
Abstract
Electrochemical and chemical oxidation methods are the two common methods used for the preparation of poly(pyrrole). The two methods have been acknowledged greatly and extensively studied because of their feasibility in manipulating the properties of poly(pyrrole) according to the desired application. However, chemical oxidation method is considered the best method for the preparation of poly(pyrrole) in larger quantities. There are other methods through which poly(pyrrole) can be synthesized such as plasma polymerization and photopolymerization, which have so far received less attention in the literature. For this paper, chemical oxidation was used to prepare poly(pyrrole) by oxidizing pyrrole with CuCl2 under different emulsifying conditions. The surfactants used were sodium dodecyl sulfate and/or p-toluenesulfonic acid. Additionally, photopolymerization was also exploited to prepare poly(pyrrole) under similar emulsifying conditions. In this method, poly(pyrrole) was synthesized with the oxidizing ability of AgNO3 under UV radiation. All samples were investigated by Fourier Transform-Infrared Spectroscopy (FT-IR), X-ray Photoelectron Spectroscopy (XPS), and Powder X-ray Diffraction (XRD). Scanning electron microscope was used to compare the morphological differences, which occurred due to different experimental conditions. The thermal stability was studied using thermogravimetric analysis (TGA).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Since the discovery of conducting polyacetylene by Shirakawa et al. in 1977, electronically conducting polymers have gained much interest in the field of electronic devices due to their intrinsic electrical and physical properties.1 The common feature of conducting polymers and a distinct property, which distinguishes them from normal insulating polymers, is the extended conjugated backbone. To achieve significant conductivity, charge carriers, such as bipolarons, should be generated by means of either electrochemical or chemical oxidation, which produces the conducting polymer in its oxidized form.
Poly(pyrrole) is considered one of the most intriguing conducting polymers for commercial applications because of its good environmental stability, relatively moderate conductivity, and easy synthetic process.2 In the last few decades, a great effort has been given in both the academic and industrial areas toward the improvement of its processability and environmental stability. This will enable the development of its technological applications such as molecular devices, sensors and actuators, batteries, capacitors, antistatic coatings, corrosion protection of active metals, polymer solar cells, and electrodes.3–12
Although there are various methods for the preparation of poly(pyrrole), it is quite often synthesized using one of two major methods: chemical or electrochemical oxidative polymerization. However, poly(pyrrole) obtained from the electrochemical method normally displays better conducting properties due to lower degree of over oxidation. The main advantage of chemical oxidative polymerization over the electrochemical method is that the poly(pyrrole) can be easily produced in large amounts with low cost.13 Poly(pyrrole) was first synthesized by Angeli et al. in 1916 via chemical oxidation of pyrrole with H2O2 to produce a black amorphous powder.14 The product was found to be insoluble. Subsequently, many researchers have used various oxidizing agents such as FeCl3, Fe(NO3)3, Fe(ClO4)3, HNO3, PbO2, CuCl2, and CuBr2 to prepare poly(pyrrole) and have reported the conductivity of poly(pyrrole) and the reaction rates.14 Among all the oxidizing agents, Fe(III) salts produced highly conductive poly(pyrrole); however, Cu(II) salts produced poly(pyrrole) with comparable conductivity next to Fe(III) salts.14
The physical properties of the conducting polymer are highly influenced by the method of preparation, the characteristics of other additives in the reaction mixture, and the reaction conditions such as time and temperature. For instance, the effect of surfactant on the morphology, conductivity, and thermal stability of the chemically synthesized poly(pyrrole) has been reported by Omastova et al.15 In the investigation, various surfactants of anionic, cationic, and nonionic types were used and the best results were obtained for the yield, conductivity, and thermal stability of the final doped poly(pyrrole) when the surfactants were dodecylbenzenesulfonic acid (DBSA) and sodium dodecylsulfate (SDS). It was proposed that the improvement in the conductivity could be due to the incorporation of bulky anionic surfactants acting as dopants. Similarly, Kudoh et al. reported that the use of surfactant increased the rate of polymerization.16 Further, it was found that the presence of phenolic derivatives containing electron withdrawing groups such as 3-nitrophenol when used along with the anionic surfactant displayed enhanced conductivity, thermal, and air stability. This is most likely due to the synergistic interaction of phenol derivative with other reactants. However, the solubility of surfactants containing bulky hydrophobic groups such as DBSA and naphthalenesulfonic acid (NSA) was found to be very low. This required a longer time of ultra-sonication to attain homogenous surfactant solutions. Kim et al. synthesized rod-type poly(pyrrole) doped via micelle formation with p-toluenesulfonic acid (pTSA), which has higher water solubility than DBSA and NSA.17 Doped poly(pyrrole) samples were prepared with different concentrations of pTSA. It was found that the best result could be obtained when the ratio of pTSA to pyrrole monomer was 2:0. It was also concluded that at this ratio, poly(pyrrole) would exhibit high crystalinity, dispersity, and thermal stability. In another study for the preparation of soluble poly(pyrrole), the increase in the concentration of surfactants such as DBSA, which contain bulky alkyl groups, was determined to lead to an increase in the doping level. This was found to cause poly(pyrrole) to be soluble in m-cresol, NMP, and chloroform depending on the conditions.18 Soluble poly(pyrrole) was also prepared by functionalizing poly(pyrrole) with functional substituents via the insertion of chlorosulfonyl and sulfonic acid groups.19
Colloidal particles of poly(pyrrole) were prepared via microemulsion–polymerization using several emulsifiers by Moon et al. to understand the relationship between the morphology and photoluminescence (PL).20,21 It was observed that a clear trend in the morphology of poly(pyrrole) particles with the increase in the concentration of cationic surfactants occurred. It was also determined that the morphology was greatly influenced by the oxidant chosen for the polymerization. Finally, it was concluded that the highest PL intensity could be obtained when the particles were small and uniformly dispersed in the medium. Using a novel oxidizing agent, benzoyl peroxide (BPO) in conjunction with inverted-emulsion–polymerization, poly(pyrrole) was synthesized by Saravanan et al. under the presence of both pTSA and SDS.22 The results indicated that the poly(pyrrole) displaying the highest conductivity was obtained when the ratios of concentrations of BPO, SDS, and pTSA with pyrrole are 1.2:1, 1:3, and 2:1 respectively. According to the authors, the increase in the conductivity would be because of the incorporation of both pTSA and SDS as dopants with the optimized doping level.22
Recent studies on photopolymerization of poly(pyrrole) in the presence of AgNO3 as an electron acceptor and UV radiation have shown interesting results for the incorporation of metal nanoparticles into poly(pyrrole).23,24 It has been suggested that this would be an appropriate route to prepare humidity sensors by coating polyester-based substrate with poly(pyrrole)/TiO2 composites and studying its effect on the electrical and humidity sensing properties.25,26 Previously, there were reports on the multiphoton-sensitized polymerization of pyrrole, self-sensitized photopolymerization of pyrrole and photopolymerization of pyrrole using ruthenium complexes, cobalt complexes, and copper complexes as electron acceptors.27,28 However, few reports on photopolymerization were found during a comprehensive literature survey.
In this study, poly(pyrrole) was synthesized using CuCl2 as the oxidizing agent through a chemical oxidation method in the presence of pTSA and/or SDS. Additionally, photochemical oxidation was performed using AgNO3 and UV radiation with the similar emulsifying conditions as applied in the aforementioned chemical oxidation method. With the help of chemical, structural, and morphological studies, the products were compared to understand the effect of the method and its experimental conditions on the properties of the final conducting polymer.
Experimental details
Materials
Pyrrole received from Sigma-Aldrich Company, USA, was distilled under vacuum, and stored in a refrigerator prior to every reaction. Copper chloride (CuCl2), silver nitrate (AgNO3), SDS, and pTSA purchased from Sigma-Aldrich Company, USA, were used as received. Super Spot MK III high intensity Ultraviolet Light spot curing system (Total UV output: 25,000 mW/cm2 @UVA, B, Visible) obtained from Lesco, Inc., USA was used for the photopolymerization. Methanol distributed by VWR International, USA, was used as solvent in the photopolymerization.
Sample preparation
Reactions were divided into two categories: chemical oxidation reactions and photopolymerization reactions. All of the reactions were performed under a N2 atmosphere to avoid any side reactions with oxygen. For the chemical synthesis of poly(pyrrole) in the absence of a surfactant, pyrrole was initially dispersed in the distilled water for 15 min after which time the oxidant solution was slowly added for 30 min. For the chemical synthesis with surfactant, pyrrole was added into distilled water and the surfactant solution was introduced into the flask. The reaction mixture was stirred for 30 min to achieve a homogenous solution. Then, the solution of oxidizing agent was slowly added to reaction mixture while under stirring. After 24 h, the reaction was ended by the addition of acetone. The black precipitate was washed thoroughly with acetone and distilled water. The product was dried at 80°C overnight.
For the photopolymerization, after the addition of all the reactants, the reaction mixture was irradiated with UV radiation every 15 min on and 5 min off for 160 min (2 h exposure of UV radiation). The reaction product was washed with acetone and water and dried at 80°C overnight. Table 1 displays the reaction details. A similar procedure was followed with surfactants for PPY-6, PPY-7, and PPY-8. Ratios are given in molar concentrations.
Characterization
All of the samples were ground into fine powders prior to the characterization. The samples were prepared for Fourier Transform-Infrared Spectroscopy (FT-IR) by making KBr pellets using 95% of KBr and 5% of the respective poly(pyrrole) sample. Nicolet FT-IR spectrometer was used for the FT-IR characterization. For scanning electron microscopy (SEM) characterization, a JEOL JSM-6300 scanning electron microscope was used to obtain the images for the samples. The samples for SEM were prepared by sprinkling the ground powder onto carbon tape, which was attached to aluminum mounts. The magnification (×20,000), accelerating voltage (15 kV), and the scale (1 μm) were specified on each image.
The XPS measurements were performed on an SSX-100 system (Surface Science Instruments) equipped with a monochromated Al K α X-ray source, a hemispherical sector analyzer (HSA), and a resistive anode detector. The samples were mounted on a sample stage using adhesive Al tapes on top of double-side carbon tapes. The base pressure of the XPS system was 2.0 × 10−10 Torr. During the data collection, the pressure was 4.0 × 10−9 Torr. The X-ray spot size was 1 × 1 mm2, which corresponded to an X-ray power of 200 W. The survey spectra were obtained with 15–25 scans at 150 eV using 1 eV/step.
Powder X-ray diffraction (XRD) data were collected in Bragg-Brentano geometry using a Rigaku Ultima IV multipurpose XRD instrument. Prior to the experiment, all samples were ground to a fine powder. A Cu Kα X-ray radiation was used running at 40 kV with current at 44 mA. Phase identification was achieved with the help of software, JADE 9.0.
Thermal degradation of samples was performed using a Thermogravimetric Analysis instrument TGA Q 500 of TA Instruments. The samples were heated from room temperature up to 500°C at a heating rate of 20°C/min. The obtained results were analyzed using the software, Universal Analysis 2000.
Results and discussion
The morphology of the poly(pyrrole) obtained from the above-mentioned experiments was analyzed using SEM. Chemically prepared poly(pyrrole), prepared in the absence of surfactant, did not show any specific morphological order, as shown in Fig. 1a for PPY-1. The PPY-1 sample displayed particulate masses and flakes with irregular shapes. It is suggested that when no surfactant was in the system, random nucleation and chain growth of polymer were possible.

Poly(pyrrole) chemically synthesized (a) without surfactant (PPY-1), (b) SDS as surfactant (PPY-2), (c) pTSA as surfactant (PPY-3), and (d) both SDS and pTSA (PPY-4)
With the introduction of SDS as surfactant in experiment PPY-2, as shown in Fig. 1b, polymer nucleation and growth would have occurred inside the micelles of SDS via emulsion polymerization. This caused the formation of poly(pyrrole) as three-dimensional regular rectangular-shaped particles. As there was a large variation in the particle size, it would be difficult to specify the average particle size. Along with these regular-shaped poly(pyrrole) particles, a few irregular-shaped particulate masses were also observed. This may indicate that the formation of particles could occur either inside or outside of the micelles.
When SDS was replaced with pTSA, in experiment PPY-3, the poly(pyrrole) particles appeared with a spherical granular shape, as shown in Fig. 1c. It is interesting to note that pTSA has a smaller hydrophobic chain than SDS; therefore, smaller micelles would be produced. However, this is probably not the only reason for the smaller particles that were derived from reaction PPY-3 as the difference in micelle size is not significant enough to explain the difference in particle size of the products arising from the two reactions. Further experimentation is required to elucidate completely the mechanisms in both reactions.
An interesting morphology was observed for PPY-4 as shown in Fig. 1d where pTSA and SDS were used in equal quantities. During the synthesis, perhaps the pyrrole monomer was initially protonated by pTSA and then absorbed into the micelles formed by the combination of pTSA and SDS. The outward appearance of this morphology could be possible with the agglomeration of particles when there were close interactions between the polymer chains.29
Morphological investigation on poly(pyrrole) samples prepared via photopolymerization displayed very distinctly different results from aforementioned chemically synthesized poly(pyrrole), as shown in Fig. 2. Even in the absence of surfactant, the polymer particles were obtained with regular and spherical shape, and with uniform size. Interestingly, the reduction of silver ion led to the development of hexagonal silver metal particles as shown in Fig. 2a.

Poly(pyrrole) photopolymerized (a) without surfactant (PPY-5), (b) SDS as surfactant (PPY-6), (c) pTSA as surfactant (PPY-7), and (d) both SDS and pTSA (PPY-8)
When SDS was used in experiment PPY-6, there appeared to be no development of the hexagonal silver metal particles as shown in Fig. 2b. The poly(pyrrole) consisted of small agglomerated particles. In the presence of pTSA in experiment PPY-7, a spherical shape of the poly(pyrrole) particles resulted; again, no hexagonal silver particles were found. When both surfactants were used in experiment PPY-8, no definite morphology was found for silver metal particles or for poly(pyrrole).
XPS characterization
XPS spectra of chemically synthesized poly(pyrrole) samples in a low resolution survey scan are shown in Fig. 3. The binding energies for C 1s at 285 eV, N 1s at 400 eV, O 1s at 530 eV, Cl 2p at 200 eV, and Cu 2p at 935 eV were identified in all spectra with differences in the intensities observed. In the spectra of PPY-2, PPY-3, and PPY-4, the binding energy for S 2p at 169 eV was recognized as the sulfate ion from SDS (CH3(CH2)11OSO4 −) and sulfite ion from pTSA (CH3C6H4SO3 −).

XPS spectra of chemically synthesized poly(pyrrole) samples: (a) PPY-1, (b) PPY-2, (c) PPY-3, and (d) PPY-4
From the XPS data, the estimated values of dopant level in each sample are mentioned in Table 2. The results obtained from the XPS for elemental composition revealed a gradual decrease in the percentage of chlorine and increase in the percentage of sulfur from PPY-1 to PPY-2 and PPY-3, and then to PPY-4. The S/N ratio for PPY-2 and PPY-3 were 0.251 and 0.177, respectively. This implies that the percentage of sulfur content in PPY-2 was slightly higher than that in PPY-3. This may indeed indicate that the dopant level of SDS in PPY-2 was greater in poly(pyrrole) than pTSA in PPY-3. This is supported by FT-IR characterization also, which will be discussed in the next section.
The same result was observed in the case of photopolymerized poly(pyrrole) samples. The higher percentage of nitrogen in PPY-5 implies the presence of the nitrate ion as a dopant. The decrease in its percentage with respect to carbon is compensated for by the increase in the percentage of sulfur in PPY-6, PPY-7, and PPY-8. Figure 4 shows the XPS spectra of these poly(pyrrole) samples. Although both pTSA and SDS were used in PPY-4 and PPY-8, the S/N ratio in both cases shows that there was no significant increase in the doping level of surfactant from PPY-2 to PPY-4 and from PPY-6 to PPY-8, respectively. When XPS spectra in both methods are compared, it should be noted that the higher values for S/N ratio found in the case of photopolymerized polyprroles would be the result of higher doping level of surfactants.

XPS spectra of photopolymerized poly(pyrrole) samples: (a) PPY-5, (b) PPY-6, (c) PPY-7, and (d) PPY-8
FT-IR characterization
Two types of amine vibrational stretching modes were identified in PPY-1 and PPY-2 at 3444 and 3335 cm−1 depending on the surrounding environments, as represented in Fig. 5.30 These two peaks were broad in the case of PPY-3 and PPY-4, probably due to hydrogen bonding with the water molecules that absorbed along with pTSA molecules.

Possible structures as determined via FT-IR
The FT-IR spectra of chemically synthesized poly(pyrrole) samples, as identified in Table 1 were investigated by comparing the major differences as shown in Fig. 6. The spectra of all of the samples displayed the characteristic peaks of poly(pyrrole) with noticeable differences due to shifts to either higher or lower frequencies, depending on the experimental conditions.

FT-IR spectra of chemically synthesized poly(pyrrole) samples: PPY-1, PPY-2, PPY-3, and PPY-4
The band at 1702 cm−1 is due to the formation of a carbonyl group by the nucleophilic attack of water molecules on pyrrole.22 This peak displayed a higher intensity in the spectrum of PPY-3. This could be attributed to the catalytic nature of pTSA, which may protonate pyrrole and cause the increase in nucleophilic attack. However, this band was reduced to lower intensity in the case of PPY-4, when both pTSA and SDS were used. One reason might be the occurrence of micelle formation by SDS molecules around protonated pyrrole molecules that would have minimized the nucleophilic attack of water molecules.
The band at 1558 cm−1 in the spectrum of PPY-1 corresponds to the ring stretching vibrations of mostly C–C and C=C bonds. This band was slightly red-shifted to 1555 cm−1 in the spectra of PPY-2, where SDS was used as an anionic surfactant. In the case of PPY-3, where pTSA was used as dopant, this band was found at 1575 cm−1 with a blue-shift. However, this band was shifted to 1552 cm−1 when both SDS and pTSA were used. These shifts in the frequencies could be interpreted in terms of the nature of dopant and its influence on the ring vibrations of poly(pyrrole).31 When pTSA is incorporated into poly(pyrrole), its effect on the bond lengths of C=C and C–C during the vibrational motion is reflected in the change in the vibration frequency. In the case of SDS, an exactly opposite effect on ring vibrations is observed. Nevertheless, the spectrum of PPY-4 displays this band at 1553 cm−1, indicating that the band was more affected by SDS as dopant rather than PTSA. This may be because of a higher affinity of SDS anion toward positively charged poly(pyrrole) where pTSA is a good leaving group and may serve as a protonating catalyst by producing an increase in the number of sites for doping. Even though the spectrum of PPY-4 displays the presence of SDS and pTSA as dopants, it closely resembles the spectra of PPY-2, indicating the probable higher doping level of SDS than pTSA in PPY-4.
The band at 1464 cm−1 in the spectra of PPY-2, PPY-3, and PPY-4 arises from the combination of mainly C–N and C=C stretching modes. This band was not clearly resolved in the spectra of PPY-1. As mentioned in the work of Tian and Zerbi, the ratio of the total intensities at 1465 and 1561 cm−1 gives the approximate conjugation length.31,32 When the spectra of these four poly(pyrrole) samples were visually compared, this ratio was apparently increased from PPY-1 to PPY-4, implying that PPY-4 has the longest conjugation length in this range of samples. A weak broad band at 1296 cm−1 in the spectrum of PPY-1 was assigned to the C–H and C–N in-plane deformation modes. This band appeared with lower intensity in the spectra of PPY-2, PPY-3, and PPY-4 probably due to the restriction of in-plane deformation occurring with the incorporation of bulky dopants. The characteristic band of ring breathing vibration at 1184 cm−1, which could be seen in PPY-1 as a broad peak, was observed in the spectra of PPY-2, PPY-3, and PPY-4 as being overlapped with the characteristic asymmetric stretching band of the S=O bond, which occurs at 1183 cm−1.15 Though the peak at 1045 cm−1 in all spectra can be considered as the characteristic C–H deformation in poly(pyrrole), it would be difficult to detect any shift in its frequency in PPY-2, PPY-3, and PPY-4.31 As a result of the incorporation of dopant into poly(pyrrole), the dopants such as pTSA and SDS also display the characteristic C–H deformation peaks at this position.
In Fig. 7, the FT-IR spectra of photopolymerized poly(pyrrole) samples PPY-5, PPY-6, PPY-7, and PPY-8 are shown. The characteristic bands of poly(pyrrole) were identified as 1545, 1467, 1292, 1184, 1038, 930, and 781 cm−1.31,32 The peaks at 2920 and 2855 cm−1 in the spectra of PPY-2, PPY-3, PPY-4, PPY-6, PPY-7, and PPY-8 were attributed to the asymmetric and symmetric stretching vibrations of alkyl C–H bonds in the dopants, indicating that the poly(pyrrole) was doped with pTSA and SDS. The peak at 3100 cm−1 in PPY-3 and PPY-7 belongs to the aromatic C–H group stretching frequency.15,17 The reason for the absence of the stretching frequencies at 3446 and 3340 cm−1 in the photopolymerized poly(pyrrole) samples, at this point, is unclear. Further experimentation will be conducted to provide a basis for understanding the cause of the absence of the aforementioned frequencies.

FT-IR spectra of photopolymerized poly(pyrrole) samples: PPY-5, PPY-6, PPY-7, and PPY-8
As discussed earlier, in the case of chemically synthesized poly(pyrrole) samples, it was assumed that the conjugation length would have reached the maximum number in PPY-8. This was confirmed as the ratio of 1467 and 1545 cm−1 from PPY-5 to PPY-8 was found to increase. In the spectrum of PPY-5, the peak at 1383 cm−1 was assigned to the stretching vibration of N=O in the nitrate ion, which is gradually diminished in the spectra of PPY-6, PPY-7, and PPY-8, suggesting the replacement of NO3 − dopant in PPY-5 by SDS in PPY-6, pTSA in PPY-7, and both surfactants in PPY-8.33
Although there were several differences between the spectra of chemically polymerized poly(pyrrole) and photopolymerized poly(pyrrole), the spectra of poly(pyrrole) samples, PPY-4 and PPY-8, match very closely as shown in Fig. 8. When comparing PPY-4 and PPY-8, it appears that the concentration of the carbonyl group that occurred, due to higher oxidation, was higher in the case of chemically prepared poly(pyrrole), PPY-4. In order to verify whether AgNO3 can oxidize pyrrole without UV radiation, a chemical oxidation experiment was performed using AgNO3 with a pyrrole: AgNO3 ratio of 1:1. While it took almost 48 h for this reaction to give approximately a 5–10% yield, the reaction in the presence of UV radiation produced approximately 30% yield of poly(pyrrole) within 2 h of UV exposure.

FT-IR spectra of PPY-4 and PPY-8
TGA characterization
A gradual weight loss was observed for all of the samples from ambient temperature to 200°C as shown in Figs. 9–11. However, some small differences were observed between all of the poly(pyrrole) samples, for example, the rate of weight loss in PPY-4 is less than that in PPY-2 and PPY-3. Perhaps this is due to a lesser amount of physically adsorbed water and residual monomer or a stronger interaction of the physically adsorbed water and residual monomer in the PPY-4 sample. As the temperature approached 200°C, the samples converged with each other, supporting the latter case as at this temperature no water or monomer was expected to be present.

TGA curves of PPY-1, PPY-2, PPY-3, and PPY-4

TGA curves of PPY-5, PPY-6, PPY-7, and PPY-8

(a) TGA curves of PPY-1 and PPY-5, (b) PPY-2 and PPY-6, (c) PPY-3 and PPY-7, and (d) PPY-4 and PPY-8
It is suggested that the slope over the temperature range from 200 to 275°C would be the beginning of degradation of doped polypyrrole chains. The degradation behavior of PPY-3 was less than the other chemically polymerized samples as it is more stable over this temperature range. Perhaps the presence of an aromatic ring in the PTSA dopant anion increased the thermal stability of polypyrrole chains in PPY-3 as compared to the chloride dopant in PPY-1 and SDS dopant anion in PPY-2.34 PPY-4 displayed the least amount of thermal stability over this same temperature range than all of the samples. As discussed in aforementioned FT-IR, the doping level of SDS may have been higher than the other samples and this, therefore, reduced its thermal stability even further.
A similar trend was observed for the photopolymerized poly(pyrrole) samples as shown in Fig. 10. However, it does appear that the amount of either physically adsorbed water or residual monomer were less than that observed for the chemically synthesized poly(pyrrole). When the poly(pyrrole) samples containing the same dopant but synthesized via the two different methods were compared individually, as shown in Fig. 11, the poly(pyrrole) samples made via the photochemical oxidation method displayed the better thermal stability behavior. This could be due to differences in the chemical structure and dopant level or, perhaps, the molecular weight.35,36
XRD characterization
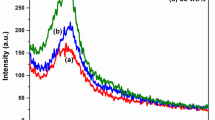
The XRD spectra of the samples display the presence of a broad peak, which occurred due to the overlap of two characteristic peaks of polypyrrole located in between the angles 2θ = 12° to 35° as shown in Figs. 12, 13, and 14. The peaks at approximately 2θ = 20° and 26° are believed to occur because of the interplanar Vander Waals arrangement of the pyrrole–pyrrole rings in polypyrrole chains and pyrrole–counterion or inter–counterion interactions, respectively. The d-spacing for this peak gives the distance between pyrrole–pyrrole stacking planes.37,38 It was found from powder diffraction file (PDF) data that the sharp crystalline peaks indicate the formation of copper chloride hydroxides (Cu2Cl(OH)3 and Cu2Cl2(OH)3). The shift in the main peak in the case of PPY-2 toward higher d-spacing might be due to increase in the size of dopant anion from Cl− in PPY-1 to SDS anion in PPY-2.

XRD spectra of PPY-1 and PPY-2

XRD spectra of PPY-2, PPY-3, and PPY-4 for 2θ = 2°–35°

XRD spectra of PPY-5, PPY-6, PPY-7, and PPY-8 for 2θ = 2°–35°
When the XRD spectra of PPY-2, PPY-3, and PPY-4 are compared from Fig. 13, the same peak was shifted more to higher d-spacing in the case of PPY-3 due to the interference of rigid aromatic benzene ring of pTS anion in between pyrrole–pyrrole interplanar arrangement.38 This increase or decrease of d-spacing may depend on the orientation of benzene ring in pTS anion. Conversely, this peak in PPY-4 remained almost at the same d-spacing as in SDS, indicating the predominating nature of SDS as dopant in PPY-4. Interestingly, the formation of copper chloride hydroxides was prevented in both PPY-3 and PPY-4 where pTSA was present.
The crystalline peaks of Ag metal shown in Fig. 15 were identified in the XRD spectra of PPY-5, PPY-6, PPY-7, and PPY-8 using PDF data. Further research may be needed on this to understand the mechanisms of these reactions.

Silver metal peaks in XRD spectra of PPY-5, PPY-6, PPY-7, and PPY-8
The peak with a d-spacing of about 10 Å at around 2θ = 8–10° in the spectra of all samples represents the enhancement of the structural order of counterions in the polymer chain. As reported by Carrasco et al., the poly(pyrrole) having highest intensity of this peak showed highest conductivity due to improved order of counterion in the material.37 It can be speculated upon comparison of the spectra of chemically synthesized and photopolymerized poly(pyrrole)s shown in Fig. 14 that the degree of the structural order of counterions in the polymer chain would have been improved in the case of photopolymerized poly(pyrrole)s. The peak at around 2θ = 8–10° in these samples is more sharp and intense, indicating the possibility of higher conductivity.
Conclusions
Poly(pyrrole) was synthesized using two methods: (i) a chemical oxidation method and (ii) a photopolymerization method, with and without surfactants. The changes in morphologies were studied using SEM in both cases with respect to the changes in the emulsifying conditions. It was understood that the morphology of poly(pyrrole) particles could be manipulated by choosing an appropriate surfactant at an appropriate concentration. Even though the poly(pyrrole) that was synthesized with the combination of PTSA and SDS by chemical and photochemical methods showed similar chemical structure in FT-IR, different morphologies were observed via SEM. XPS studies showed a gradual decrease in the dopant concentration of chloride ion in the chemically prepared poly(pyrrole) samples and nitrate ion in the photochemically prepared poly(pyrrole) samples with the incorporation of surfactants. FT-IR analysis showed that the use of surfactant could cause an increase in the conjugation length of the poly(pyrrole). It was also confirmed by XPS and FT-IR that SDS has higher affinity than pTSA as a dopant toward positively charged poly(pyrrole). TGA analysis showed a reduction in the thermal stability of poly(pyrrole) with the incorporation of surfactants. Additionally, thermal stability of photopolymerized poly(pyrrole) was higher than that of chemically prepared poly(pyrrole). XRD analysis indicated improved counterion–counterion structural order in photochemically prepared samples.
References
Shirakawa, H, Louis, EJ, MacDiarmid, AG, Chiang, CK, Heeger, AJ, “Synthesis of Electrically Conducting Organic Polymers: Halogen Derivatives of Polyacetylene, (CH) x .” J. Chem. Soc., Chem. Commun., 578–580 (1977). doi:10.1039/C39770000578
Roncali (1992) Conjugated Poly(thiophenes)—Synthesis, Functionalization, and Applications. J Chem Rev 92, 711. doi:10.1021/cr00012a009
Mirfakhrai T, Madden J D W, Baughman R. H, Polymer Artificial Muscles. Materials Today, 10(4), 30-38
Hwang, L. S.; Ko, J. M.; Rhee, H. W.; Kim, Y. C.; A Polymer Humidity Sensor. Synth. Met. 57(1), 3671-3676 (1993)
Baughman, R. H.; Shacklette, L. W.; Murthy, N. S.; Miller, G. G.; Elsenbaumer, R. L.; The Evolution of Structure during the Alkali Metal Doping of Polyacetylene and Poly(p-Phenylene). Mol Cryst Liq Cryst 253-261, 1985, 118
Naegele, D.; Bittihn, R.; Electrically Conductive Polymers as Rechargeable Battery Electrodes. Solid State Ionics 28, 983-989, 1988
HK Song, GTR Palmore (2006) Redox-Active Polypyrrole: Toward Polymer-Based Batteries. Adv. Mater, 18, 1764–1768. doi:10.1002/adma.200600375
Kudoh, Y, Nishino, A, “Recent Development in Electrolytic Capacitors and Electric Double Layer Capacitors.” Electrochemistry, 69 (6) 397-406 (2001).
Stanke, D.; Hallensleben, M. L.; Toppare, L.; Graft Copolymers and Composites of Poly(Methyl Methacrylate) and Polypyrrole Part I. Synth. Met. 1993, 89-94, 72
He, J, Gelling, VJ, Tallman, DE, Bierwagen, GP, Wallace, GG, Scanning Vibrating Electrode Studies of Electroactive Conducting Polymers on Active Metals, Vol. 843, pp. 228–253. Electroactive Polymers for Corrosion Control, ACS Symposium Series, American Chemical Society (ACS), Washington, DC (2003)
Y Saito, N Fukuri, R Senadeera, T Kitamura, Y Wada, S Yanagida, Solid State Dye Sensitized Solar Cells Using In Situ Polymerized PEDOTs as Hole Conductor. Electrochemistry Communications 6 (2004) 71–74. doi:10.1016/j.elecom.2003.10.016
L-Z Fan, J Maier; High-Performance Polypyrrole Electrode Materials for Redox Supercapacitors. Electrochem. Commun. 8, 6, 2006, 937-940
Skotheim, TA, Reynolds, J, “Conjugated Polymers: Theory, Synthesis, Properties, and Characterization” CRC Press Tayor & Francis Group, AZ, USA, (2006).
Nalwa, HS, “Hand Book of Organic Conductive Molecules and Polymers: Conductive Polymers: Synthesis and Electrical Properties.” John Wiley & Sons, New York, 1997.
Maria Omastova, Miroslava Trchova, Jana Kovarova, Jaroslav Stejskal; Synthesis and Structural Study of Polypyrroles Prepared in the Presence of Surfactants. Synth. Met. 138 (2003) 447–455. doi:10.1016/S0379-6779(02)00498-8
Kudoh Y, Akami K, Matsuya Y; Properties of Chemically Prepared Polypyrrole with an Aqueous Solution Containing Fe2(SO4)3, a Sulfonic Surfactant and a Phenol Derivative. Synth. Met. 95 (1998) 191-196. doi:10.1016/S0379-6779(98)00054-X
DK Kim, KW Oh, HJ Ahn, SH Kim; Synthesis and Characterization of Polypyrrole Rod Doped with p-Toluenesulfonic Acid via Micelle Formation. J. Appl. Polym. Sci., Vol. 107, 3925–3932 (2008). doi:10.1002/app.27509
G. J. Lee, S. H. Lee, K. S. Ahn, K. H. Kim; Synthesis and Characterization of Soluble Polypyrrole with Improved Electrical Conductivity. J. Appl. Polym. Sci., Vol. 84, 2583–2590 (2002). doi:10.1002/app.10281
KS Jang, H Lee, B Moon; Synthesis and Characterization of Water Soluble Polypyrrole Doped with Functional Dopants. Synth. Met. 143 (2004) 289–294. doi:10.1016/j.synthmet.2003.12.013
AJR Son, H Lee, B Moon; Morphology and Photoluminescence of Colloidal Polypyrrole Nanoparticles. Synth. Met. 157 (2007) 597–602. doi:10.1016/j.synthmet.2007.04.018
KS Jang, HC Ko, B Moon, H Lee; Observation of Photoluminescence in Polypyrrole Micelles. Synth. Met. 150 (2005) 127-131. doi:10.1016/j.synthmet.2005.01.013
C Saravanan, RC Shekhar, S Palaniappan; Synthesis of Polypyrrole Using Benzoyl Peroxide as a Novel Oxidizing Agent. Macromol. Chem. Phys. 2006, 207, 342–348. doi:10.1002/macp.200500376
C. R. Martins, Y. M. de Almeida, G. C. do Nascimento, W. M. de Azevedo; Metal Nanoparticles Incorporation During the Photopolymerization of Polypyrrole. J Mater Sci (2006) 41:7413–7418. doi:10.1007/s10853-006-0795-z
Breimer, MA, Yevgeny, G, Sy, S, Sadik, OA, “Incorporation of metal nanoparticles in photopolymerized organic conducting polymers: A mechanistic insight.” Nano Lett., 1 305 (2001).
Su, PG, Wang, CP, “Flexible Humidity Sensor Based on TiO2 Nanoparticles-Polypyrrole-Poly[3-(Methacrylamino)Propyl] Trimethyl Ammonium Chloride Composite Materials.” Sens. Actuators B: Chem., 129 538 (2007), 10.1016/j.snb.2007.09.011
PG Su, LN Huang; Humidity Sensors Based on TiO2 Nanoparticles/Polypyrrole Composite Thin Films. Sensors and Actuators B 123 (2007) 501–507. doi:10.1016/j.snb.2006.09.052
Yamada, K, Kimura, Y, Suzuki, S, Sone, J, Chen, J, Urabe, S, “Multiphoton-sensitized polymerization of pyrrole.” Chem. Lett., 35 (8) 908 (2006). doi:10.1246/cl.2006.908
Rodriguez, I, Gonzalez-Velasco, J, “Self-sensitized photopolymerization of pyrrole.” J. Chem. Soc. Chem. Commun., 5 387 (1990). 10.1039/c39900000387
Xing S, Zhao G; Morphology, Structure, and Conductivity of Polypyrrole Prepared in the Presence of Mixed Surfactants in Aqueous Solutions. J. Appl. Polym. Sci., Vol. 104, (2007) 1987–1996. doi:10.1002/app.25912
L. Birladeanu, “Infrared Spectroscopy-Applications in Organic Chemistry”, Wiley-Interscience, New York, 1966
Tian B, Zerbi G; J.Chem.Phys.92 (6), Lattice Dynamics and Vibrational Spectra of Pristine and Doped Polypyrrole: Effective Conjugation Coordinate. 1990, p 3886. doi:10.1063/1.457794
Tian B, Zerbi G; J.Chem.Phys.92 (6), Lattice Dynamics and Vibrational Spectra of Polypyrrole, 1990, p 3892. doi:10.1063/1.457795
Sarac AS, Ustamehmetoglu B, Mustafaev MI, Erbril C, Uzelli G (1995) Oxidative Polymerization of Pyrrole in Polymer Matrix. J Polym Sci A33:1581. doi:10.1002/pola.1995.080331004
Ormond-Prout, J, Dupin, D, Armes, SP, Foster, NJ, Burchell, MJ, “Synthesis and Characterization of Polypyrrole-Coated Poly(Methyl Methacrylate) Latex Particles.” J. Mater. Chem., 19 1433-1442 (2009).
VT Truong, BC Ennis, TG Turner, CM Jenden; Thermal Stability of Polypyrroles. Polymer International Vol. 27 Iss.2, 2007, 187–195
Lee BH, Don SY, Shim JM, Kim WS (1997) Effects of Molecular Weight on Thermal Degradation of ABS. Applied Chemistry, 1(2), 698-701
Pedro M. Carrasco, Hans J. Grande, Milagros Cortazar, Juan M. Alberdi, Javier Areizaga, Jose A. Pomposo (2006) Structure–Conductivity Relationships in Chemical Polypyrroles of Low, Medium and High Conductivity. Synth. Met. 156, 420–425. doi:10.1016/j.synthmet.2006.01.005
K. Cheah, M. Forsyth, V.-T. Truong, Ordering and Stability in Conducting Polypyrrole. Synth. Met. 94 (1998) 215-219. doi:10.1016/S0379-6779(98)00006-X
Acknowledgment
The authors would like to thank the US Army Research Laboratory (Contract #: W911NF-04-2-0029) for sponsoring this research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented at the 2008 FutureCoat! Conference, sponsored by FSCT, October 15–16, 2008, in Chicago, IL.
Rights and permissions
About this article
Cite this article
Kasisomayajula, S.V., Qi, X., Vetter, C. et al. A structural and morphological comparative study between chemically synthesized and photopolymerized poly(pyrrole). J Coat Technol Res 7, 145–158 (2010). https://doi.org/10.1007/s11998-009-9186-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11998-009-9186-0