
Opinion statement
We postulate that the frequently encountered grouping of different Parkinson disease (PD) variants into a single pathogenetic concept—rather than differentiation into its molecular subtypes—has hindered progress toward curative interventions. Parkinsonism is a clinical syndrome that in rare cases can be explained by a single genetic event or by a single environmental cause, thereby leading to monogenic PD and secondary parkinsonism, respectively. Under the former category, mutations in both alleles of the Parkin-encoding PARK2 gene leads to young-onset, autosomal recessive PD, in which neurodegeneration is restricted to dopamine-producing cells of the brainstem. Under the latter category, exposure to one of several environmental factors with neuroanatomic selectivity can cause rapid-onset, secondary parkinsonism most likely irrespective of the patient’s age and genetic makeup. Sandwiched between these two extreme and rare types, the most common variant is referred to as late-onset, idiopathic PD. In extension of a disease model first proposed by Braak et al., we consider idiopathic PD the result of an encounter between one or several environmental triggers and one or more susceptibility alleles. Importantly, this interaction produces a pre-motor syndrome followed by the typical PD phenotype over a period of decades. In our opinion, this pathophysiological process should thus be viewed as a “complex disease.” As is true for many complex human disorders, successful intervention for the common PD variant will likely occur when genetic leads as well as environmental contributors are targeted in parallel. However, successful proof-of-concept studies could arrive sooner, namely for select PD variants that can be attributed to a single genetic event and that are neuropathologically restricted. Therefore, the authors decided to focus the second portion of their review on treatment considerations regarding autosomal recessive PD cases that are caused by Parkin deficiency. We briefly draw attention to aspects of existing pharmacological and surgical therapies as they relate to the PARK2-linked variant; thereafter, we comment on new research avenues that are aimed at future therapeutic interventions to eventually slow or arrest the progression of a first variant of PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A series of excellent articles has been published in this journal on topics related to the therapy of typical Parkinson disease (PD). These include reviews of its initial treatment [1], its long-term medical management [2], treatment of motor fluctuations [3]; the management of levodopa-induced dyskinesias [4]; and a review of surgical options [5], to name a few. Here, the authors wish to draw attention to known differences in the etiopathogenesis of at least three variants of parkinsonism, highlight where these differences play a role in the treatment approach to autosomal recessive PD versus idiopathic PD, and focus on the rationale behind recent research efforts that target PARK2-linked PD for cause-directed intervention in the future.
What constitutes typical Parkinson disease?
Parkinsonism is a motor syndrome that is diagnosed based on the recognition of neurological signs that include bradykinesia, rigidity, tremor, and loss of postural reflexes. More than 20 medical conditions are known to alter the function and/or integrity of neural structures from the midbrain to the forebrain, thus producing clinical parkinsonism [6]. The clinical diagnosis of definite PD is made when the motor syndrome steadily progresses over the years and the patient shows a sustained response to levodopa therapy [1]; however, the correlating pathological substrate that generates clinical PD can vary substantially [7]. The smallest common denominator that is essential to produce clinical PD (among various neuropathological changes recorded) includes a >70 % loss of dopamine-producing neurons in the pars compacta of the substantia nigra of the midbrain and an associated (or independent) innervation loss of its target nuclei (ie, caudate and putamen) [8]. Three of the most striking insights made over the past 15 years are 1) that clinically typical PD can start in the brainstem and remain largely restricted to the loss of dopamine neurons in the substantia nigra and locus coeruleus, as in PARK2-linked PD (see below) [9]; 2) that clinically typical PD can represent one element of a systemic illness—albeit a prominently visible one owing to its typical movement abnormalities—that begins elsewhere in the body and affects the brainstem en route during a trans-synaptic spread across the neuroaxis [10, 11]; and 3) the fact that the neuropathological changes seen in the midbrain, which lead to clinically recognizable PD, can vary substantially, even when mutational events affect the same gene locus [12, 13]. In summary, we acknowledge the fact that the illness we generally recognize as clinically typical PD mirrors the complexity and heterogeneity of other human disorders, such as myocardial infarction, diabetes, and breast cancer. Therefore, as of 2012, irrefutable evidence demonstrates that 1) the clinical syndrome of PD encompasses multiple look alikes; 2) the neuropathological substrate underlying the clinical syndrome of typical PD is pleomorphic; and 3) even when PD is caused by genetic alterations in one and the same gene, it can feature more than one endophenotype. In accordance, we postulate that in order to halt the progression of clinical PD, we have to precisely delineate the molecular mechanisms that lead to the expression of each of its variants [14, 15]. Of note, the focus on differentiating an illness under investigation and stratifying patients based on unique molecular features and objective markers (rather than relying on subjective, clinical exam-based criteria for the diagnosis) allowed scientists to finally make substantive inroads into the treatment of other complex human disorders, such as breast cancer, myocardial infarctions, and diabetes.
Parkinson disease encompasses both rare and common variants
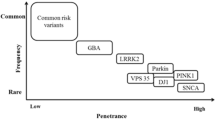
Rare cases of parkinsonism can be explained by a single genetic event or by a single environmental cause, thereby leading to monogenic PD and secondary parkinsonism, respectively (Fig. 1). It is estimated that strictly monogenic events underlie about 3 % (or less) of all cases among patients that present with clinically typical PD to a movement disorder center [16]; subjects with secondary parkinsonism due to an identifiable environmental cause, such as exposure history to a known toxin, a drug, or virus are equally rare [17]. Monogenic PD usually manifests before age 45 years and is progressive. In contrast, the onset of secondary parkinsonism is more closely timed with actual exposure to the noxious agent rather than the subject’s age [17]. Under the category of monogenic PD variants (Fig. 1), it is believed that autosomal dominant SNCA (PARK1/PARK4) gene mutations (specifically, triplication of its gene on one chromatid or point mutations in exons of one allele) [18], together with aging but most likely without a necessary trigger by the environment, promote widespread neurodegeneration and cellular inclusion formation. There, alpha-synuclein [SNCA] production inversely correlates with the age of disease onset [18]. In contrast, monogenic events that lead to young-onset autosomal recessive PD appear neuropathologically restricted and are likely caused by a metabolic defect that manifests as a result of aging-related processes in post-mitotic neurons that synthesize dopamine (see below). Under these conditions, the impact of genetics is considered very high (up to 100 % in our model) to explain the phenotype, whereas the role of environmental co-factors is likely more negligible, thereby shifting monogenic variants of PD to the far left in the area under the curve in our graph (Fig. 1). At the other extreme, a singular environmental “hit” (eg, exposure to a neurotoxin with high tropism for midbrain or striatum, or both) may precipitate parkinsonism. There, the subject’s genetic susceptibility (and age) likely plays a lesser role in the expression of the phenotype, thereby shifting that variant of parkinsonism to the far right under the curve depicted in the graph of our model (Fig. 1). Once the phenotype of secondary parkinsonism is fully expressed, it may (or may not) progress; if it does, it is believed that it will not advance at a fast rate if exposure to the noxious agent has indeed ceased.

Parkinson disease is a heterogenous disorder. Our model utilizes a bell shaped curve to display the known etiologies that underlie clinically diagnosed PD. It contains three main categories: a rare cases of secondary parkinsonism that are caused by exposure to one or more environmental trigger(s) encountered by an affected individual at any given age. These cases are grouped to the right of the bell shaped curve, where a high degree (<100%) of environmental contribution is inferred; b rare monogenic variants of PD that lead to heritable PD with an onset before the age 45 years. These are grouped to the left end of the curve, where a high degree (<100%) of genetic contribution is inferred; and c complex PD. This common, typically late-onset form of parkinsonism (ie, after age 55 years) is the product of an interaction between environmental contributors and (epi)genetic risk factors carried by the host. Several known susceptibility alleles are displayed in the model. The area under the curve also includes recently discovered risk alleles from genome wide association studies and speculates as to a possible role for microbial pathogen exposure in the host's medical history. We postulate that in these common cases of complex PD, the contribution by both factors is essential. The model does not incorporate the passage of time (likely decades) between the environmental trigger and the expression of the clinical syndrome (ie, onset of the motor syndrome in PD). This review discusses the current management of autosomal recessive PD (red box) and possible approaches in the future for its cause-directed therapy.
A bell-shaped curve for typical (complex) Parkinson disease
In contrast, late-onset “idiopathic” PD, which is the most common among all human PD variants, usually manifests after age 55 years and behaves etiopathogenetically like a complex disease. In accordance, these PD cases occupy >85 % of the area depicted under the bell-shaped curve in our model (Fig. 1). We interpret the existing body of literature on the nature of idiopathic PD (ie, clinically typical, and considered sporadic due to the absence of any readily recognizable heredity or other primary cause) in the following manner, as inspired by numerous human conditions (such as stroke, breast cancer, hypertension, or diabetes): The interaction between one or more susceptibility loci of the host and one or several environmental modifiers is essential to express the observed PD phenotype (Fig. 1) [16]. Braak et al. [10] hypothesized a neurotoxin or infectious pathogen as the elusive environmental trigger in their case series. These authors also proposed a unique staging model for the development of idiopathic PD, and placed evidence of early SNCA misprocessing near the interface(s) of host and environment, specifically at the level of neurons in the digestive tract and the olfactory system [10], which conceptually is in agreement with a possible role for the environment in the expression of a PD phenotype [17].
The subsequent trans-synaptic progression of the disease process in this complex PD model from the enteric nervous system to areas of association cortices in the host is thought to take approximately four decades [11]. In support of such a slow, initially retrograde and then trans-synaptic spread of the disease, researchers found that neuropathological changes can be transmitted from a PD neuron located in the host’s striatum to a grafted cell harvested from a fetus over a process that takes >11 years [20, 21]. Various cellular events can alter the processing of wild-type SNCA in neurons, as facilitated by its abundance, its long half-life, and its innate propensity to misfold, thereby promoting the formation of Lewy body-type inclusions. However, many other neurological conditions have been associated with intracellular Lewy body formation, thereby demonstrating their relative lack of specificity. Nevertheless, it is estimated that >80 % of all patients carrying the clinical diagnosis of PD show Lewy inclusion-associated neurodegeneration in the substantia nigra at autopsy [7]. To date, three important pathogenetic events have emerged in the metabolism of SNCA that can confer toxicity, (ie, an elevated steady state, enhanced oligomer formation, C-terminal truncation, and possibly, its exocytosis by neighboring cells) [22]. Recently identified SNCA risk alleles may contribute to one or more of these pathogenetic events. However, according to our integrated two-hit model, these genetic susceptibilities (including the carrier status of a SNCA duplication) may not suffice on their own to produce the clinically typical PD phenotype [18] without an environmental trigger (or co-factor) (Fig. 1). The mechanisms by which the elusive environmental co-factor inhibits and propagates disease in the neuro-axis of the host remain enigmatic.
Intriguingly, heterozygous mutations in the GBA1 gene, which encodes the lysosomal enzyme acid β -glucocerebrosidase, represent a frequent genetic risk factor for complex PD (Fig. 1) as well as for dementia with Lewy bodies [23]. The penetrance rate for carriers of a GBA1 mutation to express a PD phenotype is not yet known, but the mechanisms underlying this strong association remain under intense investigation [24]. Importantly, results from multiple studies demonstrated that virtually all patients with definite PD who carry a mutant GBA1 allele show evidence of neural SNCA misprocessing at autopsy [eg, 25], thereby suggesting that GBA1 plays an important regulatory role in the steady-state of SNCA in vivo [24]. Of note, the metabolism of SNCA as a logical platform for the identification of future targets in the treatment of SNCA-linked PD, GBA1-linked neurological illnesses, and possibly, of related Lewy body disorders was the subject of two recent reviews by the authors [24, 26]. Monogenic, SNCA-linked PD and GBA1-associated complex PD will therefore not be discussed further below.
Association studies identify multiple additional contributors to complex Parkinson disease
Aside from the role of genotypic variants of SNCA, allelic changes in at least ten additional loci have been identified as likely modulators of the risk to develop “complex PD” [33]. Among them, dominantly inherited mutations in the leucine-rich repeat kinase 2-encoding LRRK2 (PARK8) gene can be found in >2 % of all late-onset PD cases [12]. Intriguingly, the mean penetrance rate across all age groups of mutation carriers measures >25 %. However, it varies considerably with geography and ethnicity, and rises progressively with each decade lived. These observations are suggestive of, but do not yet prove, that an additional hit may be required to express the PD phenotype. LRRK2 mutations that promote the development of complex PD typically lead to SNCA-positive Lewy body inclusions, but they can also be associated with tau inclusions or occur without any detectable inclusion formation (revealing only cell loss in the substantia nigra), thus demonstrating the pleomorphic variability of LRRK2-linked PD (“endophenotypes”) [12]. Most investigators have postulated an intra-neuronal gain-in-kinase-function effect for mutant LRRK2 [27], but its physiological function in the brain remains elusive. Recently, several reports have identified a significant role for distinct LRRK2 alleles in human susceptibility to Crohn’s disease, leprosy, and cancer. Moreover, related studies have begun to implicate LRRK2 genotypes in the modulation of the innate immune response as executed by mononucleated cells following their exposure to inflammatory stimuli under both ex vivo and in vivo conditions [28]. Importantly, these stimuli included several conferred by microbial structures such as bacterial lipopolysaccharides and viral particles [29, 30]. Given these results, we speculate that LRRK2-associated disease actually represents a group of endophenotypes under the rubric of complex PD. There, the affected, genetically susceptible host may have previously encountered a single environmental trigger once, or has seen multiple environmental factors together, or was exposed on multiple occasions to one or more pathogens over the course of decades (Fig. 1). In extending the Braak hypothesis, these environmental co-factors may have interacted with the host (and his/her genome) via the gastrointestinal system or the olfactory epithelium [10, 31]. Importantly, the still elusive environmental trigger(s) required as co-factors could be microbial in nature [29]. By implication, a carrier of LRRK2 mutations without exposure to the elusive environmental co-factor, or without the passage of time required for disease propagation across the neuroaxis, may not express the motor syndrome of PD during his/her lifespan [32].
Results from a recent meta-analysis of several, large genome-wide association studies (GWAS) conducted in Europe and North America have revealed five new loci (ie, ACMSD, STK39, LAMP3, SYT11, and CCDC62) as risk modulators for clinically typical PD to the previously known list of six genes (MAPT, SNCA, HLA-DRB5, LRRK2, BST1, and GAK) [33]. Interestingly, risk modulation in association with distinct GBA1 genotypes was not picked up in these studies. From a complex PD perspective, four genes (ie, HLA-DRB5, BST1, STK39, LRRK2) have been associated to date with the immune system per se or with inflammatory responses by the host (or both), which could be interpreted as possibly relevant in a two-hit model of etiopathogenesis (Fig. 1). It remains to be determined in future studies whether the other seven genes play a role in PD development by modulating the host’s primary susceptibility to disease, or his/her inflammatory response to environmental triggers, or the downstream neuronal effects triggered by the encounter of host and his/her environment (Fig. 1). Of note, the SYT11 gene product synaptotagmin was previously reported as a Parkin-interacting protein; however, variants of the three known autosomal recessive, monogenic PD loci (PARK2, DJ-1, PINK1) were not detected by GWAS analyses. The combined population-attributable risk conferred by single nucleotide polymorphisms in the 11 loci identified by Nalls et al. [33] measured 60.3 %. The latter finding suggests that additional alleles, with possibly even smaller individual contributions to overall PD risk modulation, await discovery.
Recessive variants of monogenic Parkinson disease
The first description of a clinical motor syndrome, initially referred to as “autosomal recessive, juvenile parkinsonism” (due to its onset as early as age < 20 years) was provided by Yamamura et al. [9] in 1973. The gene responsible for the syndrome (PARK2, gene locus on chromosome 6) was identified in 1998, and its gene product was named Parkin by Kitada et al. [34]. Parkin is thought to be a component of a neuronal E3 ubiquitin ligase complex that mediates the targeting of substrate proteins for proteasomal degradation [35], and to be essential in the removal of impaired mitochondria by autophagy [36]. Nevertheless, irrefutable in vivo evidence for either role has not yet been provided. As of 2012, mutations affecting both alleles of PARK2, PINK1 (PARK6) or DJ-1 (PARK7) underlie the majority of typical, autosomal recessive PD cases (Fig. 1) [16].
Characteristics of PARK2-linked Parkinson disease
In the original report, Yamamura et al. [9] described signs of typical PD in four families with recessive inheritance. Today, the PARK2-linked phenotype of PD is recognizable by up to 10 additional pathological features, although each carries relatively low specificity. These include 1) early age at onset (usually < 45 years) and a possible (recessive) family history; 2) asymmetric dystonic gait disturbance rather than tremor as the initial sign; 3) foot dystonia upon awakening; 4) hyperreflexia on examination; 5) autonomic signs that, when present, are usually milder than those seen in SNCA-positive PD; 6) a paradoxical sleep benefit (ie, diurnal fluctuation of symptoms and signs); 7) a strong and sustained response to levodopa treatment; 8) the frequent occurrence of levodopa-induced dyskinesias and daily motor fluctuations; 9) overall slow progression with some cases reported of >40 years in duration; and finally 10), despite the occurrence of psychiatric abnormalities in some patients, a relatively infrequent occurrence of dementia in advanced cases of the illness [9, 13, 37].
According to some estimates, as many as 50 % of these recessive PD variants may be attributable to PARK2 mutations [38]. To date, over 100 distinct PARK2 genotypes have been associated with the development of heritable PD (familial and sporadic). However, the overall contribution of Parkin-deficient PD cases to the entire spectrum of patients presenting with clinical PD remains relatively small (Fig. 1) [16]. At autopsy examination of fewer than 10 cases, PARK2-linked cases reveal dopamine cell loss in the substantia nigra and locus coeruleus, mostly without inclusion formation; two cases have been described that revealed Lewy bodies and Lewy neurites in surviving midbrain cells, and one compound heterozygote PD patient showed tau-positive inclusions [13], thereby demonstrating that, as is the case in LRRK2-linked disease, more than one endophenotype likely exists for this PD variant. It has been suggested that full-length Parkin and truncated Parkin proteins (but not Parkin deficiency) may be essential in the formation of Lewy bodies in the brain [13]. No case of DJ-1-deficient PD pathology has been published, whereas the brains of four patients with heterozygous and one subject with compound heterozygous PINK1 mutations have come to autopsy; the latter revealed Lewy inclusion formation [39]. Importantly, the relatively restricted neuropathology of PARK2-linked PD, which is supported by the notion that even patients who have had disease for >10 years seem to lack the nervous system-wide degeneration encountered in SNCA-associated variants, may make this variant of PD a logical candidate for causative intervention in the future (see below).
Genetic diagnosis of Parkin-associated Parkinson disease
The diagnosis of PARK2-associated PD requires confirmation of disease-causing mutations in both alleles [34]. Many PD patients have been described that show an age of onset of >45 years but carry a mutation in only one PARK2 allele (ie, a heterozygous mutation) [40, 41]. The comprehensive search for compound heterozygous or homozygous mutation carrier status in affected individuals is complicated by three main factors: 1) the large size of the gene’s locus (> 1.5 megabases); 2) the high number of point mutation and deletion mutation rates described to date (> 100); and 3) the increasing detection of exon copy number variants [16]. Whether bona fide heterozygosity at the PARK2 locus by itself represents a risk factor for late-onset, complex PD in select individuals is an intriguing but hotly debated issue in the field, with compelling arguments provided both in favor of and against such a concept [19]. Nevertheless, this topic also bears great relevance with respect to possible targeted treatment intervention in the future (see below).
Current therapy of PARK2-linked autosomal recessive Parkinson disease
As of 2012, monogenic and complex PD, as well as most cases of secondary parkinsonism, remain frustratingly incurable. Because PARK2-linked recessive PD shows typical motor signs of complex PD, it may remain indistinguishable from the latter in individual patients for several years when judged on clinical grounds alone [13]. Furthermore, because it also features a strong and sustained response to pharmacological therapy with levodopa and dopamine agonists, the basic treatment regimen for Parkin-deficient PD does not significantly differ from that of other variants of typical PD. A series of comprehensive articles have been published in this journal on the therapy of PD. In lieu of restating their content, we specifically like to refer the reader to the informative reviews on PD’s initial treatment [1], its long-term medical management [2], the treatment of motor fluctuations in PD [3], the management of levodopa-induced dyskinesias [4], and a review of surgical options for patients with typical PD [5].
Furthermore, in late 2011, a panel of experts in the field of PD provided a comprehensive, evidence-based medicine review update on behalf of the International Movement Disorder Society. In it, Fox et al. [42] outlined the pharmacological treatment options for the motor symptoms of PD, including its monotherapy, for adjunct treatment to levodopa therapy, the delay of motor complications in PD, the treatment of motor fluctuations, their efficacy and side effect profile, as well as practice implications for all currently approved drugs that were available in at least one country (as of 2010). These authors also reviewed the range and indication of surgical interventions for motor control in PD treatment, the efficacy and safety of these procedures, their adjunct treatment effect on motor complications, the pros and cons surrounding surgical target selection, the range of non-motor side effects, as well as overall practice implications [42]. In parallel, Seppi et al. [43] have published an equally comprehensive evidence-based medicine review update on behalf of the same professional body on the treatment of non-motor symptoms in PD. Individual countries have followed suit in issuing related guidelines for the comprehensive care of their PD patients [44]. In addition to drawing attention to these authoritative review articles, which were aimed at the entire spectrum of PD variants, we wish to point out three issues that we deem relevant in the context of PARK2-linked PD. These include the occurrence and management of motor fluctuations in pharmacologically treated patients, the possible exploitation of the paradoxical sleep benefit, and the question of whether PARK2-linked PD patients show a superior response to surgical intervention. The vast majority of Parkin-deficient PD patients develop levodopa-induced dyskinesias (as well as an increase in often painful dystonia episodes) as a result of treatment, thereby reflecting neurological signs of unwanted motor fluctuations; these occur relatively early during the course of their illness (ie, >5 years after the start of levodopa therapy) and are often difficult to manage. They represent a source of ongoing distress, leading to anxiety, panic attacks, and mood dysregulation. The evidence-based guidelines for the prevention and delay of motor complications [42] are therefore of particular relevance to PARK2 PD patients. To avoid steep rises in dopamine concentrations in the striatum, which are thought to underlie the development of motor fluctuations down the road, levodopa alone (ie, without a peripheral decarboxylase inhibitor) is sometimes utilized in select cases. Furthermore, regularly scheduling and taking short rest periods, such as after lunchtime, is frequently recommended for those PARK2 PD patients who suffer from disrupted sleep architecture (as a result of neuronal loss in the locus coeruleus) and who experience a sleep benefit (ie, a reduction in severity of their symptoms after periods of sleep). This can enhance their performance level in activities of daily living during the second half of the day.
With respect to the question of any altered response to brain surgery, we wish to draw the reader’s attention to two studies that addressed this issue specifically. In the first, Lohmann et al. [45] assessed genotyped PD patients 12 to 24 months after bilateral deep brain stimulation surgery. There, patients carried either one PARK2 mutation (n = 7), two mutant alleles (n = 7), or had no evidence of any PARK2 mutation (n = 39) [45]. At follow-up, the authors found that the “levodopa equivalent daily dose” required for an optimal treatment effect was significantly lower in patients carrying two PARK2 mutations (in this small cohort after a relatively short observation period), which is consistent with reports in the literature that had previously suggested Parkin-deficient PD patients may require smaller daily doses of levodopa. However, the overall rate and severity of post-operative motor fluctuations in Parkin-deficient patients was similar to the other two groups. Intriguingly, PARK2 heterozygous patients did not fare any different from PD patients with wild-type PARK2 alleles [45]. Another group reported the effects of surgery in 11 affected PARK2 mutation carriers (six with homozygous, five with heterozygous PARK2 mutations), and in one patient with homozygous PINK1mutations, as measured by the Unified Parkinson Disease Rating Scale, 12 months after bilateral deep brain stimulation surgery. There, PD signs improved by only 34 % in patients with recessive PD compared to 56 % improvement in mutation-negative PD. However, at 3- to 6-year follow-up examinations, both groups showed highly similar outcomes regarding clinical improvement [46]. Therefore, given that the observation periods were shorter than 10 years, no superiority in post surgical (DBS) outcomes can be ascribed to PARK2 PD patients at this time.
In addition, physiotherapy remains an essential part of treatment in all PD patients in order to maintain and improve their motor performances [42]. Moreover, voice therapy, swallowing assessment, music therapy, and occupational interventions [43, 47] are of particular importance in Parkin-deficient patients given the fact that this variant of PD often affects employed professionals during young adulthood, is of long duration, and is frequently complicated by significant motor fluctuations.
Considerations concerning future therapeutic strategies in Parkin-deficient Parkinson disease
As described, currently available PD therapeutics offer symptomatic relief but do not address the root cause of the disease. Individuals with monogenic forms of Parkin-linked PD are unique in two specific aspects that make them perhaps ideal candidates to benefit from tailored, cause-directed therapies in the future: 1) the cause is known and is limited to a genetic event (ie, loss of Parkin protein), and 2) the neuropathology is restricted to two specific nuclei in the brainstem unlike idiopathic (complex) forms of the disease. Implementation of genetic screening practices to identify PD patients with young-onset disease due to a highly likely monogenic cause offers the possibility to identify and then recruit individuals with two mutant PARK2 alleles into future intervention trials [15]. These patients could uniquely benefit from therapies that specifically target the genetic mutation in PARK2, which underlies their disease, and/or downstream molecular targets of Parkin that are dysregulated in its absence. Some of these therapeutic approaches are highlighted below. As is true for all variants of PD, treatment options for three related indications are desperately needed: neuroprevention (to delay the onset in at-risk patients), neuroprotection (to protect remaining neurons upon diagnosis), and neurorestoration (to reverse neuronal loss) [48].
PARK2 gene replacement therapy
Given that loss-of-function mutations in Parkin cause nigral degeneration in recessive PD, virus-mediated PARK2 gene replacement (or repair) holds great promise as a cause-directed therapy; it potentially offers both neuroprotective and neuropreventive benefits. Proof-of-concept studies in animals have indeed shown that virus-mediated delivery of Parkin is protective in laboratory models of parkinsonism as explored in primates, mice, rats, and fruit flies [49]. Of note, there are currently four clinical trials underway that test the efficacy of specific virus-mediated delivery of a human gene (other than PARK2) to treat idiopathic (complex) PD [50]; collectively, the evidence provided to date suggests that virus-mediated delivery of certain genes may be a safe and potentially effective therapeutic option in the future. However, several considerations and limitations must first be addressed prior to the exploration of PARK2 delivery in humans: First, the suitability of the ideal PARK2 construct and the sustained duration of Parkin protein expression need to be determined. Fewer than 30 % of nigral neurons may remain in the corresponding hemi-midbrain at the time of symptom onset in the contralateral limb. Second, the stereotaxic approach to deliver the gene construct to the substantia nigra in the midbrain needs to be optimized to ensure maximum delivery efficiency and safety. As an alternative approach to delivery to the midbrain, investigators are trying to optimize gene delivery via the striatum to dopamine cell bodies in the substantia nigra by utilizing viral vehicles that undergo retrograde axonal transport. For example, an HIV-based vector pseudotyped with a rabies virus glycoprotein-encoding gene has been developed that provides highly efficient retrograde axonal transport to the midbrain in mice and monkeys [51]. Finally, current animal studies have been performed using PARK2 cDNA cassettes driven by an ectopic neuronal promoter. However, the long-term effects of high, unregulated levels of PARK2 cDNA expression in dopaminergic neurons remain unknown. In our opinion, a more suitable, potentially therapeutic PARK2 minigene would contain endogenous regulatory elements that modulate the expression of Parkin to near-homeostatic normal levels in remaining neurons, thus retaining the cellular regulatory mechanisms that govern its expression in the nigrostriatal pathway. Of note, the indication for Parkin-directed (gene) therapy may prove to extend beyond PARK2-linked variants of PD as over-expression of the wild-type protein has been shown to be neuroprotective in several animal models of nigral degeneration, including in SNCA- and tau-induced models of disease as well as in LRRK2-, Pael-R- and Pink1-dependent paradigms of neurodegeneration [50]. Similarly, patients with other variants of PD may benefit from small molecule therapeutics that specifically increase the expression of endogenous Parkin from one (or both) wild-type alleles. Drug screens to identify such molecules are actively being carried out in some laboratories.
Patient-derived induced pluripotent stem cell (iPSC)-based cell replacement strategies
New technologies to generate dopaminergic neurons from patient-derived induced pluripotent stem cells (iPSCs) offer hope for the possibility of future transplant-based neurorestorative therapies in PD. One could envision treating an individual with corrected neurons derived from their own source of primary cell, a form of “personalized medicine” that has gained much attention in many human disease models including PD. Of note, neuronal transplantation in PD has been extensively reviewed and will not be described in detail here [52, 53]. Of particular relevance to monogenic forms of PD is the recent demonstration by Soldner et al. [54] that it is possible to genetically “correct” mutations in iPSCs, thereby opening the possibility of one day transplanting PARK2-linked PD individuals with dopaminergic neurons derived from their fibroblasts that carry a repaired now wild-type PARK2 gene. When combining these molecular advances with steadily evolving progress in neurosurgical techniques, the concept of iPSC-based grafting emerges as an exciting platform to further explore for future applications. However, iPSC-based technologies are in their infancy and must be rigorously tested as well as optimized for their safety and efficacy in non-human primates before they can be moved to clinical trials. We would also caution that the occurrence of significant side effects observed in response to ventral fetal mesencephalic grafts, which were previously performed in PD patients and included “runaway” dyskinesias and painful dystonias [42], may possibly herald similar adverse events in iPSC-based experimental approaches; these unintended and difficult to manage side effects remain poorly understood as to their molecular causes.
Other pharmaceutical interventions
Dysregulated pathways and molecular targets downstream of loss-of-Parkin function represent key therapeutic targets that, if successfully engaged, offer both neuroprotective and neuropreventive opportunities. Although the essential function of Parkin activity in dopaminergic cells during aging remains to be determined, there is convincing in vivo and in vitro evidence that mitochondrial deficits occur downstream of loss-of-Parkin expression. These include, but are not limited to, dysregulated intracellular and mitochondrial calcium homeostasis, mitochondrial enzyme activity deficits, and altered mitochondrial membrane potential [55]. Improving mitochondrial integrity and their performance therefore represents a desirable therapeutic target. In accordance, calcium channel blockers including isradipine [56] and zonisamide [57] are now being actively investigated as potential PD drugs. Of note, zonisamide also inhibits quinoprotein formation and increases glutathione S-transferase levels in the striatum [58]. Likewise, the effects of coenzyme Q10, an antioxidant, which increases complex I activity, improves mitochondrial respiration, and protects against cell death in experimental paradigms from tissue culture models to laboratory animals of parkinsonism, have emerged as a potential target. Unfortunately, a recent placebo-controlled, randomized clinical trial examining various doses of coenzyme Q10 has revealed no convincing benefit in patients with idiopathic PD [59]. Importantly, one could argue that this drug still has the potential to benefit PARK2-linked patients, who we consider to suffer from an etiopathologically distinct variant of PD (see above; Fig. 1). A potential treatment benefit may not have been observed in this clinical trial, which was designed without an objective, molecular stratification-based selection process for any subtype of PD [14, 15].
Summary
Cause-directed therapy of one or more PD variants will likely arrive when three pre-requisites have been met: 1) the successful delineation of the molecular, step-by-step events that elicit the phenotype in one or more subtypes of PD (we postulate that the greatest chance for a breakthrough lies within the monogenic forms of recessive PD); 2) the definition of specific targets, (as informed by the rigorous analysis of patient pathology-derived concepts), which will translate into a greater likelihood of success in subsequent drug development efforts and in the preclinical validation of new compounds; and 3) the differentiation of clinically typical PD variants for stratification purposes that is based on the employment of objective molecular tools and biological markers (rather than on the reliance of subjective rating scales and neuroimaging modalities that remain relatively non-specific).
References and Recommended Reading
Tarsy D. Initial treatment of Parkinson’s disease. Curr Treat Options Neurol. 2006;8(3):224–35.
Bertoni JM, Prendes JL, Sprenkle P. Long-term Medical Treatment for Parkinson’s Disease. Curr Treat Options Neurol. 2001;3(6):495–506.
Hinson VK. Parkinson’s disease and motor fluctuations. Curr Treat Options Neurol. 2010;12(3):186–99.
Rao J. Treatment of Levodopa-induced Dyskinesia. Curr Treat Options Neurol. 2007;9(3):205–9.
Bronte-Stewart H. Parkinson’s Disease: Surgical Options. Curr Treat Options Neurol. 2003;5(2):131–47.
Wilkinson JR, Weintraub D, and Stern MB. Clinical Manifestations of Parkinson’s Disease. In: Watts RL, Standaert DG, and Obeso JA, editors. Movement Disorders, 3rd Edition. McGraw-Hill; 2012. p. 229–46.
Hughes AJ et al. JNNP 1992;55:181–184.
Bernheimer H, et al. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20(4):415–55.
Yamamura Y, et al. Paralysis agitans of early onset with marked diurnal fluctuation of symptoms. Neurology. 1973;23(3):239–44.
Braak H, et al. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110(5):517–36.
Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: the dual hit theory revisited. Ann N Y Acad Sci. 2009;1170:615–22.
Zimprich A, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–7.
Pramstaller PP, et al. Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. 2005;58(3):411–22.
Schlossmacher MG, Mollenhauer B. Biomarker research in Parkinson’s disease: objective measures needed for patient stratification in future cause-directed trials. Biomark Med. 2010;4(5):647–50.
Klein C, et al. Translational research in neurology and neuroscience 2011: movement disorders. Arch Neurol. 2011;68(6):709–16.
Klein C, Schlossmacher MG. The genetics of Parkinson disease: Implications for neurological care. Nat Clin Pract Neurol. 2006;2(3):136–46.
Marras C, and Tanner CM. Epidemiology of Parkinson’s disease. In: Watts RL, Standaert DG, and Obeso JA, editors. Movement Disorders, 3rd Edition, McGraw-Hill; 2012. p. 169–92.
Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–18.
Klein C, et al. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007;6(7):652–62.
Kordower JH, et al. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14(5):504–6.
Li JY, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14(5):501–3.
Mollenhauer B, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213(2):315–25.
Sidransky E, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651–61.
Sardi SP, et al. Mutant GBA1 expression and Synucleinopathy risk: First insights from cellular and mouse models. Neurodegener Dis, 2012.
Eblan MJ, Walker JM, Sidransky E. The glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2005;352(7):728–31. author reply 728–31.
Tomlinson JJ, Cullen V, and Schlossmacher MG. Identifying targets in alpha-Synuclein metabolism to treat Parkinson’s and related disorders, in protein misfolding diseases: Current and emerging principles and therapies, In: Ramirez-Alvaro M, Kelly JW, and Dobson CM, editors. Wiley & Sons 2010.
Lee BD, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med. 2010;16(9):998–1000.
Liu Z, et al. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol. 2011;12(11):1063–70.
Hakimi M, et al. Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm. 2011;118(5):795–808.
Moehle MS, et al. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci. 2012;32(5):1602–11.
Salat-Foix D, et al. Gastrointestinal symptoms in Parkinson disease: clinical aspects and management. Can J Neurol Sci. 2011;38(4):557–64.
Mutez E, et al. Transcriptional profile of Parkinson blood mononuclear cells with LRRK2 mutation. Neurobiol Aging. 2011;32(10):1839–48.
Nalls MA, et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377(9766):641–9.
Kitada T, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–8.
Shimura H, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25(3):302–5.
Narendra DP, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):e1000298.
Khan NL, et al. Parkin disease: a phenotypic study of a large case series. Brain. 2003;126(Pt 6):1279–92.
Lucking CB, et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342(21):1560–7.
Gandhi S, et al. PINK1 protein in normal human brain and Parkinson’s disease. Brain. 2006;129(Pt 7):1720–31.
Klein C, et al. Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: Expanding the phenotype. Ann Neurol. 2000;48(1):65–71.
Oliveira SA, et al. Parkin mutations and susceptibility alleles in late-onset Parkinson’s disease. Ann Neurol. 2003;53(5):624–9.
Fox SH, et al. The movement disorder society evidence-based medicine review update: Treatments for the motor symptoms of Parkinson’s disease. Mov Disord. 2011;26 Suppl 3:S2–41.
Seppi K, et al. The movement disorder society evidence-based medicine review update: Treatments for the non-motor symptoms of Parkinson’s disease. Mov Disord. 2011;26 Suppl 3:S42–80.
Grimes DA. Canadian guidelines on Parkinson’s disease (on behalf of the Canadian Parkinson’s Research Alliance). Can J Neurol Sci, 2012 (in press).
Lohmann E, et al. Are parkin patients particularly suited for deep-brain stimulation? Mov Disord. 2008;23(5):740–3.
Moro E, et al. Bilateral subthalamic stimulation in Parkin and PINK1 parkinsonism. Neurology. 2008;70(14):1186–91.
de Bruin N, et al. Walking with music is a safe and viable tool for gait training in Parkinson’s disease: the effect of a 13-week feasibility study on single and dual task walking. Parkinsons Dis. 2010;2010:483530.
Horstink M, et al. Review of the therapeutic management of Parkinson’s disease. Report of a joint task force of the European Federation of Neurological Societies and the Movement Disorder Society-European Section Part I: early (uncomplicated) Parkinson’s disease. Eur J Neurol. 2006;13(11):1170–85.
Ulusoy A, Kirik D. Can overexpression of parkin provide a novel strategy for neuroprotection in Parkinson’s disease? Exp Neurol. 2008;212(2):258–60.
Witt J, Marks Jr WJ. An update on gene therapy in Parkinson’s disease. Curr Neurol Neurosci Rep. 2011;11(4):362–70.
Kato S, et al. Efficient gene transfer via retrograde transport in rodent and primate brains using a human immunodeficiency virus type 1-based vector pseudotyped with rabies virus glycoprotein. Hum Gene Ther. 2007;18(11):1141–51.
Preynat-Seauve O, et al. Pluripotent stem cells as new drugs? The example of Parkinson’s disease. Int J Pharm. 2009;381(2):113–21.
Allan LE, Petit GH, Brundin P. Cell transplantation in Parkinson’s disease: problems and perspectives. Curr Opin Neurol. 2010;23(4):426–32.
Soldner F, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146(2):318–31.
Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008;7(1):97–109.
Chan CS, et al. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature. 2007;447(7148):1081–6.
Murata M. The discovery of an antiparkinsonian drug, zonisamide. Rinsho Shinkeigaku. 2010;50(11):780–2.
Murata M. Zonisamide: a new drug for Parkinson’s disease. Drugs Today (Barc). 2010;46(4):251–8.
Storch A, et al. Randomized, double-blind, placebo-controlled trial on symptomatic effects of coenzyme Q(10) in Parkinson disease. Arch Neurol. 2007;64(7):938–44.
Disclosure
T. Kitada: none. J.J. Tomlinson: none. H.S. Ao: none. D.A. Grimes: none. M.G. Schlossmacher: none. This work has been supported by the Ottawa Hospital Research Institute (to T.K.), Parkinson Society Canada (to J.J.T.), Parkinson Research Consortium Ottawa (H.S.A.), Department of Medicine at The Ottawa Hospital (D.A.G., M.G.S.), and the Government of Canada (M.G.S.).
Tohru Kitada and Julianna J. Tomlinson contributed equally to this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Tohru Kitada and Julianna J. Tomlinson contributed equally.
Rights and permissions
About this article
Cite this article
Kitada, T., Tomlinson, J.J., Ao, H.S. et al. Considerations Regarding the Etiology and Future Treatment of Autosomal Recessive Versus Idiopathic Parkinson Disease. Curr Treat Options Neurol 14, 230–240 (2012). https://doi.org/10.1007/s11940-012-0175-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11940-012-0175-8