-1,4-glucanase components with different molecular weight of 25 and 90 kDa as well as pI values of 3.5 and 3.6, respectively. Yuuki et al. [13] found two β-1,3-1,4-glucanases from B. subtilis HL-25 as well. However, an identical molecular weight of 24 kDa with different pI values of 8.55 and 8.75 were found. In the extract of germinating barley, two β-1,3-1,4-glucanases of 28 and 33 kDa with pI values of 8.5 and greater than 10 were also detected [20]. Therefore, according to the size and pI, both J1 and J2 were different from the ones found previously. In addition, this would be the first report of bacterial β-1,3-1,4-glucanases showing the lowest pI of 3.5 and 3.6 found.
The pH optimum and stability of J1 and pJ2 were in the range of 6–6.5 and 6–9, respectively. They were similar to the other β-1,3-1,4-glucanases previously found to have optimum pH values of 6–10 [7, 9, 19, 15]. Considering the effect of temperature, both J1 and pJ2 were notably active in the temperature range of 40 to 60°C which is similar to others showing activity from 45 to 65°C [7]. Therefore, they could be classified as mesophilically active enzymes which were not stable at 60°C where the remaining activity decreases to 20–50% of the original after 2 h. These enzymes were different from the thermophilic β-1,3-1,4-glucanases from B. brevis [9] and C. thermocellum [15] which showed 5% remaining activity at 65°C for 1 h and 65% at 75°C for 24 h, respectively. However, when kinetic parameters were taken into account, the J1 did show higher V max and lower K m values than the enzyme from Streptococcus bovis JB1 having a V max of 338.3 μU/ml.min and a K m of 2.78 mg/ml [19], indicating that this would be a promising enzyme for commercial use in the future.
β-Glucan degradation by the crude enzyme preparation yielded higher levels of triose than those of J1 and pJ2. It is possible that higher activities and synergism of the ratio of proper enzyme components applied to the reaction may affect the higher yield of triose. However, considering the degradation pattern of DPG, the relative yields of trioses and pentoses were similar and that of tetroses notably lower. These results are different from the glucanase of rice which gave only two major oligosaccharides with mobility intermediate between cellobiose–cellotriose and cellotriose–cellotetrose supposedly cellotriose and cellotetrose with a 1,3 linkage at the end [22]. In addition, the product from the crude enzyme (J1+pJ2) was also different from those of Bacillus macerans and B. amyloliquefaciens where glucanase-catalyzed β-glucan degradation yielded oligosaccharide trimers, tetramers, pentamers and dimers of approximately 50–60, 30, 4–6 and <1%, respectively [30]. In our study, both monomer and dimer were not detected. Hence, it would be concluded that β-glucan degradation activity of strain GN156 preferred to attack the molecular structures higher than seven glucose units and above. In addition, enzyme activity was specific for β-d-glucan containing 1,3 and 1,4 linkages and hydrolysed to only 1,4-β-d-glycosidic linkages indicating similarity to the enzymes studied by Akiyama et al. [22] and Schimming et al. [15]. This would leave the oligosaccharide products with a β-1,3 linkage at the end. With this high level of substrate specificity, it would be expected that J1 and pJ2 provide some useful and interesting oligosaccharides for the other microorganisms involved in grass or spent-grain fermentation for silage production.
References
Brett, C. T. Z., & Waldron, K. W. (1996). Physiology and Biochemistry of Plant Cell Walls. 2nd edn. London: Chapman & Hall.
Buckeridge, M. S., Vergara, C. E., & Carpita, N. C. (2001). Insight into multi-site mechanisms of glycosyl transfer in (1–4) β-glycans provided by the cereal mixed-linkage (1-3), (1-4) β-d-glucan synthase. Phytochemistry, 57, 1045–1053.
Carpita, N. C., & Gibeaut, D. M. (1993). Structural models of primary cell walls in flowering plants: consistency of molecular structure with the physical properties of the walls during growth. The Plant Journal, 3, 1–30.
Roulin, S., & Feller, U. (2001). Reversible accumulation of (1–3, 1–4)-β-glucan endohydrolase in wheat leaves under sugar depletion. Journal of Experimental Botany, 52, 2323–2332.
Santos, M., Jimènez, J. J., Bartolomè, B., & GÒmez-Cordovès, C. (2002). Variability of brewer’s spent grain within a brewery. Food Chemistry, 80, 17–21.
Shindo, S., & Tachibana, T. (2004). Production of L-lactic acid from spent grain, a by-product of beer production. Journal of the Institute of Brewing, 110, 347–351.
Planas, A. (2000). Review: Bacterial 1,3-1,4-β-glucanase:structure, function and protein engineering. Biochimica et Biophysica Acta, 1549, 361–382.
Dixon, M., & Webb, E. C. (1979). Enzyme. 3rd. New York: Academic.
Louw, M. E., Reid, S. J., & Watson, T. G. (1993). Characterization, cloning and sequencing of a thermostable endo-1,3-1,4-glucanase-encoding gene from an alkalophilic Bacillus brevis. Applied Microbiology Biotechnology, 38, 507–513.
Malet, C., Jiménez-Barbero, J., Bernabé, M., Brosa, C., & Planas, A. (1993). Stereochemical course and structure of the products of the enzymic action of endo-1,3-1,4-beta-d-glucan 4-glucanohydrolase from Bacillus licheniformis. Biochemecal Journal, 296, 753–758.
El-Helow, E. R., & El-Ahawany, A. M. (1999). Lichenase production by catabolite repression-resistant Bacillus subtilis mutants: Optimization and formulation of an agro-industrial by-product. Enzyme and Microbial Technology, 24, 325–331.
Tang, X. - J., He, G. - Q., Chen, Q. - H., Zhang, X. - T., & Ali, M. A. M. (2004). Medium optimization for the production of thermal stable β-glucanase by Bacillus subtilis ZJF-1A5 using response surface methodology. Bioresource Technology, 93, 175–181.
Yuuki, T., Tezuka, H., & Yabuuchi, S. (1989). Purification and some properties of two enzymes from a β–glucanase hyperproducing strain, Bacillus subtilis HL-25. Agricultural and Biological Chemistry, 53, 2341–2346.
Erfle, J. D., Teather, R. M., Wood, P. J., & Irvin, J. E. (1988). Purification and properties of a 1,3-1,4-beta-d-glucanase (lichenase,1,3-1,4-beta-d-glucan-4-glucanohydrolase, EC 3.2.1.73) from Bacteroides succinogenes cloned in Escherichia coli. The Biochemical Journal, 255, 833–841.
Schimming, S., Schwarz, W. H., & Staudenbauer, W. L. (1991). Properties of a thermoactive β-1,3-1,4-glucanase (lichenase) from Clostridium thermocellum expressed in Escherichia coli. Biochemical and Biophysical Research Communications, 177, 447–452.
Chen, H., Li, X. L., & Ljungdahl, L. G. (1997). Sequencing of a 1,3-1,4-beta-d-glucanase (lichennase) from anaerobic fungus Orpinomyces strain PC-2: properties of the enzyme expressed in Escherichia coli and evidence that the gene has a bacterial origin. Journal of Bacteriology, 179, 6028–6034.
Kitamura, E., Myouga, H., & Kamei, Y. (2002). Polysaccharolytic activities of bacterial enzymes that degrade the cell walls of Pythium prophyrae, a causative fungus of red rot disease in Porphyra yezoensis. Fisheries Science, 68, 436–445.
Flint, H. J., Mcpherson, C. A., & Bisset, J. (1989). Molecular cloning of genes from Ruminococcus flavefaciens encoding xylanase and β-1,3-1,4-glucanase activities. Applied and Environmental Microbiology, 55, 1230–1233.
Ekinci, M. S., McCrae, S. I., & Flint, H. J. (1997). Isolation and overexpression of a gene encoding extracellular β–1,3-1,4-glucanase from Streptococcus bovis JB1. Applied and Environmental Microbiology, 63, 3752–3756.
Woodward, J. R., & Fincher, G. B. (1982). Purification and chemical propertied of two β-1,3-1,4-glucan endohydrolases from germinating barley. European Journal of Biochemistry/FEBS, 121, 663–669.
Lai, D. M., Hoj, P. B., & Fincher, G. B. (1993). Purification and characterization of β-1,3-1,4-glucan endohydrolases from germinated wheat (Triticum aestivum). Plant Molecular Biology, 22, 847–859.
Akiyama, T., Kaku, H., & Shibuya, N. (1996). Note: Purification and partial characterization of an endo-1,3-1,4-β-glucanase from rice Oryza sativa L. Bioscience, Biotechnology, and Biochemistry, 60, 2078–2080.
Apiraksakorn, J., Buwjoom, T., & Nitisinprasert, S. (2006). Characterization of grass degrading bacteria active on β–1,3-1,4-glucans from Bacillus subtilis GN156 protential use for grass silage-making. Kasetsart Journal (Nat. Sci.), 40, 136–147.
Okeke, B. C., & Obi, S. K. C. (1995). Saccharification of agro–waste materials by fungal cellulases and hemicellulases. Bioresource Technology, 51, 23–27.
Miller, G. L. (1959). Use of dinitrosalicylic acid reagent for determination of reducing sugar. Analytical Chemistry, 31, 426–428.
Lowry, O. H., Rosembrough, N. J., Farr, A. L., & Randall, P. L. (1951). Protein measurement with the folin phenaol reagent.. Journal of Biological Chemistry, 193, 265–275.
Laemmli, U. K. (1970). Cleavage of structural protein during the assembly of the head of bacteriophage T4. Nature, 227, 680–685.
Akita, M., Kayatama, K., Hatada, Y., Ito, S., & Horikoshi, K. (2005). A novel -β-glucanase from Bacillus haoldurans C-125. FEMS Microbiology Letters, 248, 9–15.
Johansson, L., Virkki, L., Anttila, H., Esselström, H., Tuomainen, P., & Sontag-Strohm, T. (2006). Hydrolysis of β-glucan. Food Chemistry, 97, 71–79.
Olsen, O., Borriss, R., Simon, O., & Thomson, K. K. (1991). Hybrid Bacillus β-1,3-1,4-glucanase: engineering thermostable enzymes by construction of hybrid genes. Molecular and General Genetics, 225, 177–185.
Acknowledgement
This work was supported by Commission on Higher Education, Ministry of education, Royal Thai Government and Research Grant of Graduate School, Kasetsart University, Bangkok, Thailand.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Apiraksakorn, J., Nitisinprasert, S. & Levin, R.E. Grass Degrading β-1,3-1,4-d-glucanases from Bacillus subtilis GN156: Purification and Characterization of Glucanase J1 and pJ2 Possessing Extremely Acidic pI . Appl Biochem Biotechnol 149, 53–66 (2008). https://doi.org/10.1007/s12010-007-8058-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-007-8058-2