
Abstract
Recently, magnesium (Mg) and its alloys have attracted more attention because of their biodegradability and fascinating mechanical properties in the medical field. However, their low corrosion resistance and high degradability in the body have a great effect on mechanical stability and cytocompatibility, which hinders its clinical applications. Therefore, here we introduce a bifunctional composite coating composed of polycaprolactone and synthesized hydroxyapatite nanoparticles (HA-NPs) loaded with simvastatin deposited on the AZ31 alloy via electrospinning technique. The synthesized HA-NPs and composite nanofibers layer were characterized using TEM, FE-SEM, FTIR, and XRD to understand the physiochemical properties of the composite nanofibers compared to pristine polymer and bare alloy. Corrosion resistance was evaluated electrochemically using potentiodynamic polarization and EIS measurements, and biodegradability was evaluated in terms of pH and Mg ions release in SBF solution. The as-prepared coating was found to retard the corrosion and increased the osteocompatibility as resulted in cell culture test, a higher cell attachment and proliferation on the implant biointerface, in addition to releasing simvastatin in a controlled platform.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Magnesium (Mg), a light metal with appealing properties such as a low density of 1.74 g/cm3 and good mechanical properties, is suitable for different applications, such as aerospace,1 the automotive industry,2 telecommunications,3 and medical applications.4,5 Recently, the demand for a biomaterials substitute has attracted more attention to Mg, a biodegradable metal that might be a promising candidate for bone fixation and tissue regeneration.6 Mg and its alloys show a unique advantage in repairing damaged tissues in orthopedic application, as Mg could be a temporary implant because of its biodegradability in the body.7,8 Moreover, Mg ions are essential in living cells, for instance, about 50% of Mg ions in the human body are found in bone. Due to its biodegradability, it can eliminate the second surgery requirement as it eventually degrades and corrodes in the body after healing the surrounding tissues.9,10,11 Another advantage is that Mg ions are nontoxic and can be excreted from the body via the kidneys.12 On the other hand, implantation of Mg in the body is limited by its poor corrosion resistance which affects its mechanical properties for long-term implantation. Up to date, surface modification could be a promising way to improve corrosion resistance and biocompatibility of the implant.13,14 There is a different biofunctional coating made of polymers, ceramics, or composites that can be applied to Mg and its alloys.14
Electrospinning is a versatile technique that can produce nanofibers with web-like architecture that mimics extracellular matrix (ECM), improving cell adhesion and attachment.15,16,17,18 For hard tissue applications, synthetic biodegradable polymers, such as poly (lactide-co-glycolide) (PLGA),19 poly (ε-caprolactone) (PCL), and poly (l-lactic acid) (PLLA),20,21,22,23 and their copolymers, have been used on a large scale because of their favorable biocompatibility and biodegradability. Among these polymers, PCL is an FDA-approved biomaterial characterized with its biodegradability, low cost, flexibility, and high biocompatibility with osteoblasts, which makes it an excellent candidate for durable implants.24 However, most of the polymers, including PCL, have restrictions in orthopedic applications because they are hydrophobic, not stiff enough, and lack bioactivity.25 A combination of these polymers with bioactive inorganic phases, such as hydroxyapatite (HA), bioactive glass (BG), and tricalcium phosphate, might overcome their low biodegradability.25,26 HA is highly bioactive which helps to form a mineral-like apatite phase on the material surface that ultimately induces a direct bond with the bone tissues.27,28,29 Much effort has been devoted to using HA abilities in different composites incorporated in different kinds of polymers, such as PLGA/HA, PLLA/HA, PCL/HA, poly (3-hydroxybutyrate) (P (3HB))/HA, among others.29,30 Compared with microfillers, the larger specific surface area of HA nanofillers affects organic-based composites much better with more bioactivity. To improve composite coating bioactivity on the Mg implant loading drugs into these biomaterials was performed.31 Recently, biodegradable materials functionalized with antibiotics, growth factors, chemotherapeutic agents, and antiinflammatory drugs have been introduced into drug delivery systems, showing significant improvement.32,33
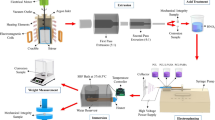
In the present study, the major objective is to enhance corrosion resistance and improve osteointegration using HA/SIM composite coating. Figure 1 is a schematic diagram that summarizes the experimental procedure of our study. Bone tissue engineering ECM contains collagen nanofibers and HA nanoparticles. Piskin et al. found that application of SIM drug has a positive effect, as it can significantly increase bone formation.34 We hypothesized that the combination of HA/SIM incorporated with PCL polymer in composite nanofiber could promote apatite formation on the implant biointerface by biomineralization capability and bone formation. On the other hand, corrosion products on Mg alloy surface rich of apatite labile ions could improve the biological response. Moreover, SIM release could improve bone formation and MC3T3-osteoblast cells proliferation.

Illustrative diagram explains the route steps of Mg alloy coated with electrospinning technique to the outer surface of Mg alloy using the different polymeric and composite nanofibers
Materials and methods
Materials
The materials used in this study were as follows: Poly (ε-caprolactone) PCL (average Mw: 80,000), ammonium phosphate dibasic (NH4)2HPO4, Hank’s balanced solution, and simvastatin were purchased from the Sigma-Aldrich Company (South Korea). Calcium nitrate tetrahydrate (Ca(NO3)2.4H2O) (Alfa Aesar Company, South Korea), aqueous ammonia solution, chloroform and methanol, ethanol were purchased from Samchun Pure Chemical, South Korea. Mg, Al, and Zn alloy with design AZ31 were obtained from Alfa Aesar Company, South Korea, then resized with dimensions of 12 × 12 × 6.35 mm3, and further polished with 200 and 2000 grit SiC paper. The samples were degreased in acetone for 10 min, then sonicated for 15 min, then immersed in distilled water, and finally dried at 60°C.
Synthesis of Hydroxyapatite Nanoparticles (HA-NPs)
Hydroxyapatite nanoparticles (HA-NPs) were synthesized using wet chemical precipitation, as reported previously.28 Briefly, a 0.6 M (NH4)2HPO4 aqueous solution was added dropwise to a 1 M Ca(NO3)2.4H2O aqueous solution with strong stirring overnight. The ratio of Ca/P was 1.67 when these solutions were mixed to produce HA-NPs; to adjust the pH of the solution, ammonium solution was added until the pH reached 11. Thereafter, the solution was stirred for 4 h at room temperature (RT) and then was heated to 90°C for 1.5 h under continuous stirring. Then, the resulting precipitate was kept at RT under vigorous stirring for 24 h to produce a homogeneous solution. Finally, the solution was filtered by using distilled water to wash the precipitate several times, then by alcohol, and finally by a mixture of alcohol and distilled water (volume ratio = 1:1). The final precipitated HA-NPs were dispersed in alcohol to modify the agglomeration of the obtained powder. The powder was dried under vacuum for 24 h at 70°C and then calcined at 650°C for 4 h.
Preparation of PCL/HA-NPs/simvastatin nanocomposite coating
Nanocomposite fibers were prepared by electrospinning. Prior to mixing nanoparticles and polymer, HA-NPs were dispersed in a mixture of chloroform/methanol with ratios of 3:1 v/v and stirred for 6 h with a magnetic stirrer.35,36 To this mixture, a weighed amount of PCL was added to obtain 8 wt% of polymer composite solutions, which were then stirred overnight. A 10 wt% of the nanoparticles was added to the mixture of the PCL. Then, the prepared solution was sonicated for 15 min for further dispersion, after which SIM (5 wt% of the PCL) was added before electrospinning the composite solution. The electrospinning was set up at a DC voltage of 16 kV for 10 min with a flow rate of 1.45 ml/h for each surface to keep all samples at the same conditions, upon applying high voltage, a fluid jet was ejected, as the jet accelerated toward the collector, the solvent evaporated and the nanofibers were collected on the Mg alloy surface, and thereafter samples were vacuum-dried. Hereafter, in this study, the samples coated with PCL, PCL with HA-NPs, and PCL with HA-NPs/SIM are referred to as PCL, PCLH, and PCLHS, respectively.
Surface characterization
The different morphologies of the composite nanofibers on the Mg alloy surface were characterized by using a field emission scanning electron microscope (FE-SEM, Carl Zeiss supra-40 VP Germany). The X-ray diffraction (XRD, Rigaku, Japan) measurements were performed in a continuous scanning mode at a rate of 5°/min. Fourier transform infrared (FTIR) spectra of the different samples were directly measured using an FTIR, ABB Bomen MB100 spectrometer, scan range 4000–400 cm−1 in transmittance mode (PerkinElmer, Spectrum GX, USA), and characterization for the synthesized HA-NPs was performed using transmission electron microscopy (BIO-TEM HITACHI H-7650 Japan).
Electrochemical corrosion test
The electrochemical corrosion test was performed using a potentiostat device (ZIVE SP1, South Korea). The test was performed on a cell containing SBF solution at 37°C, pH = 7.4. The potentiodynamics were measured by using a linear sweep on Ag/AgCl saturated in KCl as the reference electrode and platinum as the counter electrode. During the measurements, the initial potential (E0) was 2.5 V and the final potential (E1) was 1.5 V. A STEP-PSTAT control was used, and the ranges of the current and voltage were 10 µA and 10 V, respectively; the scan rate was set at 5.0 mV/s. The exposed specimen area measured was 0.875 cm2. Potentiostatic EIS was measured with the same setup of the cell, with initial, middle, and final frequencies at 1 kHz, 100 MHz, and 0.1 Hz, respectively. All samples were exposed to the solution before testing until a steady state of the open-circuit potential was reached. The EIS spectra were analyzed and fitted in terms of an equivalent circuit using the commercial software ZMAN.
Immersion test
According to the ASTM standard G31, the immersion test was performed where samples were immersed in SBF solution at 37°C for 1, 3, and 7 days; the ratio of the surface area to the solution volume was 1:30 cm2/mL.9 The SBF solution was prepared as in our previous work.37 Thereafter, the surface of the bare and coated samples was observed by using FE-SEM after the immersion test. In addition, the samples biodegradability was analyzed in terms of the pH of the extract at different intervals of time. Furthermore, ICPS (inductively coupled plasma spectroscope) was used to measure the amount of Mg ions released from the bare and coated samples, and the extraction solution was diluted with DI water.
Drug release test
Simvastatin loaded with a PCL/HA-NPs nanocomposite on AZ31 alloy was soaked in 15 ml PBS at 37°C for predetermined times; 3 ml of PBS was removed for sampling and replaced with fresh PBS. Using a UV spectrophotometer, the amount of drug release was calculated at a wavelength 238 nm of maximum absorbance of simvastatin in PBS. The calibration curve of the simvastatin in PBS solution was calculated by measuring the absorbance versus different known concentrations of simvastatin.38 For this interval, the calibration curve fits the Lambert and Beer’s law: A = 0.0253 + 0.53621 × C, where A is absorbance and C is concentration (mg/ml). In this test, samples were triplicated and an average value was reported.
Cytocompatibility and cell proliferation
Cellular response in terms of cytotoxicity and cell compatibility of the composite nanofiber-coated AZ31 alloy was investigated with CCK-8 and compared with the bare sample, with mouse preosteoblast cells (MC3T3-E1). The cells were cultured at 37°C under 5% CO2 in alpha-MEM (α-MEM; Gibco, Tulsa, OK, U.S.) with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin–streptomycin. The extract was prepared by immersing the samples in 20 ml α-MEM for 24 h at 37°C. Cells were grown on the extract for 1, 3, and 5 days, and the proliferation was evaluated by using Dogindo’s cell-counting kit-8; 500 μL of MC3T3-E1 osteoblast cell suspension (7.500 cells/well, alpha MEM with 10% FBS, and 1% penicillin–streptomycin) was dispensed in a preincubated 48-well plate containing an amount of extract for the designated time. Cell proliferation of the different samples was observed at 1, 3, and 5 days, where 100 μL of cell suspension extract, added to100 μL of Alpha-MEM from each well, was transferred to 96-well plates, and 20 μL of cck-8 solution was added to each well, followed by 4-h incubation at 37°C. Absorbance of each well’s content was measured by using a microplate reader (Tecan, Austria) at a wavelength of 450 nm. To observe cell attachment, the cell morphology of the different samples was observed by using FE-SEM after 1 and 3 days. This was performed by cell fixation with 3.5% glutaraldehyde for 1.5 h, with dehydration of the sample by regular increases of the concentration of ethanol from 25%, 50%, 75%, and 100%, then dried overnight and mounted for FE-SEM imaging.
Results and discussion
Materials characterization
Hydroxyapatite is a bioceramic material that has excellent bioactivity behavior. Figure 2 shows the morphology and characterization of the synthesized HA-NPs. Figure panels show the TEM image, where it has nanorod-like shapes. To clearly assess the assembling structure of the HA nanorods, XRD peaks of HA show a higher crystal structure at 2θ = 26° and 2θ = 32° as shown in Fig. 2c. Fourier transform infrared (FTIR) spectroscopy was employed to characterize the different functional groups of HA-NP Ca10 (PO4)6(OH)2. The first indication for formation of HAp is in the form of a strong complex broad FTIR band centered at about 1000-1100 cm−1 due to asymmetric stretching mode of vibration for the PO4 group. The band at 560 cm−1 corresponds to symmetric P-O stretching vibration of the PO4 group. FTIR spectrum shows a transmission at 560 cm−1 assigned to the characteristic phosphate (v4) band, at 1024 cm−1 assigned to the phosphate (v3) band, and at 3570 cm−1 assigned to the (OH) stretching group of the fabricated hydroxyapatite nanoparticles.39

Characterizations of synthesized HA-NPs: (a) TEM image, (b) FTIR spectrum, (c) Wide XRD patterns, and (d) narrow XRD patterns
The resulting PCL nanofibers and nanocomposites are shown in Fig. 3. FE-SEM images show a uniform distribution of the nanofibers on the AZ31 alloy surface among the coated samples. The PCLH nanocomposite morphology was affected by nanoparticles substitute; for instance, increasing the fiber diameters affects the nanocomposite fibers in the drug loading. The successful electrospinning of PCL, PCL dispersion with HA-NPs, and simvastatin formed dense, well-adhered nanocomposite fibers on the AZ31 alloy surface. The average fiber diameter was 354 ± 238 nm for the PCL solution, 367 ± 175 nm for the PCLH composite nanofiber, followed by 387 ± 215 nm for the PCLHS fiber. The electrostatic force generated between the nozzle and AZ31 Mg alloy directly affected the fiber morphology.40

FE-SEM images of the electrospun plain and composite nanofibers morphology and fiber diameters distribution
The crystal structure and phase composition of the different samples, as characterized by using X-ray diffraction, are illustrated in Fig. 4. In the pattern of the bare Mg alloy, peaks of Mg found by X-ray were observed. When we looked at the samples coated with PCL, PCLH, and PCLHS, peaks of PCL appeared at 2θ = 21.6 and at 2θ = 23.8.41 In the PCLH samples, the HA peak appeared at 2θ = 32,28 which indicated formation of the nanocomposite on the AZ31 alloy interface; the inset figure in Fig. 3 shows a low peak intensity compared with the Mg peaks of the substrate. Moreover, there were no characteristic peaks for the drug, because of the low drug concentration compared to that of the other materials.

XRD patterns of bare AZ31 alloy and different coated Mg alloy samples with pristine and composite nanofibers (Coding refer: 1 = Mg, 2 = PCL, 3 = HA)
The chemical bonding of the different samples was characterized with FTIR analysis, as shown in Fig. 5. The absorption bands on the PCL coating related to stretching vibrations were explained for both plain PCL polymer and the composite coating. Peaks at 2944 and 2865 cm−1 were assigned to the asymmetric CH2 stretching. The stretching peaks at 1722 cm−1 were assigned to the ester and lactone carbonyl; the one at 1293 cm−1 was assigned to C–O and C–C stretching in the crystalline phase; and the one at 1241 cm−1 was assigned to asymmetric C–O–C stretching. There was a significant decrease in the C–O and C–C crystalline structure on the composite fibers compared to that of the PCL coating. The composite fiber samples’ characteristic (PO4)3 absorption bands attributed to hydroxyapatite nanoparticles were found at 564 and 603 cm−1. The FTIR spectrum further confirmed the presence of HA and PCL in the composite coating.

FTIR absorption spectra of pristine and composite nanofiber of PCL, PCL-loaded HA-NPs and SIM drug samples on AZ31 alloy
Sample biodegradability
Potentiodynamic polarization test was used to detect the formation of a protective film on the alloy surface and to see how the electrospun nanofibers and composites affected the corrosion protection in SBF solution. The related polarization curves are presented in Fig. 6a, and the resulting parameters are summarized in Table 1. Basically, high corrosion resistance shows a low corrosion current density, high metal potential, and high polarization resistance. Here, the resulting corrosion potentials (Ecorr) of the bare Mg alloy, PCL, PCLH, and PCLHS samples were −1.44, −1.39, −1.42, and −1.41 V, respectively. On the other hand, the corrosion current densities (Icorr) values are 6.26, 0.017, 5.33, and 0.812 µA/cm2 for bare, PCL, PCLH, and PCLHS, respectively; these values are summarized along with anodic Tafel slopes (ba), cathodic Tafel slopes (bc), and corrosion rate (CR) in Table 1. Current results show improvement on corrosion current density values which were found to be significantly decreased, as a result, a lower corrosion rate was obtained.5,17 These results could show an increase in the corrosion potentials and decrease in corrosion current densities compared to the bare sample, which indicates higher corrosion resistance as mentioned previously. The improvement in the corrosion resistance indicates that the nanofiber film on the surface could prevent a direct reaction with chloride ions in SBF solution with the inner surface of the alloy.42 The substrates coated with only PCL show more corrosion resistance than did the bare sample; however, the corrosion potential decreased after adding the HA-NPs as compared to PCL only, as shown in Fig. 6a, perhaps because the added hydrophilic nanoparticles, such as HA, are more susceptible to reaction with SBF, forming CaCl2 and other corrosion products. The value of the polarization resistance (Rp), which is inversely proportional to the corrosion current density, was also calculated using Stern–Geary equation (1).37
The polarization resistance of PCL, PCLH, and PCLHA coatings was 10.71, 8.38, and 93.25 (Ω/cm2), respectively. These values are significantly higher than that of bare alloy, 5.7 (Ω/cm2).

(a) Potentiodynamic polarization curves of the different samples in terms of working electrode potential and current density. (b) Nyquist plot and equivalent circuit for bare and coated samples. (c) Equivalent circuit of the different samples
Electrochemical impedance spectroscopy (EIS) is an efficient way to better explain the corrosion process at AC voltage compared to DC potentiodynamics. The resulting Nyquist curves of the bare alloy, PCL, PCLH, and PCLHS samples are shown in Fig. 6b. The results were calculated and fitted after immersion of the samples in SBF solution (Table 2). The coated samples showed a single capacitive loop at all high frequencies, while the loops of bilayered PCLH, PCLHS, and monolayered PCL coating have significantly larger diameters than the bare alloy. The presence of only one capacitive loop in the coated samples indicated that the electrochemical reaction area at the metal/coating interface was still small in this immersion period. This phenomenon made it hard to separate the time relaxation of the physical impedance of the coating from that of the electrochemical reaction impedance at the Mg alloy/coating interface.43 The EIS results of the bare sample can be fitted well using a simple equivalent circuit as indicated in Fig. 6c which was composed of Rs, representing the electrolyte resistance, capacitance Q1, and resistance R1 associated with the compact oxide layer of the bare sample. However, after coating, the circuit increased two resistance elements because of the coating film; Q2 is the coating capacitance, and R2 is the coating pore resistance and charge transfer. In general, the higher is the charge transfer resistance, the higher is the sample corrosion resistance. The charge transfer resistances (R2) of the PCL coating and of the bilayer PCLH and PCLHS coatings are 21.12, 19.07, and 33.51 kΩ, respectively, which are all significantly higher than that of the bare sample, which has a resistance of 1.16 kΩ. This improvement in corrosion results can be attributed to the formation of a thin polymer layer on the surface, which provides sufficient protection for the substrate against corrosion. Moreover, the coating resistance of PCL nanofiber is the highest compared to that of the other coated samples. It is revealed from our results that a composite has a higher average fiber diameter because of the nanoparticles, which affect the porosity, permitting the SBF solution to penetrate into the substrate, as well as surface hydrophobicity, which mainly affects the resistance of the substrate.27 Corrosion products are another important issue, because they affect the surrounding tissues when biomaterials are implanted into the human body. Figure 7 shows the FE-SEM images of the corroded surfaces after immersion of the different samples in SBF solution. The surface of the bare sample shows cracks, which look small after 1 day of immersion and increase after 7 days of immersion because of the distortion of the outer surface and high reactivity.44 The PCL coating samples show small pores and cracks, which increased with immersion time. HA nanoparticles on the nanofibers play a role in forming HA as a biomineralization and corroding the implant interface.

FE-SEM images indicate surface morphology of bare and coated samples after immersion in SBF solution for 1, 3, and 7 days at 37°C
Biodegradability of Mg implant could be represented in chemical equation (2) in terms of pH value and Mg ions release. Hydroxide group represents the alkalinity of the sample when immersed in the SBF solution. Figure 8a shows the changes of pH of the SBF solutions containing bare alloy, PCL, PCLH, and PCLHA. Because of the degradation process, all samples had an alkaline solution. However, compared to the AZ31 alloy, PCL, PCLH, and PCLHA had lower pH after 7 days of immersion. The pH of the bare alloy reached 11.5, whereas that of the coated samples was 9.6. Higher pH can lead to complications, like hemolysis; so controlling the local pH is very important for bone implant applications.15

(a) Mg ion release from the coated substrates compared with the bare alloy in SBF solution. (b) pH value change in SBF solution containing different samples conditions in different intervals of times
Mg ions are another factor to indicate biodegradability of the Mg implant. In Fig. 8b, more Mg ions are released from the bare alloy than from the coated samples. The PCL coating showed the lowest ion release among the coating samples, perhaps because the hydrophilic effect of hydroxyapatite can increase the dissolution of Mg, since hydrophilic particles facilitate ion penetration.31
Drug release test
Figure 9 shows the drug release profile of nanocomposite fibers containing 5 wt% of the drug. Two phases can be observed in the release behavior, an initial rapid release followed by a sustained release. After the first day, 65.43% of the drug was released into the PBS medium as the first phase, and 15.85% of the loaded drug was released in the second phase. PCL is a hydrophobic and semicrystalline polymer. As shown in the previous section, it exhibits a low degradation rate, and simvastatin is also a hydrophobic drug. (Its solubility in water is about 0.03 g/l.) We hypothesize that the drug may be incorporated into the amorphous part of the semicrystalline polymer; so the drug is released first by diffusion from these composites. Then, the release rate becomes slower as the drug is released by degradation of the polymer, showing a uniform and slow degradation. The results of our study show that this nanocomposite fiber web on AZ31 alloy can release simvastatin in a controlled manner, making it a suitable drug delivery device.

Cumulative in vitro release profile of simvastatin from the coated substrate in PBS solution. The data are calculated as the mean ± SD (n = 3 and p < 0.05)
Cytocompatibility and cell proliferation
In vitro cytocompatibility evaluation is a basic biological test for biomaterials. Cell proliferation is the first sign of biocompatibility using materials extraction, as well as cell attachment to the biomaterials interface. Figure 10a shows CCK assay data, and PCLH samples show higher cell proliferation compared to bare alloy of AZ31 after 24 h, while PCLHS shows the highest cell growth among the samples. Significant cell proliferation was noticed after 3 and 5 days of incubation for PCLHS, and it was clear that the composite coating resulted in higher proliferation and continuous cell growth revealing the osteogenic effect of simvastatin on cell proliferation. These results confirm that HA-NPs and SIM play an important role in cell proliferation and bone cell differentiation.15 In contrast, the extract of the bare samples showed lower cell proliferation than with the coated samples but does not show any toxicity to the cells. We predicted that the number of cells would statistically increase more for the coated AZ31 alloy at each time interval than for the bare samples, meaning that all the coated samples have a significantly better initial biocompatibility than the bare alloy group. To confirm this hypothesis, the FE-SEM images show morphologies of the MC3T3 mouse osteoblast cell line after 1 and 3 days of the culture test. Figure 10b shows cell attachment to the interface. For the bare alloy after 1 day of incubation, the morphology of the cells does not appear to be normal and healthy because of the presence of cracks and combined hydrogen release, which results in the absence of cells. On the other hand, incorporation of HA-NPs and SIM into the polymer matrix improved the biocompatibility according to cell attachment. This behavior may result from better adsorption of extracellular matrix (ECM) proteins, such as fibronectin, to the composite nanofibers than to the plain polymers, perhaps because the fiber web mimics natural ECM and HA as well as the drug incorporation.45 Hence, the PCLHS composite coating was highly compatible with bone regeneration.

(a) Cell proliferation through 1, 3, and 5 days of incubation. The data are reported as the mean ± SD (n = 3 and p < 0.05). (b) FE-SEM images show cell attachment with direct contact assay after 1 day and 3 days for the bare AZ31 alloy and different coated samples
Conclusions
To sum up, our results show that synthesized HA resulted in crystal structure analysis with XRD and FTIR techniques. Electrospinning of Mg coating with a composite PCL/HA/SIM nanofiber is an effective strategy to retard degradation and improve osteoblast cell proliferation. Results indicate that nanofibers loaded with HA and simvastatin works as a dual-functional coating toward enhancing bone tissue regeneration and controlling the degradation process. Samples degradation was found to be controlled by means of nanocomposite coating that was evaluated through electrochemical polarization and EIS techniques. Drug loaded within the composite matrix shows excellent bioactivity, inducing biomineralization formation on the SBF solution in an in vitro test. In vitro cell culture test revealed that using HA and simvastatin improved cell adhesion and proliferation more than bare alloy. Therefore, nanofiber composite with PCL/HA/SIM by means of electrospinning could have a potential role in drug delivery and hard tissue engineering.
References
Czerwinski, F, “Controlling the Ignition and Flammability of Magnesium for Aerospace Applications.” Corros. Sci., 86 1–16 (2014)
Kulekci, MK, “Magnesium and its Alloys Applications in Automotive Industry.” Int. J. Adv. Manuf. Technol., 39 (9) 851–865 (2008)
Hu, BH, Tong, KK, Niu, XP, Pinwill, I, “Design and Optimisation of Runner and Gating Systems for the Die Casting of Thin-Walled Magnesium Telecommunication Parts through Numerical Simulation.” J. Mater. Process. Technol., 105 (1) 128–133 (2000)
Waizy, H, Seitz, J-M, Reifenrath, J, Weizbauer, A, Bach, F-W, Meyer-Lindenberg, A, Denkena, B, Windhagen, H, “Biodegradable Magnesium Implants for Orthopedic Applications.” J. Mater. Sci., 48 (1) 39–50 (2013)
Mousa, HM, Hussein, KH, Woo, HM, Park, CH, Kim, CS, “One-Step Anodization Deposition of Anticorrosive Bioceramic Compounds on AZ31B Magnesium Alloy for Biomedical Application.” Ceram. Int., 41 (9, Part A) 10861–10870 (2015)
Mordike, BL, Ebert, T, “Magnesium: Properties—Applications—Potential.” Mater. Sci. Eng. A, 302 (1) 37–45 (2001)
Kotoka, R, Yamoah, NK, Mensah-Darkwa, K, Moses, T, Kumar, D, “Electrochemical Corrosion Behavior of Silver Doped Tricalcium Phosphate Coatings on Magnesium for Biomedical Application.” Surf. Coat. Technol., 292 99–109 (2016)
Zhao, MJ, Cai, C, Wang, L, Zhang, Z, Zhang, JQ, “Effect of Zinc Immersion Pretreatment on the Electro-Deposition of Ni onto AZ91D Magnesium Alloy.” Surf. Coat. Technol., 205 (7) 2160–2166 (2010)
Mousa, HM, Hussein, KH, Pant, HR, Woo, HM, Park, CH, Kim, CS, “In Vitro Degradation Behavior and Cytocompatibility of a Bioceramic Anodization Films on the Biodegradable Magnesium Alloy.” Colloids Surf. A, 488 82–92 (2016)
Xue, D, Yun, Y, Schulz, MJ, Shanov, V, “Corrosion protction of biodgradable Magnesium Implants using Anodization.” Mater. Sci. Eng. C, 31 (2) 215–223 (2011)
Mousa, HM, Park, CH, Kim, CS, Surface Modification of Magnesium and its Alloys Using Anodization for Orthopedic Implant Application. Magnesium Alloys, InTech (2017)
Luo, X, Cui, XT, “Electrochemical Deposition of Conducting Polymer Coatings on Magnesium Surfaces in Ionic Liquid.” Acta Biomater., 7 (1) 441–446 (2011)
Wong, HM, Yeung, KWK, Lam, KO, Tam, V, Chu, PK, Luk, KDK, Cheung, KMC, “A Biodegradable Polymer-Based Coating to Control the Performance of Magnesium Alloy Orthopaedic Implants.” Biomaterials, 31 (8) 2084–2096 (2010)
Staiger, MP, Pietak, AM, Huadmai, J, Dias, G, “Magnesium and Its Alloys as Orthopedic Biomaterials: A Review.” Biomaterials, 27 (9) 1728–1734 (2006)
Hanas, T, Sampath Kumar, TS, Perumal, G, Doble, M, “Tailoring Degradation of AZ31 Alloy by Surface Pre-Treatment and Electrospun PCL Fibrous Coating.” Mater. Sci. Eng. C, 65 43–50 (2016)
Soujanya, GK, Hanas, T, Chakrapani, VY, Sunil, BR, Kumar, TSS, “Electrospun Nanofibrous Polymer Coated Magnesium Alloy for Biodegradable Implant Applications.” Proc. Mater. Sci., 5 817–823 (2014)
Kim, J, Mousa, HM, Park, CH, Kim, CS, “Enhanced Corrosion Resistance and Biocompatibility of AZ31 Mg Alloy using PCL/ZnO NPs via Electrospinning.” Appl. Surf. Sci., 396 249–258 (2017)
Yasin, AS, Mohamed, IMA, Mousa, HM, Park, CH, Kim, CS, “Facile Synthesis of TiO2/ZrO2 Nanofibers/Nitrogen Co-Doped Activated Carbon to Enhance the Desalination and Bacterial Inactivation via Capacitive Deionization.” Sci. Rep., 8 (1) 541 (2018)
Jose, MV, Thomas, V, Johnson, KT, Dean, DR, Nyairo, E, “Aligned PLGA/HA Nanofibrous Nanocomposite Scaffolds for Bone Tissue Engineering.” Acta Biomater., 5 (1) 305–315 (2009)
Mousa, HM, Abdal-hay, A, Bartnikowski, M, Mohamed, IMA, Yasin, AS, Ivanovski, S, Park, CH, Kim, CS, “A Multifunctional Zinc Oxide/Poly(Lactic Acid) Nanocomposite Layer Coated on Magnesium Alloys for Controlled Degradation and Antibacterial Function.” ACS Biomater. Sci. Eng., 4 (6) 2169–2180 (2018)
Kim, H-W, Lee, H-H, Chun, G-S, “Bioactivity and Osteoblast Responses of Novel Biomedical Nanocomposites of Bioactive Glass Nanofiber Filled Poly(Lactic Acid).” J. Biomed. Mater. Res. Part A, 85A (3) 651–663 (2008)
Bianco, A, Di Federico, E, Moscatelli, I, Camaioni, A, Armentano, I, Campagnolo, L, Dottori, M, Kenny, JM, Siracusa, G, Gusmano, G, “Electrospun Poly(ε-caprolactone)/Ca-Deficient Hydroxyapatite nanohybrids: Microstructure, Mechanical Properties and Cell Response by Murine Embryonic Stem Cells.” Mater. Sci. Eng. C, 29 (6) 2063–2071 (2009)
Fabbri, P, Bondioli, F, Messori, M, Bartoli, C, Dinucci, D, Chiellini, F, “Porous Scaffolds of Polycaprolactone Reinforced with In Situ Generated Hydroxyapatite for Bone Tissue Engineering.” J. Mater. Sci. Mater. Med., 21 (1) 343–351 (2010)
Tian, P, Xu, D, Liu, X, “Mussel-Inspired Functionalization of PEO/PCL Composite Coating on a Biodegradable AZ31 Magnesium Alloy.” Colloids Surf. B, 141 327–337 (2016)
Bakhsheshi-Rad, HR, Hamzah, E, Kasiri-Asgarani, M, Jabbarzare, S, Iqbal, N, Abdul Kadir, MR, “Deposition of Nanostructured Fluorine-Doped Hydroxyapatite–Polycaprolactone Duplex Coating to Enhance the Mechanical Properties and Corrosion Resistance of Mg Alloy for Biomedical Applications.” Mater. Sci. Eng. C, 60 526–537 (2016)
Zomorodian, A, Santos, C, Carmezim, MJ, Silva, TME, Fernandes, JCS, Montemor, MF, “ ‘In-Vitro’ Corrosion Behaviour of the Magnesium Alloy with Al and Zn (AZ31) Protected with a Biodegradable Polycaprolactone Coating Loaded with Hydroxyapatite and Cephalexin.” Electrochim. Acta, 179 431–440 (2015)
Mousa, HM, Tiwari, AP, Kim, J, Adhikari, SP, Park, CH, Kim, CS, “A Novel In Situ Deposition of Hydroxyapatite Nanoplates using Anodization/Hydrothermal Process Onto Magnesium Alloy Surface Towards Third Generation Biomaterials.” Mater. Lett., 164 144–147 (2016)
Abdal-hay, A, Vanegas, P, Hamdy, AS, Engel, FB, Lim, JH, “Preparation and Characterization of Vertically Arrayed Hydroxyapatite Nanoplates on Electrospun Nanofibers for Bone Tissue Engineering.” Chem. Eng. J., 254 612–622 (2014)
Shikinami, Y, Okuno, M, “Bioresorbable Devices Made of Forged Composites of Hydroxyapatite (HA) Particles and Poly l-lactide (PLLA). Part II: Practical Properties of Miniscrews and Miniplates.” Biomaterials, 22 (23) 3197–3211 (2001)
Fang, R, Zhang, E, Xu, L, Wei, S, “Electrospun PCL/PLA/HA Based Nanofibers as Scaffold for Osteoblast-Like Cells.” J. Nanosci. Nanotechnol., 10 (11) 7747–7751 (2010)
Dong, H, Li, Q, Tan, C, Bai, N, Cai, P, “Bi-Directional Controlled Release of Ibuprofen and Mg2 + from Magnesium Alloys Coated by Multifunctional Composite.” Mater. Sci. Eng. C, 68 512–518 (2016)
Yoo, HS, Kim, TG, Park, TG, “Surface-Functionalized Electrospun Nanofibers for Tissue Engineering and Drug Delivery.” Adv. Drug Deliv. Rev., 61 (12) 1033–1042 (2009)
Rezk, AI, Rajan Unnithan, A, Hee Park, C, Sang Kim, C, “Rational Design of Bone Extracellular Matrix Mimicking Tri- Layered Composite Nanofibers for Bone Tissue Regeneration.” Chem. Eng. J., 350 812 (2018)
Thylin, MR, McConnell, JC, Schmid, MJ, Reckling, RR, Ojha, J, Bhattacharyya, I, Marx, DB, Reinhardt, RA, “Effects of Simvastatin Gels on Murine Calvarial Bone.” J. Periodontol., 73 (10) 1141–1148 (2002)
Tang, ZG, Black, RA, Curran, JM, Hunt, JA, Rhodes, NP, Williams, DF, “Surface Properties and Biocompatibility of Solvent-Cast Poly[ε-Caprozlactone] Films.” Biomaterials, 25 (19) 4741–4748 (2004)
Gautam, S, Dinda, AK, Mishra, NC, “Fabrication and Characterization of PCL/Gelatin Composite Nanofibrous Scaffold for Tissue Engineering Applications by Electrospinning Method.” Mater. Sci. Eng. C, 33 (3) 1228–1235 (2013)
Mousa, HM, Lee, DH, Park, CH, Kim, CS, “A Novel Simple Strategy for In Situ Deposition of Apatite Layer on AZ31B Magnesium Alloy for Bone Tissue Regeneration.” Appl. Surf. Sci., 351 55–65 (2015)
Kouhi, M, Morshed, M, Varshosaz, J, Fathi, MH, “Poly (ε-Caprolactone) Incorporated Bioactive Glass Nanoparticles and Simvastatin Nanocomposite Nanofibers: Preparation, Characterization and In Vitro Drug Release for Bone Regeneration Applications.” Chem. Eng. J., 228 1057–1065 (2013)
Rehman, I, Bonfield, W, “Characterization of Hydroxyapatite and Carbonated Apatite by Photo Acoustic FTIR Spectroscopy.” J. Mater. Sci. Mater. Med., 8 (1) 1–4 (1997)
Fujihara, K, Kotaki, M, Ramakrishna, S, “Guided Bone Regeneration Membrane Made of Polycaprolactone/Calcium Carbonate Composite Nano-fibers.” Biomaterials, 26 (19) 4139–4147 (2005)
Lebourg, M, Suay Antón, J, Gomez Ribelles, JL, “Characterization of Calcium Phosphate Layers Grown on Polycaprolactone for Tissue Engineering Purposes.” Compos. Sci. Technol., 70 (13) 1796–1804 (2010)
Mousa, HM, Hussein, KH, Raslan, AA, Lee, J, Woo, HM, Park, CH, Kim, CS, “Amorphous Apatite Thin Film Formation on a Biodegradable Mg Alloy for Bone Regeneration: Strategy, Characterization, Biodegradation, and In Vitro Cell Study.” RSC Adv., 6 (27) 22563–22574 (2016)
Gu, Y, Bandopadhyay, S, Chen, C-F, Guo, Y, Ning, C, “Effect of Oxidation Time on the Corrosion Behavior of Micro-Arc Oxidation Produced AZ31 Magnesium Alloys in Simulated Body Fluid.” J. Alloy. Compd., 543 109–117 (2012)
Wen, C, Guan, S, Peng, L, Ren, C, Wang, X, Hu, Z, “Characterization and Degradation Behavior of AZ31 Alloy Surface Modified by Bone-Like Hydroxyapatite for Implant Applications.” Appl. Surf. Sci., 255 (13) 6433–6438 (2009)
Ayukawa, Y, Yasukawa, E, Moriyama, Y, Ogino, Y, Wada, H, Atsuta, I, Koyano, K, “Local Application of Statin Promotes Bone Repair Through the Suppression of Osteoclasts and the Enhancement of Osteoblasts at Bone-Healing Sites in Rats.” Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod., 107 (3) 336–342 (2009)
Acknowledgments
This paper was supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea (NRF) by Ministry of Education, Science and Technology (Project No. 2016R1A2A2A07005160) and also partially supported by the program for fostering next-generation researchers in engineering of the National Research Foundation of Korea (NRF) funded by the Ministry of Science, (Project No. 2017H1D8A2030449).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Rezk, A.I., Mousa, H.M., Lee, J. et al. Composite PCL/HA/simvastatin electrospun nanofiber coating on biodegradable Mg alloy for orthopedic implant application. J Coat Technol Res 16, 477–489 (2019). https://doi.org/10.1007/s11998-018-0126-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11998-018-0126-8